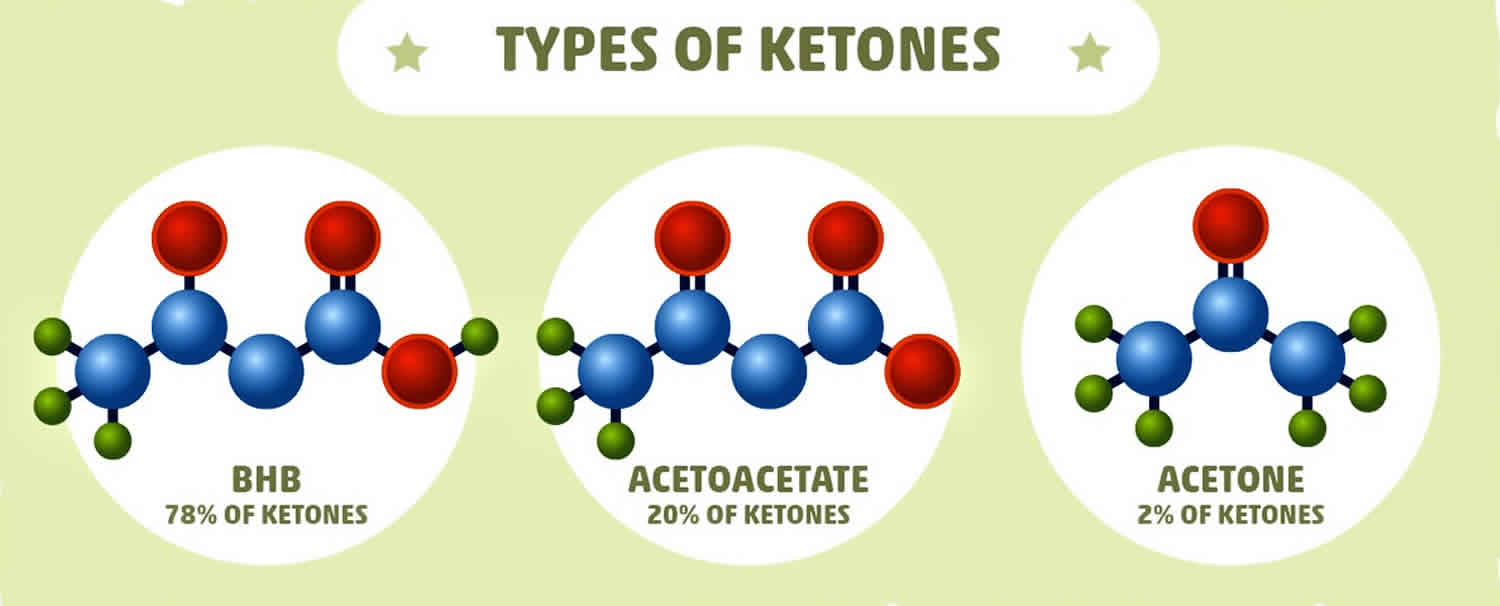
What is beta hydroxybutyrate
Beta-hydroxybutyrate (β-hydroxybutyrate or β-hydroxybutyric acid) is 1 of 3 sources of ketone bodies (acetoacetate, beta-hydroxybutyrate, and acetone). Beta-hydroxybutyrate (β-hydroxybutyrate) relative proportion in the blood (78%) is greater than the other 2 ketone bodies, acetoacetate (20%) and acetone (2%). During carbohydrate deprivation (starvation, digestive disturbances, frequent vomiting), alcoholism, decreased carbohydrate utilization (diabetes mellitus), diabetic ketoacidosis (DKA), glycogen storage diseases, high-fat, low-carbohydrate diets and alkalosis, acetoacetate production increases. The increase may exceed the metabolic capacity of the peripheral tissues. As acetoacetate accumulates in the blood, a small amount is converted to acetone by spontaneous decarboxylation. The remaining and greater portion of acetoacetate is converted to beta hydroxybutyrate (see Figure 1). Beta-hydroxybutyrate is the predominant ketone body present in severe diabetic ketoacidosis (DKA). Ketosis and ketoacidosis may be induced on purpose in some children with epilepsy who have frequent seizures and do not respond to available medications or other treatments.
Recent studies have shown that serum ketones and beta-hydroxybutyrate testing are both effective in diagnosing diabetic ketoacidosis (DKA). Some healthcare practitioners prefer beta-hydroxybutyrate. In acute diabetic ketoacidosis (DKA), the ketone body ratio (beta-hydroxybutyrate/acetoacetate ratio) rises from normal (1:1) to as high as 10:1. In response to insulin therapy, beta-hydroxybutyrate levels commonly decrease long before acetoacetate levels. However, beta-hydroxybutyrate is not available in all laboratories.
Diabetic ketoacidosis (DKA), is a severe life threatening complication of type 1 diabetes mellitus caused by an absolute or relative deficiency of insulin in the body that often requires intensive treatment 1. Diabetic ketoacidosis (DKA) is associated with sudden and severe high blood glucose (acute hyperglycemia), a severe insulin deficiency, and a disruption of the body’s acid-base balance. Excess ketones and glucose are dumped into the urine by the kidneys in an effort to flush them from the body. This causes increased urination, thirst, dehydration, and a loss of electrolytes. The affected person may also experience symptoms such as rapid breathing, shortness of breath, a fruity scent to the breath, nausea, vomiting, fatigue, confusion, and eventually coma. The intensity of treatment is therefore variable and depends on the severity of clinical signs and the degree of metabolic derangement. Most diabetic ketoacidosis (DKA) patients require intensive, in-hospital treatment.
Beta-hydroxybutyrate (β-hydroxybutyrate or β-hydroxybutyric acid) is a convenient carrier of energy from fat cells (adipocytes) to peripheral tissues during fasting or exercise 2. However, beta-hydroxybutyrate is more than just a metabolite, having important cellular signaling roles as well. Beta-hydroxybutyrate is an endogenous inhibitor of histone deacetylases (HDACs) and a ligand for at least two cell surface receptors and indirectly by altering the levels of other regulatory metabolites including acetyl-CoA, succinyl-CoA, and NAD+ 2. In addition, the downstream products of beta-hydroxybutyrate metabolism including acetyl-CoA, succinyl-CoA, and NAD+ (nicotinamide adenine dinucleotide) themselves have signaling activities. These regulatory functions of beta-hydroxybutyrate serve to link the outside environment to cellular function and gene expression, and have important implications for the pathogenesis and treatment of metabolic diseases including type 2 diabetes 2. Perhaps most intriguing is that beta-hydroxybutyrate is an endogenous inhibitor of histone deacetylases (HDACs) 3, joining a small but growing list of metabolic intermediaries that affect gene expression via chromatin modifications 4.
Histone deacetylase (HDAC) inhibition by beta-hydroxybutyrate might affect the pathogenesis of type 2 diabetes in at least two ways: through direct regulation of HDAC-dependent glucose metabolism, and by promoting resistance to oxidative stress 2. Studies in knockout mice have shown that class I HDACs have a key role in regulating metabolic disease. HDAC3 regulates expression of gluconeogenic genes 5 and HDAC3 knockout mice have reduced fasting glucose and insulin levels 6. In fact, chronic treatment with beta-hydroxybutyrate, a broad HDAC inhibitor that might be expected to phenocopy loss of function of HDAC3, keeps mice essentially metabolically normal on a high-fat diet 7. Butyrate treatment is associated with lower glucose and insulin levels, better glucose tolerance, prevention of weight gain, and improved respiratory efficiency; butyrate also provides some of these benefits even to mice already obese from being fed a high-fat diet 7. The functional consequences to the cell and organism of HDAC inhibition by beta-hydroxybutyrate are only just beginning to be cataloged. Given the widespread nature of gene regulation by HDACs, and the important role of HDACs in regulating dynamic changes of gene expression in response to stimuli, scientists may have only scratched the surface of the clinical and pathophysiological consequences of this signaling function of beta-hydroxybutyrate.
The ability of beta-hydroxybutyrate to regulate HDAC activity and thereby epigenetic gene regulation is particularly notable for potentially implicating a wide variety of genes as regulatory targets of beta-hydroxybutyrate 2. Beta-hydroxybutyrate is already known to induce resistance to oxidative stress via HDAC inhibition, and other HDAC inhibitors regulate gluconeogenesis 2. The unique effects of beta-hydroxybutyrate may help explain the therapeutic benefit of low-carbohydrate and ketogenic diets. However, teasing apart the specific role of beta-hydroxybutyrate in the effects of a ketogenic diet is a challenging task 2. Ketogenic diets inextricable combine reduced carbohydrate consumption, reduced glucose utilization, reliance on beta-oxidation of lipids for energy, reduced insulin signaling, and increased glucagon signaling, along with increased ketone body levels 8. New experimental tools are required to permit the safe manipulation of beta-hydroxybutyrate levels outside of the confines of a ketogenic diet in order to understand the full spectrum of action of beta-hydroxybutyrate in amelioration of metabolic disease 2.
The first step in beta-hydroxybutyrate utilization in peripheral tissues is its conversion to acetoacetate by mitochondrial β-hydroxybutyrate dehydrogenase (BDH1), which uses NAD as a cofactor and generates NADH. Next, acetoacetate is converted to acetoacetyl-CoA by succinyl-CoA:3-ketoacid coenzyme A transferase (OXCT1, also known as SCOT), which uses succinyl-CoA as a CoA donor, releasing free succinate. Acetoacetyl-CoA is then split into two acetyl-CoA that are burned in the TCA cycle to generate ATP in peripheral tissues. Utilization of beta-hydroxybutyrate therefore might increases acetyl-CoA levels, decrease succinyl-CoA levels, and alter the NAD/NADH ratio in peripheral tissues such as muscle.
Increasing the intracellular pools of acetyl-CoA should indirectly increase protein acetylation. This effect is complementary to HDAC inhibition by beta-hydroxybutyrate, but may have broader effects in multiple cellular compartments. Increasing the acetyl-CoA substrate for both enzymatic and non-enzymatic protein acetylation should drive these reaction equilibria towards acetylation. This may particularly increase protein acetylation in mitochondria. For example, CR, fasting and high-fat diets, states associated with increased lipid utilization and therefore high acetyl-CoA throughput, all cause increased mitochondrial protein acetylation—even though neither acetyltransferases nor the HDACs that are inhibited by beta-hydroxybutyrate are known to enter mitochondria 9. Acetylation – and deacetylation by SIRT3 – is very widespread in mitochondria 10 and regulates the function of several mitochondrial enzymes including HMGCS2 11 and the long-chain acyl-CoA dehydrogenase (LCAD) 12. Increased acetyl-CoA pools also affect nuclear protein acetylation. Mitochondrial acetylcarnitine is known to be a source of acetyl-CoA for histone acetylation 13. Export of acetyl-CoA from the mitochondria is accomplished via a citrate shuttle mediated by citrate synthase inside mitochondria and ATP citrate lyase in the cytoplasm 14. ATP citrate lyase is a key enzyme in fatty acid biosynthesis, but its role in facilitating acetyl-CoA export from mitochondria is also required for the increase in histone acetylation that occurs with growth factor stimulation 14. An alternative pathway for acetyl-CoA export from mitochondria is via the enzymes carnitine acetyltransferase (CAT) and carnitine/acylcarnitine translocase 15. Indeed, a muscle-specific knockout of CAT in mice compromises glucose tolerance and decreases metabolic flexibility 15, demonstrating the importance of intracellular acetyl-CoA transport to overall metabolic health.
Consumption of succinyl-CoA by OXCT1 during utilization of beta-hydroxybutyrate in peripheral tissues may also have broad regulatory consequences on cellular metabolism. Lysine succinylation is very widespread in mitochondria 16, and present across diverse organisms 17. A substantial fraction of these succinylation sites are regulated by the mitochondrial desuccinylase, the sirtuin SIRT5 16. HMGCS2 has long known to be succinylated, a modification that reduces its activity 18; HMGCS2 activity in the liver is restored by desuccinylation by SIRT5. The mechanism of lysine succinylation is not clearly understood; an enzymatic succinyl-transferase is not known to exist in mammalian cells, and as both liver succinyl-CoA abundance and succinylation of HMGCS2 are reduced after treatment of rats with glucagon 18, it is possible that succinylation is primarily a non-enzymatic process dependent upon local concentrations of succinyl-CoA. HMGCS2 is not expressed in peripheral tissues, which do not engage in ketogenesis in any event; but the enzymes in many key mitochondrial pathways are heavily succinylated and regulated by SIRT5, including fatty acid oxidation, branched-chain amino acid catabolism, and the TCA cycle. By analogy with acetylation (and the effect of succinylation on HMGCS2), these pathways may be activated by a reduction in succinylation. Consumption of succinyl-CoA during beta-hydroxybutyrate utilization and consequent reduction in mitochondrial protein succinylation may therefore regulate many of these crucial mitochondrial pathways in peripheral tissues.
Cellular NAD balance is emerging as a crucial mediator of metabolic disease and aging 19. NAD utilization by beta-hydroxybutyrate differs from that of glucose in two important respects. First, beta-hydroxybutyrate consumes fewer NAD+ per acetyl-CoA produced. Metabolism of one molecule of glucose to two molecules of acetyl-CoA involves conversion of four molecules of NAD+ into NADH. Two of these are converted in the cytosol during glycolysis, and two in the mitochondrion by pyruvate decarboxylase. The cytosolic NADH are shuttled into mitochondria, potentially leading to depletion of the cytoplasmic NAD pool with high glucose utilization. By contrast, metabolism of one beta-hydroxybutyrate molecule into the same two molecules of acetyl-CoA involves conversion of only two molecules of NAD+ into NADH, both in the mitochondrion by BDH1 and thereby preserving the cytoplasmic NAD pool 20. The cytoplasmic and mitochondrial NAD pools are relatively distinct, so the preservation of cytoplasmic NAD+ by beta-hydroxybutyrate may have important cellular effects. NAD+ is a cofactor for sirtuin deacylases (such as nuclear/cytoplasmic SIRT1) as well as poly-ADP-ribose polymerase (PARP) 19. Consumption of NAD+ by PARP or overproduction of NADH may promote age-related diseases by inhibiting the activity of sirtuins 21. Conversely, repletion of NAD+ by exogenous feeding with nicotinadmide mononucleotide improves glucose tolerance in both high-fat diet-fed and aged mice 22. The relative sparing of NAD+ by utilization of beta-hydroxybutyrate vis a vis glucose may therefore have important consequences for metabolic diseases and diabetes.
Figure 1. Beta hydroxybutyrate production
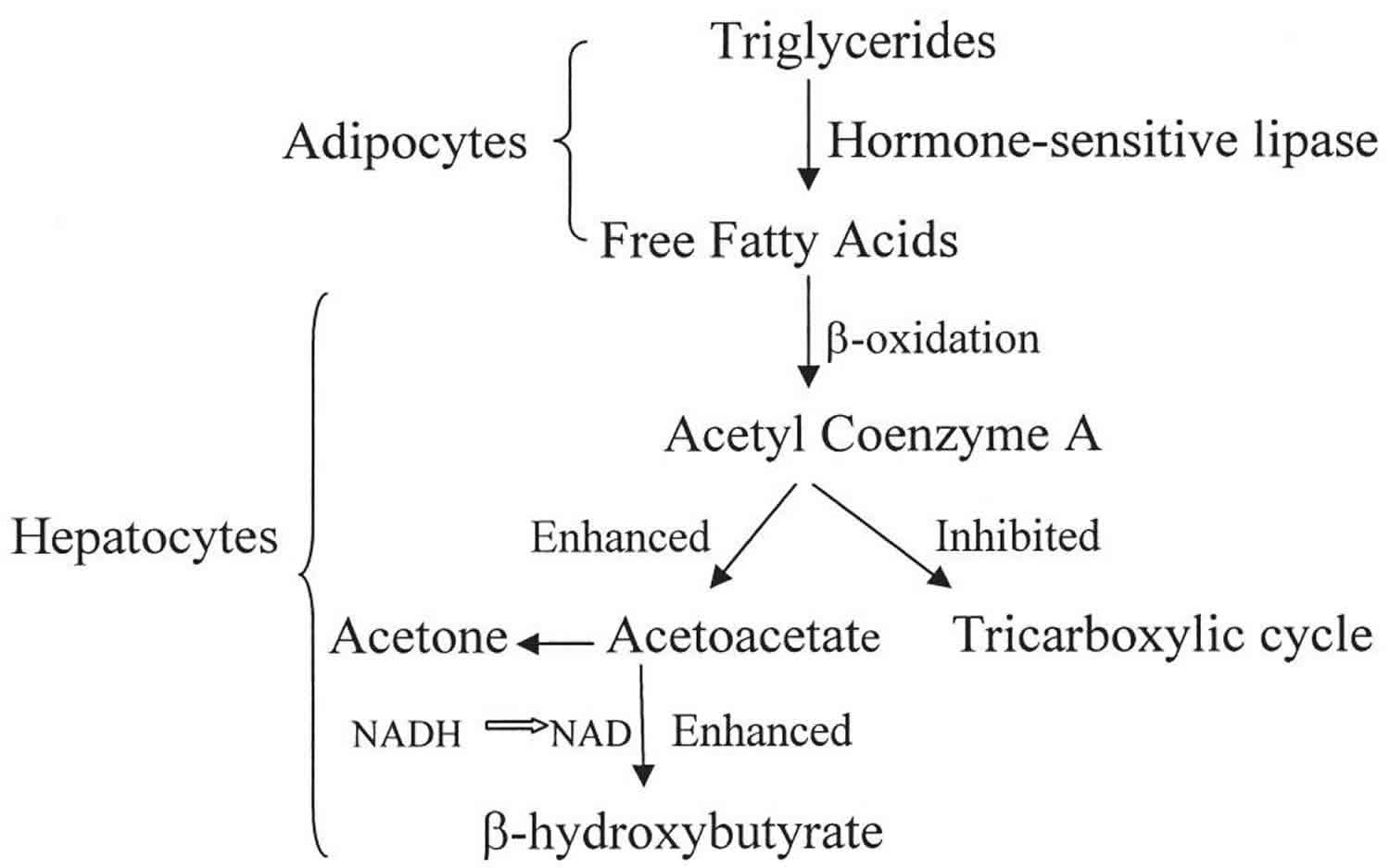
Figure 2. Beta hydroxybutyrate formation from acetyl-CoA (acetyl coenzyme A).
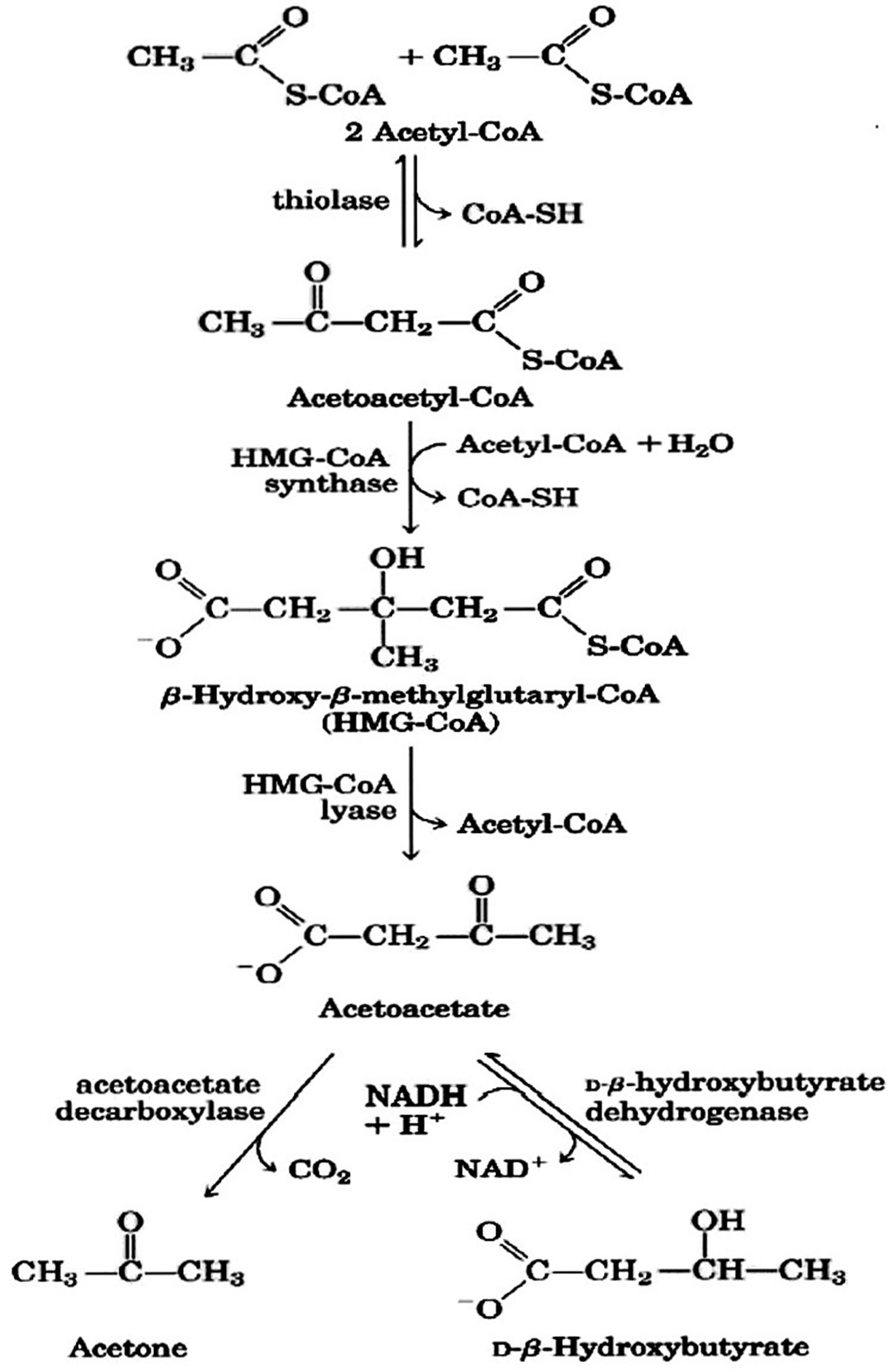
Footnote: Ketone bodies are used by tissues as a source of energy through a pathway that involves firstly that beta hydroxybutyrate (β-hydroxybutyric acid) is converted back to acetoacetate (AcAc) this is then transformed into acetoacetyl-CoA and, finally, two molecules of acetyl-CoA are formed from acetoacetyl-CoA which are used in the Krebs cycle.
Metabolism of beta-hydroxybutyrate increases cellular levels of acetyl-CoA (acetyl coenzyme A) and reduces levels of succinyl-CoA and NAD+. These secondary effects can further increase mitochondrial protein acetylation and reduce mitochondrial protein succinylation, potentially regulating the function of many metabolic enzymes. The relative sparing of cytoplasmic NAD levels with utilization of beta-hydroxybutyrate rather than glucose can alter the activity of NAD-dependent enzymes such as sirtuins. Finally, acetyl-CoA generated in mitochondria can be transported into the nucleus via the citrate shuttle to serve as substrate for histone acetyltransferases, a secondary mechanism by which beta-hydroxybutyrate might increase histone acetylation and alter gene expression.
[Source 23 ]Figure 3. Metabolic pathway of ketosis and tissues ketolysis.
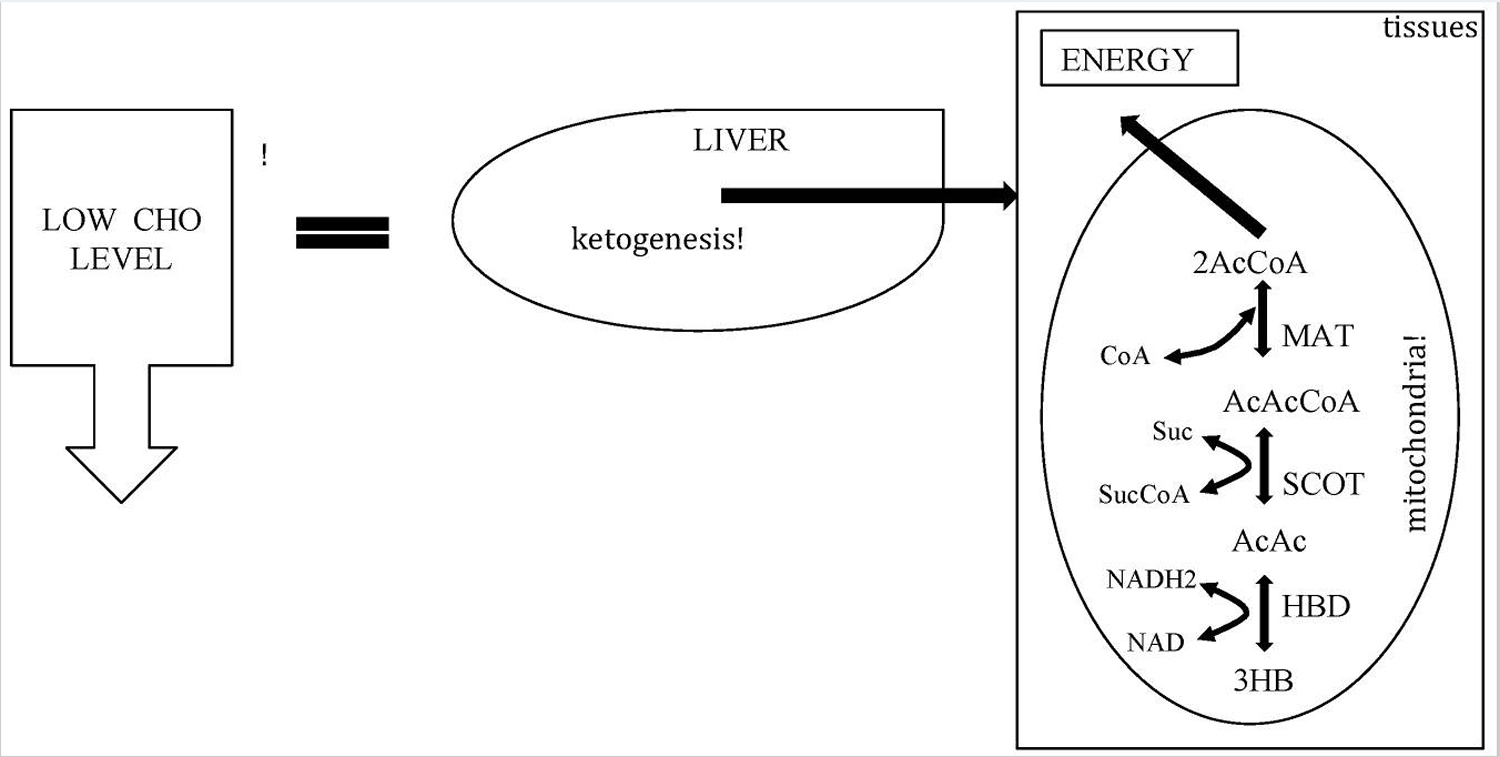
Footnote: CHO = carbohydrate.
[Source 23 ]Beta-hydroxybutyrate receptors
Many G-protein coupled receptors (GPCRs) bind to fatty acid ligands, and have important roles in metabolism and metabolic disease 24. At least two GPCRs that bind short-chain fatty acids also bind beta-hydroxybutyrate. HCAR2 (also known as HCA2, PUMA-G, or Gpr109), is a Gi/o-coupled GPCR that was first identified as a nicotinic acid receptor 25. Recently HCAR2 was shown to bind and be activated by beta-hydroxybutyrate 26. HCAR2 activation by beta-hydroxybutyrate (or other ligands) reduces lipolysis in adipocytes 27. For beta-hydroxybutyrate, this might perhaps represent a feedback mechanism to regulate availability of the fatty acid precursors of ketone body metabolism. However, elevated free fatty acids in plasma from dysregulated adipocytes are thought to contribute to insulin resistance through a variety of mechanisms including proinflammatory cytokines, oxidative stress, and ER stress 28. Pharmacological agonists of HCAR2 reduce both plasma free fatty acids and plasma glucose in humans with type 2 diabetes 29. HCAR2 also mediates the antiatherosclerotic effects of nicotinic acid in mouse models 30, probably through modulation of macrophage function 31. Thus, activation of HCAR2 by beta-hydroxybutyrate may contribute to improved glucose control as well as ameliorating some of the macrovascular complications of type 2 diabetes.
Beta-hydroxybutyrate is also a ligand for the free fatty acid receptor 3 (FFAR3, also known as GPR41). FFAR3 is another Gi/o-protein coupled receptor that is highly expressed in sympathetic ganglions. FFAR3 knockout mice have reduced basal oxygen consumption and body temperature, but are then insensitive to the usual further sympathetic depression seen during prolonged fasting 32. Antagonism of FFAR3 by beta-hydroxybutyrate suppresses sympathetic tone and heart rate, and may be responsible for sympathetic depression during fasting 32. However, an electrophysiological study in rats later reported that beta-hydroxybutyrate acts as an agonist of FFAR3 33. The physiological details of this adrenergic modulation by beta-hydroxybutyrate, and its effects on glucose homeostasis or cardiovascular disease, remain to be determined.
Ketone bodies
Ketones or ketone bodies are intermediate products of fat metabolism. Ketones are produced when glucose is not available to the body’s cells as an energy source and/or when the body cannot use glucose as a fuel source because there is no insulin or not enough insulin. Ketones can form when a person does not eat enough carbohydrates (for example, in cases of fasting, starvation, high-protein diets, high-fat and low carb diet [e.g., ketogenic diet, Atkin’s diet]) or when a person’s body cannot use carbohydrates properly (e.g., type 1 diabetes, some type 2 diabetes). When carbohydrates are not available, the body metabolizes fat instead to get the energy it needs to keep functioning. When fat is metabolized, byproducts called ketone bodies build up in the blood, causing first ketosis and then progressing to ketoacidosis, a form of metabolic acidosis. Diabetic ketoacidosis (DKA) is most frequently seen with uncontrolled type 1 diabetes and can be a medical emergency. Moreover, in non-diabetics, strenuous exercise, exposure to cold, frequent, prolonged vomiting, and several digestive system diseases can also increase fat metabolism, resulting in ketosis and ketones in urine (ketonuria).
There are three ketone bodies:
- Acetoacetate,
- Beta-hydroxybutyrate, and
- Acetone.
Acetoacetate (acetoacetic acid) is created first. Beta-hydroxybutyrate is created from acetoacetate. Acetone is a spontaneously created side product of acetoacetate. Under normal conditions of adequate dietary carbohydrate, the production of free acetoacetic acid is negligible (<0.3 mmol/l) and this compound, transported via the blood stream, is rapidly metabolized by various tissues, especially the skeletal and heart muscles. In conditions of overproduction of acetoacetic acid, it accumulates above normal levels and part of it is converted to the other two ketone bodies leading to ketonemia and ketonuria (presence of ketone bodies in the blood and urine). Beta-hydroxybutyrate is the predominant ketone body present in severe diabetic ketoacidosis (DKA). Different ketone tests measure one or more ketone bodies, and their results are not interchangeable.
Acetone (produced by spontaneous decarboxylation of acetoacetate), being a very volatile compound, is eliminated mainly via respiration in the lungs, hence the characteristic sweet “fruity breath” breath odour of ketosis is caused by acetone, which, being a very volatile compound, is eliminated mainly via respiration in the lungs. Acetone even though it does not have metabolic functions, its presence can be useful from a clinical diagnostic point of view. Thus it is to be considered that a “fruity breath” indicates a condition of ketosis that could be physiological (fasting, low carbohydrate diet, post exercise) 34.
Ketone bodies (primarily beta hydroxybutyrate and acetoacetic acid) are strong acids 35. They dissociate freely and produce a large amount of hydrogen ions 36. This overproduction of hydrogen ions overwhelms the buffering capacity of the body, quickly leading to metabolic acidosis 35. All of the described changes in metabolism contribute to the ongoing state of hyperglycemia, glucosuria, osmotic diuresis, electrolyte changes, ketosis, and acidosis which are seen with diabetic ketoacidosis (DKA) 35.
Blood ketones are primarily used to screen for, detect, and monitor a serious, sometimes life-threatening condition called diabetic ketoacidosis (diabetic ketoacidosis (DKA)) in people with type 1 and sometimes type 2 diabetes. diabetic ketoacidosis (DKA) can occur when a diabetic’s blood glucose is significantly increased, with illness, severe infection, pregnancy, and a variety of other conditions.
Beta hydroxybutyrate test
Different ketone tests measure one or more ketone bodies, and their results are not interchangeable. Ketone testing can be carried out at home. The most accurate way of testing for ketones is to use a blood glucose meter which can test for ketones as well as blood glucose levels. You can also test urine for ketone levels, however, the testing of urine means that the level you get is representative of your ketone levels up to a few hours ago.
- Blood ketone testing gives a snapshot of the status of ketone accumulation at the time that the sample was collected. Blood ketones may be measured in a laboratory or with a handheld monitor.
- The laboratory test uses serum, the liquid portion of the blood, and typically measures acetoacetate. Beta-hydroxybutyrate can be ordered as a separate blood test.
- When whole blood from a fingerstick is tested for ketones using a handheld monitor, the monitor measures beta-hydroxybutyrate. This test may be performed at a person’s bedside in a hospital or emergency room, in a doctor’s office, or performed by a person at home.
- Dipstick serum ketone determination using nitroprusside reagent is often used to estimate ketone body status, but that method has inherent problems. The dipstick does not measure beta-hydroxybutyrate, the most abundant of the physiological ketone bodies; the nitroprusside reagent only reacts with acetoacetate and acetone.
- Urine ketone testing reflects recent rather than current blood ketones. Urine testing is much more common than blood ketones testing. It may be performed by itself, with a urine glucose test, or as part of a urinalysis. The urine methods measure either acetoacetate or acetoacetate and acetone but do not usually detect beta-hydroxybutyrate.
Blood beta-hydroxybutyrate is primarily used to investigate the differential diagnosis of any patient presenting to the emergency room with hypoglycemia, acidosis, suspected alcohol ingestion, or an unexplained increase in the anion gap. Blood ketones are sometimes ordered, along with other tests such as blood gases, glucose, and electrolytes, to detect ketoacidosis in non-diabetics if they have signs and symptoms of diabetic ketoacidosis (DKA) due to, for example, ingestion of excessive amounts of alcohol.
Some signs and symptoms of ketoacidosis include:
- Increased urination, excessive thirst
- Dehydration, loss of electrolytes
- Rapid breathing, shortness of breath
- Fruity scent to the breath
- Nausea, vomiting
- Fatigue
- Confusion
- Coma (sometimes)
In non-diabetics, blood ketones are usually ordered when a person has symptoms associated with ketosis or ketoacidosis.
Monitoring of beta hydroxybutyrate may be helpful in assessing the efficacy of diabetic ketoacidosis (DKA) treatment. In order for peripheral tissues to utilize ketones as energy, insulin has to be present and beta hydroxybutyrate has to be converted to acetoacetic acid 37. Therefore, as diabetic ketoacidosis (DKA) is resolving, the concentration of beta hydroxybutyrate will decrease and the concentration of acetoacetic acid will increase 37.
Blood beta-hydroxybutyrate is also used to investigate pediatric patients, the presence or absence of ketonemia or ketones in urine (ketonuria) is an essential component in the differential diagnosis of inborn errors of metabolism
Serum beta-hydroxybutyrate is a key parameter monitored during controlled 24-hour fasts
Beta hydroxybutyrate levels
Beta hydroxybutyrate normal serum level <0.4 mmol/L
Serum beta hydroxybutyrate increases in response to fasting, but should not exceed 0.4 mmol/L following an overnight fast (up to 12 hours). If beta hydroxybutyrate levels are low or normal, then the person either does not have excess ketone production or the ketone body that is elevated is not being detected by the test method used.
If beta hydroxybutyrate levels are increased, then the person has some degree of ketosis or ketoacidosis.
The beta-hydroxybutyrate/acetoacetate ratio is typically between 3:1 and 7:1 in severe ketotic states.
In pediatric patients, a hypo- or hyper-ketotic state (with or without hypoglycemia) may suggest specific groups of metabolic disorders.
Can I test for ketones in my urine instead of my blood?
In many cases, yes, urine is tested much more frequently than blood. However, since it will not detect beta-hydroxybutyrate, the main ketone body with diabetic ketoacidosis (DKA), your healthcare provider may prefer that you monitor your blood.
Can I get diabetic ketoacidosis if I have type 2 diabetes?
Yes, although it is not as common as in type 1 diabetes. It may occur in type 2 diabetes, especially when you have a severe infection or illness. Ketosis and ketoacidosis may also be seen in non-diabetics, people with starvation, alcoholism, and with high-fat, low-carbohydrate diets. It may be induced on purpose in some children with epilepsy who have frequent seizures and do not respond to available medications or other treatments.
Beta hydroxybutyrate supplement
It has been demonstrated in animal—and/or human studies—that ketogenic diets and supplements may have metabolism-based therapeutic potential in the treatment of several diseases, such as Alzheimer’s disease 38, Parkinson’s disease 39, glucose transporter type 1-deficiency syndrome 40, amyotrophic lateral sclerosis 39, cancer 41, epilepsy 42, schizophrenia 43, anxiety 44, autism spectrum disorder 45, and depression 46.
However, there is limited evidence to support the beneficial effects of exogenous ketone supplements in psychiatric diseases at the moment 44, but the use of exogenous ketone supplements may be a viable alternative or adjuvant to pharmacotherapy in the treatment of these disorders 47.
In contrast to diabetic ketosis, which can induce pathological levels of blood beta hydroxybutyrate (ranging >25 mM) and potentially lead to life-threatening acidosis, nutritional ketosis elevates blood beta hydroxybutyrate from the normal range (0.1–0.2 mM) to a safe and—in many cases—therapeutic range (1–7 mM: therapeutic ketosis) 48. While rigorous adherence to ketogenic diets is typically difficult to follow and requires clear medical guidance and strong motivation, consumption of exogenous ketogenic agents effectively induces ketosis with little difficulty 49. Moreover, prolonged consumption of ketogenic diets may generate side effects, such as weight loss, alteration of mentation, growth retardation, nephrolithiasis, nausea, constipation, gastritis, hyperlipidemia, hypoglycemia, hyperuricemia, and ulcerative colitis 50. Consequently, developing a safer alternative method using ketone body precursors and exogenous ketone supplements, such as ketone salts (Na+/K+-beta hydroxybutyrate mineral salt) or ketone esters (R,S-1,3-butanediol—acetoacetate diester), to circumvent dietary restriction is appealing.
Recent research has demonstrated that it is possible to rapidly increase and maintain blood levels of ketone bodies in a dose-dependent manner in both animals and humans 51 for the treatment of several CNS diseases 52. Thus, it is possible that exogenous ketone supplementation-induced ketosis may be an effective therapeutic tool against psychiatric diseases. Indeed, exogenous ketone supplements have a modulatory influence on behavior and anxiolytic effect in animal studies 52. Moreover, in contrast to ketogenic diets, exogenous ketone supplements are relatively well-tolerated and can be formulated and titrated to minimize or avoid side effects 53, 54, 55.
Beta hydroxybutyrate side effects
Exogenous ketone supplements are relatively well-tolerated and can be formulated and titrated to minimize or avoid side effects 53, 54, 55. Moreover, it was demonstrated that a proper dose of ketone salt alone (Na+/K+-beta hydroxybutyrate mineral salt) 53 or in combination with other exogenous ketone supplements, such as ketone ester (R,S-1,3-butanediol—acetoacetate diester) and medium chain triglyceride (KEKS and KSMCT, respectively), may be a safe and efficacious way to achieve ketosis 53. Thus, exogenous ketone supplements may be an effective alternative to ketogenic diets for therapeutic ketosis.
- Feldman EC, Nelson RW. Canine and Feline Endocrinology and Reproduction. 3rd ed. St. Louis, Missouri: Elsevier; 2004. pp. 581–615[↩]
- Newman JC, Verdin E. β-hydroxybutyrate: much more than a metabolite. Diabetes Res Clin Pract. 2014;106(2):173–181. doi:10.1016/j.diabres.2014.08.009 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4414487[↩][↩][↩][↩][↩][↩][↩][↩]
- Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Science. 2013 Jan 11; 339(6116):211-4.[↩]
- The nexus of chromatin regulation and intermediary metabolism. Gut P, Verdin E. Nature. 2013 Oct 24; 502(7472):489-98.[↩]
- Mihaylova MM, Vasquez DS, Ravnskjaer K, Denechaud PD, Yu RT, Alvarez JG, et al. Class IIa histone deacetylases are hormone-activated regulators of FOXO and mammalian glucose homeostasis. Cell. 2011;145:607–21[↩]
- Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell. 2010;18:436–47.[↩]
- Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, et al. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58:1509–17[↩][↩]
- Kim do Y, Rho JM. The ketogenic diet and epilepsy. Curr Opin Clin Nutr Metab Care. 2008;11:113–20.[↩]
- He W, Newman JC, Wang MZ, Ho L, Verdin E. Mitochondrial sirtuins: regulators of protein acylation and metabolism. Trends Endocrinol Metab. 2012;23:467–76[↩]
- Rardin MJ, Newman JC, Held JM, Cusack MP, Sorensen DJ, Li B, et al. Label-free quantitative proteomics of the lysine acetylome in mitochondria identifies substrates of SIRT3 in metabolic pathways. Proceedings of the National Academy of Sciences. 2013[↩]
- Shimazu T, Hirschey MD, Hua L, Dittenhafer-Reed KE, Schwer B, Lombard DB, et al. SIRT3 deacetylates mitochondrial 3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body production. Cell Metab. 2010;12:654–61[↩]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature. 2010;464:121–5.[↩]
- Madiraju P, Pande SV, Prentki M, Madiraju SR. Mitochondrial acetylcarnitine provides acetyl groups for nuclear histone acetylation. Epigenetics : official journal of the DNA Methylation Society. 2009;4:399–403.[↩]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–80[↩][↩]
- Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–77[↩][↩]
- Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, et al. SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 2013;18:920–33.[↩][↩]
- Weinert BT, Scholz C, Wagner SA, Iesmantavicius V, Su D, Daniel JA, et al. Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell reports. 2013;4:842–51[↩]
- Quant PA, Tubbs PK, Brand MD. Glucagon activates mitochondrial 3-hydroxy-3-methylglutaryl-CoA synthase in vivo by decreasing the extent of succinylation of the enzyme. Eur J Biochem. 1990;187:169–74[↩][↩]
- Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–8[↩][↩]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 7. New York: W.H. Freeman; 2012[↩]
- Imai SI, Guarente L. NAD and sirtuins in aging and disease. Trends in cell biology. 2014[↩]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–36.[↩]
- Paoli A. Ketogenic Diet for Obesity: Friend or Foe? International Journal of Environmental Research and Public Health. 2014;11(2):2092-2107. doi:10.3390/ijerph110202092. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3945587/[↩][↩]
- Layden BT, Angueira AR, Brodsky M, Durai V, Lowe WL., Jr Short chain fatty acids and their receptors: new metabolic targets. Transl Res. 2013;161:131–40.[↩]
- Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med. 2003;9:352–5[↩]
- Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem. 2005;280:26649–52.[↩]
- Offermanns S, Colletti SL, Lovenberg TW, Semple G, Wise A, AP IJ. International Union of Basic and Clinical Pharmacology. LXXXII: Nomenclature and Classification of Hydroxy-carboxylic Acid Receptors (GPR81, GPR109A, and GPR109B) Pharmacol Rev. 2011;63:269–90.[↩]
- Boden G. Obesity, insulin resistance and free fatty acids. Current opinion in endocrinology, diabetes, and obesity. 2011;18:139–43[↩]
- Dobbins RL, Shearn SP, Byerly RL, Gao FF, Mahar KM, Napolitano A, et al. GSK256073, a selective agonist of G-protein coupled receptor 109A (GPR109A) reduces serum glucose in subjects with type 2 diabetes mellitus. Diabetes, obesity & metabolism. 2013;15:1013–21[↩]
- Lukasova M, Malaval C, Gille A, Kero J, Offermanns S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J Clin Invest. 2011;121:1163–73[↩]
- Zandi-Nejad K, Takakura A, Jurewicz M, Chandraker AK, Offermanns S, Mount D, et al. The role of HCA2 (GPR109A) in regulating macrophage function. Faseb J. 2013;27:4366–74[↩]
- Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41) Proc Natl Acad Sci U S A. 2011;108:8030–5[↩][↩]
- Won YJ, Lu VB, Puhl HL, 3rd, Ikeda SR. beta-Hydroxybutyrate modulates N-type calcium channels in rat sympathetic neurons by acting as an agonist for the G-protein-coupled receptor FFA3. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:19314–25[↩]
- Paoli A, Rubini A, Volek JS, Grimaldi KA. Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets. European Journal of Clinical Nutrition. 2013;67(8):789-796. doi:10.1038/ejcn.2013.116. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3826507/[↩]
- Guyton AC, Hall JE. Textbook of Medical Physiology. 11th ed. St. Louis, Missouri: Elsevier; 2006. pp. 840–976.[↩][↩][↩]
- Kerl ME. Diabetic ketoacidosis: Pathophysiology and clinical laboratory presentation. Compend Contin Educ Pract Vet. 2001;23:220–229.[↩]
- Stojanovic V, Ihle S. Role of beta-hydroxybutyric acid in diabetic ketoacidosis: a review. Can Vet J. 2011;52(4):426–430. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3058661[↩][↩]
- Broom GM, Shaw IC, Rucklidge JJ. The ketogenic diet as a potential treatment and prevention strategy for Alzheimer’s disease. Nutrition (2019) 60:118–21. 10.1016/j.nut.2018.10.003[↩]
- Veyrat-Durebex C, Reynier P, Procaccio V, Hergesheimer R, Corcia P, Andres CR, et al. How can a ketogenic diet improve motor function? Front Mol Neurosci (2018) 11:15. 10.3389/fnmol.2018.00015[↩][↩]
- Bekker YAC, Lambrechts DA, Verhoeven JS, van Boxtel J, Troost C, Kamsteeg EJ, et al. Failure of ketogenic diet therapy in GLUT1 deficiency syndrome. Eur J Paediatr Neurol (2019) (in press). 10.1016/j.ejpn.2019.02.012[↩]
- Sremanakova J, Sowerbutts AM, Burden S. A systematic review of the use of ketogenic diets in adult patients with cancer. J Hum Nutr Diet (2018) 31(6):793–802. 10.1111/jhn.12587[↩]
- Kovács Z, D’Agostino DP, Dobolyi A, Ari C. Adenosine A1 receptor antagonism abolished the anti-seizure effects of exogenous ketone supplementation in Wistar Albino Glaxo Rijswijk rats. Front Mol Neurosci (2017) 10:235. 10.3389/fnmol.2017.00235[↩]
- Kraeuter AK, van den Buuse M, Sarnyai Z. Ketogenic diet prevents impaired prepulse inhibition of startle in an acute NMDA receptor hypofunction model of schizophrenia. Schizophr Res (2018) 206:244–500. 10.1016/j.schres.2018.11.011[↩]
- Hollis F, Mitchell ES, Canto C, Wang D, Sandi C. Medium chain triglyceride diet reduces anxiety-like behaviors and enhances social competitiveness in rats. Neuropharmacology (2018) 138:245–56. 10.1016/j.neuropharm.2018.06.017[↩][↩]
- Ruskin DN, Murphy MI, Slade SL, Masino SA. Ketogenic diet improves behaviors in a maternal immune activation model of autism spectrum disorder. PLoS One (2017. b) 12(2):e0171643. 10.1371/journal.pone.0171643[↩]
- Brietzke E, Mansur RB, Subramaniapillai M, Balanzá-Martínez V, Vinberg M, González-Pinto A, et al. Ketogenic diet as a metabolic therapy for mood disorders: evidence and developments. Neurosci Biobehav Rev (2018) 94:11–6. 10.1016/j.neubiorev.2018.07.020[↩]
- Kovács Z, D’Agostino DP, Diamond D, Kindy MS, Rogers C, Ari C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front Psychiatry. 2019;10:363. Published 2019 May 23. doi:10.3389/fpsyt.2019.00363 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6543248[↩]
- Branco AF, Ferreira A, Simões RF, Magalhães-Novais S, Zehowski C, Cope E, et al. Ketogenic diets: from cancer to mitochondrial diseases and beyond. Eur J Clin Invest (2016) 46(3):285–98. 10.1111/eci.12591[↩]
- Poff AM, Ward N, Seyfried TN, Arnold P, D’Agostino DP. Non-toxic metabolic management of metastatic cancer in VM mice: novel combination of ketogenic diet, ketone supplementation, and hyperbaric oxygen therapy. PLoS One (2015) 10:e0127407. 10.1371/journal.pone.0127407[↩]
- Bostock EC, Kirkby KC, Taylor BV. The current status of the ketogenic diet in psychiatry. Front Psychiatry (2017) 8:43. 10.3389/fpsyt.2017.00043[↩]
- Stubbs BJ, Cox PJ, Evans RD, Santer P, Miller JJ, Faull OK, et al. On the metabolism of exogenous ketones in humans. Front Physiol (2017) 8:848. 10.3389/fphys.2017.00848[↩]
- Ari C, Kovács Z, Juhasz G, Murdun C, Goldhagen CR, Koutnik AP, et al. Exogenous ketone supplements reduce anxiety-related behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk rats. Front Mol Neurosci (2016) 9:137. 10.3389/fnmol.2016.00137[↩][↩]
- Stubbs BJ, Cox PJ, Evans RD, et al. On the Metabolism of Exogenous Ketones in Humans. Front Physiol. 2017;8:848. Published 2017 Oct 30. doi:10.3389/fphys.2017.00848 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5670148[↩][↩][↩][↩]
- A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s disease. Newport MT, VanItallie TB, Kashiwaya Y, King MT, Veech RL. Alzheimers Dement. 2015 Jan; 11(1):99-103.[↩][↩]
- Clarke K, Tchabanenko K, Pawlosky R, Carter E, Todd King M, Musa-Veloso K, et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul Toxicol Pharmacol (2012. a) 63(3):401–8. 10.1016/j.yrtph.2012.04.008[↩][↩]
{kind=link}