What is Chediak-Higashi syndrome
Chediak-Higashi syndrome also called Chediak-Higashi disease, is a rare genetic disorder that affects many parts of the body, as well as the immune system. Chediak-Higashi syndrome is caused by mutations in the LYST gene and is inherited in an autosomal recessive fashion 1. Chediak-Higashi disease damages immune system cells, leaving them less able to fight off invaders such as viruses and bacteria. As a result, most people with Chediak-Higashi syndrome have repeated and persistent infections beginning in infancy or early childhood. These infections tend to be very serious or life-threatening.
Chediak Higashi syndrome is also known as Begnez-Cesar syndrome, leukocyte anomaly albinism or defect in natural killer lymphocytes. Chediak-Higashi syndrome is rare only about 200 to 500 cases of Chediak-Higashi syndrome have been reported worldwide 2. The incidence is the same in males and females. Chediak Higashi syndrome affects all races, but researchers suspect its occurrence may be under-reported.
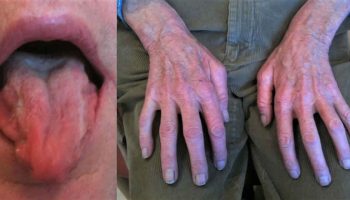
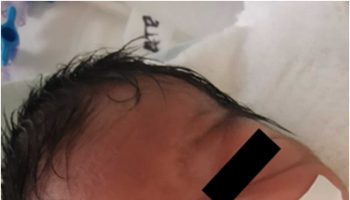
Chediak-Higashi syndrome is also characterized by a condition called oculocutaneous albinism, which causes abnormally light coloring (pigmentation) of the skin, hair, and eyes. Affected individuals typically have fair skin and light-colored hair, often with a metallic sheen. Oculocutaneous albinism also causes vision problems such as reduced sharpness; rapid, involuntary eye movements (nystagmus); and increased sensitivity to light (photophobia).
Many people with Chediak-Higashi syndrome have problems with blood clotting (coagulation) that lead to easy bruising and abnormal bleeding. In adulthood, Chediak-Higashi syndrome can also affect the nervous system, causing weakness, clumsiness, difficulty with walking, and seizures.
Signs and symptoms begin to affect children with Chediak-Higashi syndrome shortly after birth or by age five at the latest.
- Oculocutaneous albinism, or hypopigmentation of the hair, eyes and skin; hair is described as light in colour with a metallic sheen
- Prolonged bleeding and easy bruising due to platelet abnormalities
- Frequent viral, bacterial and fungal infections
- Photosensitivity
- Nystagmus: abnormal, involuntary eye movements
- Numbness/tingling in the limbs due to peripheral neuropathy
- Poor immunity due to abnormal function and shape of natural killer cells
If Chediak-Higashi disease is not successfully treated, most children with Chediak-Higashi syndrome reach a stage of the disorder known as the accelerated phase. This severe phase of the disease is thought to be triggered by a viral infection. In the accelerated phase, white blood cells (which normally help fight infection) divide uncontrollably and invade many of the body’s organs. The accelerated phase is associated with fever, episodes of abnormal bleeding, overwhelming infections, and organ failure. These medical problems are usually life-threatening in childhood.
A small percentage of people with Chediak-Higashi syndrome have a milder form of the condition that appears later in life. People with the adult form of the disorder have less noticeable changes in pigmentation and are less likely to have recurrent, severe infections. They do, however, have a significant risk of progressive neurological problems such as tremors, difficulty with movement and balance (ataxia), reduced sensation and weakness in the arms and legs (peripheral neuropathy), and a decline in intellectual functioning.
Treatment for Chediak-Higashi syndrome may include:
- Cord blood or bone marrow transplant, which is considered the treatment of choice; it is more successful if done before a child reaches the accelerated phase of the disease
- Chemotherapy is needed to put hemophagocytosis (the hemophagocytic lymphohistiocytosis phase) into remission before a transplant can take place
- Avoidance of exposure to sunlight and sun protective measures
Figure 1. Chediak-Higashi syndrome

What causes Chediak-Higashi syndrome
Chediak-Higashi syndrome is caused by mutations in the lysosomal trafficking regulator (LYST) gene or Chediak-Higashi syndrome (CHS1) gene 3. The LYST gene or the CHS1 gene is located on the long arm of chromosome 1 [1q42-43]. Around 40 different mutations have been discovered, including nonsense and missense mutations and deletions and insertions 4. This gene provides instructions for making a protein known as the lysosomal trafficking regulator. Researchers believe that lysosomal trafficking regulator protein plays a role in the transport (trafficking) of materials into structures called lysosomes and similar cell structures. Lysosomes act as recycling centers within cells. They use digestive enzymes to break down toxic substances, digest bacteria that invade the cell, and recycle worn-out cell components.
The mutation of the lysosomal trafficking regulator (LYST) gene or Chediak-Higashi syndrome (CHS1) gene disrupts the protein synthesis and affects the storage and secretory functions of lysosomal granules of leukocytes, fibroblasts, dense bodies of platelets, azurophilic granules of neutrophils, and melanosomes of melanocytes. The defects result in enlarged vesicles and non-functional lysosomes 5.
Mutations in the LYST gene impair the normal function of the lysosomal trafficking regulator protein, which disrupts the size, structure, and function of lysosomes and related structures in cells throughout the body. In many cells, the lysosomes are abnormally large and interfere with normal cell functions. For example, enlarged lysosomes in certain immune system cells prevent these cells from responding appropriately to bacteria and other foreign invaders. As a result, the malfunctioning immune system cannot protect the body from infections.
In pigment cells called melanocytes, cellular structures called melanosomes (which are related to lysosomes) are abnormally large. Melanosomes produce and distribute a pigment called melanin, which is the substance that gives skin, hair, and eyes their color. People with Chediak-Higashi syndrome have oculocutaneous albinism because melanin is trapped within the giant melanosomes and is unable to contribute to skin, hair, and eye pigmentation.
Researchers believe that abnormal lysosome-like structures inside blood cell fragments called platelets underlie the abnormal bruising and bleeding seen in people with Chediak-Higashi syndrome. Similarly, abnormal lysosomes in nerve cells probably cause the neurological problems associated with Chediak-Higashi disease.
Chediak Higashi syndrome inheritance pattern
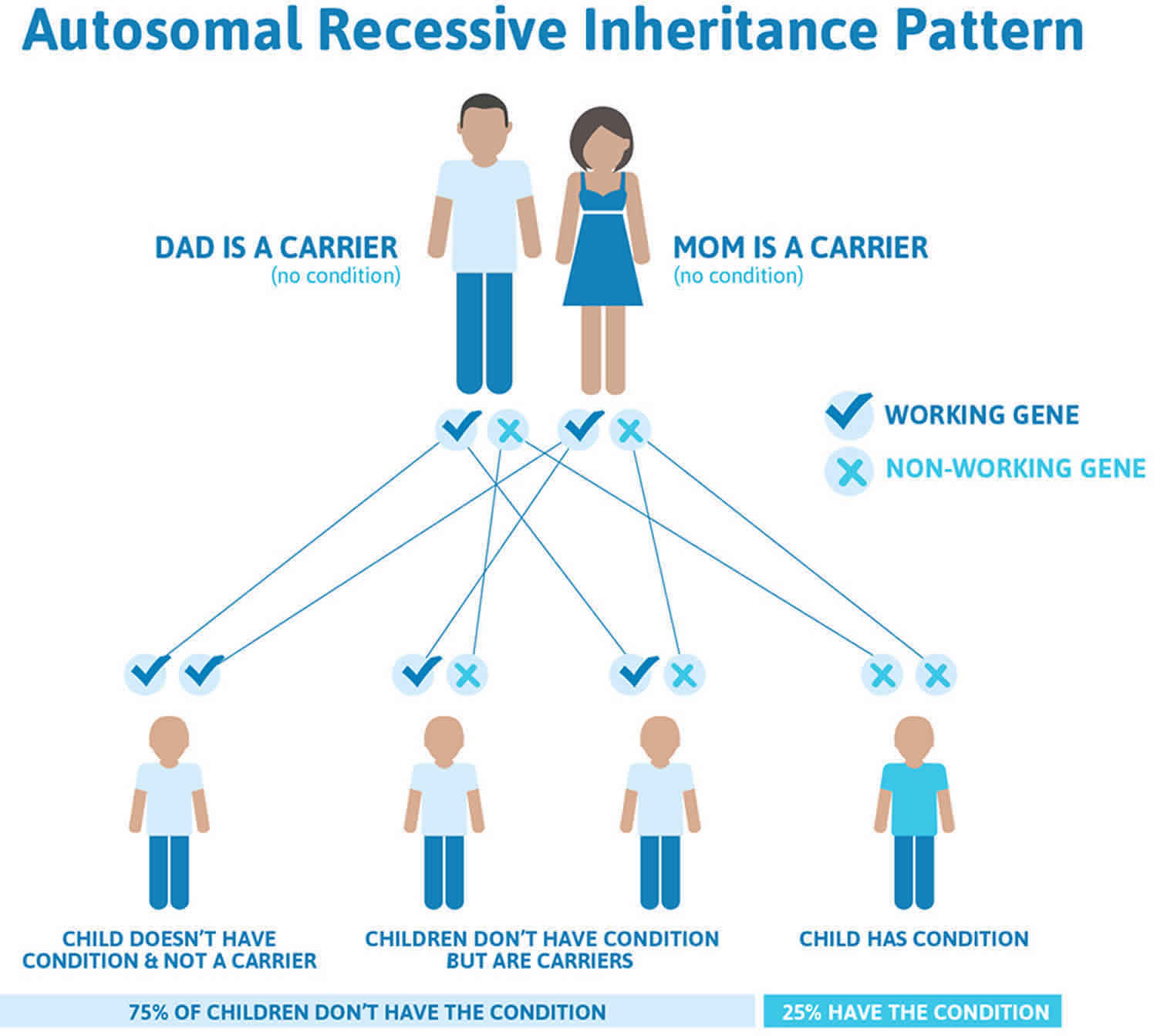
Chediak Higashi syndrome is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Chediak Higashi syndrome is inherited in an autosomal recessive pattern means that a person must inherit two changed copies of the same gene (one abnormal gene from each parent) in order to have the condition. If a person inherits one abnormal gene and one normal gene, then in most cases that person will be a healthy carrier because the normal gene compensates for the abnormal gene. Being a carrier means that you do not have the condition, but carry a changed copy of the gene on one of a pair of genes.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Chediak Higashi syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Chediak-Higashi syndrome symptoms
Chediak-Higashi syndrome presents at an early age with symptoms of immunodeficiency with recurrent infections, partial oculocutaneous albinism, and mild bleeding tendency due to coagulation defects. It has been observed that the severity of the disease correlates with the molecular phenotype. Generally, mutations resulting in a loss of function leads to a severe childhood-onset of the disease. Whereas, a missense mutation is associated with a more milder adolescent or adult-onset disease. However, exceptions to this have been reported in two cases.
Approximately 85% of affected individuals develop the accelerated phase, or hemophagocytic lymphohistiocytosis, a life-threatening, hyperinflammatory condition. All affected individuals including adolescents and adults with atypical Chediak-Higashi syndrome and children with classic Chediak-Higashi syndrome who have successfully undergone allogenic hematopoietic stem cell transplantation develop neurologic findings during early adulthood.
The severity of Chediak-Higashi disease is not only associated with molecular phenotype but also with the cellular phenotype. A range of intracellular granule enlargement in different cell types was observed from the studies of melanocytes and fibroblasts from patients with different clinical phenotype.
Chediak-Higashi syndrome signs and symptoms
- Partial oculo-cutaneous albinism: This is a prominent feature but the degree of pigment dilution varies and can either be present normally, partially, or could be absent. It can include the skin, hair, and eyes. There is a metallic or “silvery” appearance of the hair that can be seen in the classic forms of the disease. The clumping of the pigment within the hair shaft also can be visualized under a light microscope. A decrease in the pigmentation of the iris leads to a decrease in the pigmentation of the retina. The visual acuity might be affected and patients could either have a normal acuity or have some moderate impairment. Other ophthalmologic symptoms include photophobia, increasing red reflex, and a horizontal or rotating nystagmus.
- Immunodeficiency: Affected individuals have recurrent infections that are often severe and typically begin in infancy. Patients are more susceptible to bacterial and fungal infections with staphylococcal, streptococcal, pneumococcal, and beta-hemolytic species being the most predominant. Skin infections and upper respiratory tract infections are some of the most common infections. Recently, periodontitis has been identified as an important indicator of immune dysfunction and could help lead to the correct diagnosis. Patients with the atypical disease may not show symptoms of unusual or severe infections.
- Bleeding tendency: Symptoms are usually mild and include epistaxis, mucosal or gum bleeding, and easy bruising. Symptoms are subtle and generally do not require any medical intervention.
- Accelerated phase: This arises in 85% of the individuals affected and can occur at any age. It is associated with a poor prognosis and is the most common cause of mortality. The accelerated phase is characterized by fever, hepatosplenomegaly, lymphadenopathy, neutropenia, anemia, and sometimes thrombocytopenia. There is also a diffuse lymphohistiocytic infiltration of the liver, spleen, bone marrow, lymph nodes, and the central nervous system. The accelerated phase was initially thought to be caused by a malignancy such as lymphoma but is now known to be hemophagocytic lymphohistiocytosis that is associated with multi-organ inflammation. The hemophagocytic syndrome is caused by the inappropriate stimulation of the macrophages in the bone marrow and the lymphoid organs that leads to phagocytosis of blood cells and production of a large number of pro-inflammatory cytokines. The triggers of the accelerated phase remain unclear. It is believed that infections such as the Epstein-Barr virus might hasten the development, although it is yet unproven. The absence of natural killer (NK) cells also is believed to play a role in the development. Approximately 90% of patients die within the first 10 years of life.
- Neurologic manifestations: Neurological features manifest by early adulthood despite successful allogeneic hematopoietic stem cell transplantation. These changes are due to the long-term progression of the disease and include stroke, coma, ataxia, tremor, motor and sensory neuropathies, and absent deep-tendon reflexes.
- Atypical phenotype: An unknown number of individuals have atypical or milder phenotypes of the disease that are unrecognized. Features may include the following:
- Subtle or absent oculocutaneous albinism
- A decreased platelet-dense bodies with subtle bleeding symptoms
- Severe infections in childhood that become less frequent with age or infections that are insignificant.
- Progressive neurological symptoms such as intellectual disabilities, tremors, gait disturbances, parkinsonism.
- The neurological manifestations are inconsistent and nonspecific. Neurodegeneration may be the predominant symptom with only subtle alterations in the pigmentation, immune functions, and bleeding abnormalities.
- All individuals, however, have abnormal granules within the leukocyte.
Onset of Chediak-Higashi syndrome in adult life is associated with milder symptoms. These may include:
- Jaundice
- Skin infections
- Recurrent infections of the sinuses or respiratory tract
- Enlarged lymph glands
- Gum disease (gingivitis)
- Hyperhidrosis and miliaria
- Fever without infection
Chediak-Higashi syndrome complications
The majority of patients (around 80%) will undergo an accelerated phase of Chediak-Higashi syndrome. This phase is marked by the nonmalignant reproduction of white blood cells in multiple body organs. This accelerated phase can be precipitated by the presence of a viral infection, and it is often fatal if it occurs in childhood. This phase is called hemophagocytic lymphohistiocytosis.
Chediak-Higashi syndrome diagnosis
Clinically, a diagnosis should be considered in individuals that exhibit signs of immunodeficiency; pigment dilution of the skin, hair, or eyes; congenital or transient neutropenia; and signs of unexplained neurologic symptoms or neurodegeneration.
Diagnosis of Chediak-Higashi syndrome is made by:
- Examination of the white blood cells for the presence of large granules
- Light microscopy of the hair to find pigment clumping
A diagnosis can be made based on the presence of abnormally large granules in cells such as melanocytes, leukocytes and their bone marrow precursors, fibroblasts, the central and peripheral nervous tissue, and hair.
Molecular genetic testing also can be done to detect the biallelic variants in the LYST gene.
Chediak-Higashi syndrome treatment
Once the diagnosis of Chediak Higashi has been confirmed, the following can be done to assess the extent of Chediak Higashi disease:
- Assess the presence of accelerated phase
- Splenomegaly
- Any history of recurrent fever or unexplained fever
- Cytopenia affecting at least two cell lines
- Increased serum ferritin concentration
- Increased levels of soluble interleukin-2 receptor levels
- Signs of hemophagocytosis in either the bone marrow or cerebrospinal fluid (CSF)
- Evidence of liver dysfunction (hypertriglyceridemia or hypofibrinogenemia)
- Complete a thorough neurological examination
- Screen for signs of lymphoma as the hemophagocytic lymphohistiocytosis that is associated with Chediak Higashi may have a similar clinical appearance.
- Genetic consultation
Treatment of Clinical Symptoms
- Hematological and immune deficiency
- The only cure is an allogeneic hematopoietic stem cell transplantation (HSCT). Therefore, hematopoietic stem cell transplantation should be done as soon as the diagnosis is established.
- Outcomes are most favorable when the hematopoietic stem cell transplantation is done before the development of the accelerated phase. Hence, the accelerated phase must be ruled out or in remission before hematopoietic stem cell transplantation can be conducted.
- If signs of accelerated phase are evident, then hemophagocytosis should be brought into a stage of remission before hematopoietic stem cell transplantation.
- The guidelines for treatment of the accelerated phase is the same as that for familial hemophagocytic lymphohistiocytosis and includes a combination therapy of dexamethasone, cyclosporine A, and etoposide. Around 75% of individuals achieve remission by eight weeks. Relapses are not uncommon, and the treatment response declines over time.
- The 5-year survival rate was reported to be 62%. It was seen that the success was more prevalent in individuals with HLA-matched donors.
- Transplantation that took place during the accelerated phase had a higher rate of mortality. However, those in remission had better outcomes after an hematopoietic stem cell transplantation.
- Ocular symptoms
- Visual acuity might be improved by correcting the refractive errors.
- Sunglasses should be worn for protecting sensitive eyes against the UV rays.
- Hypopigmentation: Individuals must wear sunscreen to prevent skin cancers and sun damage. The degree of protection depends on the severity of hypopigmentation.
- Neurological manifestation: since the symptoms are progressive in nature, rehabilitation should be started for older patients as early as possible during the course of the disease.
Management in Pregnancy
There is limited data that is available for individuals with Chediak Higashi. However, it is reported that there is no impact on pregnancy or labor. The course of the disease also had no effect in mothers diagnosed with the disease.
Genetic Counseling
This is an autosomal recessive disease and therefore, parents of the affected individuals are heterozygotes for the disease (i.e., they are carriers of one abnormal LYST gene). Molecular genetic testing needs to be carried out to check the carrier status of the parents.
If the parents are heterozygotes, each sibling of an affected individual has a 25% chance of developing the disease, 50% chance of being a carrier and 25% chance of not developing the disease nor being a carrier.
Evaluation of At-risk Relatives:
It is necessary to assess siblings of affected individuals early. This will help in undergoing an hematopoietic stem cell transplantation before complications such as accelerated phase develop.
- Molecular testing can be done to assess the genetic status if family specific pathogen variants are known.
- If the variants are unknown, then an examination of peripheral blood can be done to detect the presence of inclusions in the white blood cells.
The best time for determining genetic risk as well as carrier status is before pregnancy. Genetic counseling including risks and complications should be offered to adults who are carriers or are at risk of being carriers or are already affected. A preimplantation genetic diagnosis is also an option for those where the pathogenic LYST genes have been identified.
DNA banking possibilities also should be offered to affected individuals where the DNA that is extracted, typically the white blood cells are stored for future use. There is a possibility that testing and understanding genes will improve in the future.
Prevention of Secondary Complications
- Protect against exposure of infectious agents as much as possible.
- Nonsteroidal anti-inflammatory drugs (NSAIDs) should also be avoided as they can exacerbate bleeding tendencies.
- Prompt and aggressive use of antibiotics to treat bacterial illnesses.
- Use of widespread antibiotic prophylaxis prior to dental or invasive procedures is controversial. However, it should be considered in those with a compromised immune system and neutropenia.
- Immunizations should be administered.
- Prior to any invasive procedures, intravenous desmopressin should be given for 30 minutes to help control bleeding. Platelet transfusion might be necessary for those with serious trauma or extensive bleeding.
Monitoring
- Classical Chediak Higashi
- There are no specific guidelines for surveillance.
- The current standard is to evaluate for hematopoietic stem cell transplantation as soon as possible once the diagnosis is established.
- An ophthalmologic examination is also necessary
- Atypical or adult-onset form of disease
- Annual screening that should include the following:
- Ultrasound of the abdomen to detect presence of hepatosplenomegaly
- A complete blood count to check for signs of cytopenias
- Signs of liver disease including hypofibrinogenemia and hypertriglyceridemia.
- Check levels of serum ferritin concentration
- Monitor levels of interleukin-2 receptor
- A bone marrow biopsy or a lumbar puncture if there is suspicion of CNS involvement or symptoms of accelerated phase
- Ophthalmologic exam.
- Annual screening that should include the following:
Chediak Higashi syndrome prognosis
Without treatment, the outlook for Chediak-Higashi syndrome is poor. Around 50–85% of children with Chediak-Higashi syndrome will enter the accelerated hemophagocytic lymphohistiocytosis phase. This process with fatal without treatment and most patients will die by the age of 10 years.
The most common cause of death in patients with Chediak Higashi syndrome results from recurrent infections or the development of accelerated phase where there is lymphoproliferation into major organs. 80% of deaths occur in the first decade of life, and those who survive into adulthood develop progressive neurological symptoms.
- Toro C, Nicoli ER, Malicdan MC, et al. Chediak-Higashi Syndrome. 2009 Mar 3 [Updated 2018 Jul 5]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK5188[↩]
- Chediak-Higashi syndrome. https://ghr.nlm.nih.gov/condition/chediak-higashi-syndrome[↩]
- Ajitkumar A, Ramphul K. Chediak Higashi Syndrome. [Updated 2019 Apr 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507881[↩]
- Bowman SL, Bi-Karchin J, Le L, Marks MS. The road to lysosome-related organelles: Insights from Hermansky-Pudlak syndrome and other rare diseases. Traffic. 2019 Jun;20(6):404-435 [↩]
- Pluthero FG, Di Paola J, Carcao MD, Kahr WHA. NBEAL2 mutations and bleeding in patients with gray platelet syndrome. Platelets. 2018 Sep;29(6):632-635.[↩]
{kind=link}