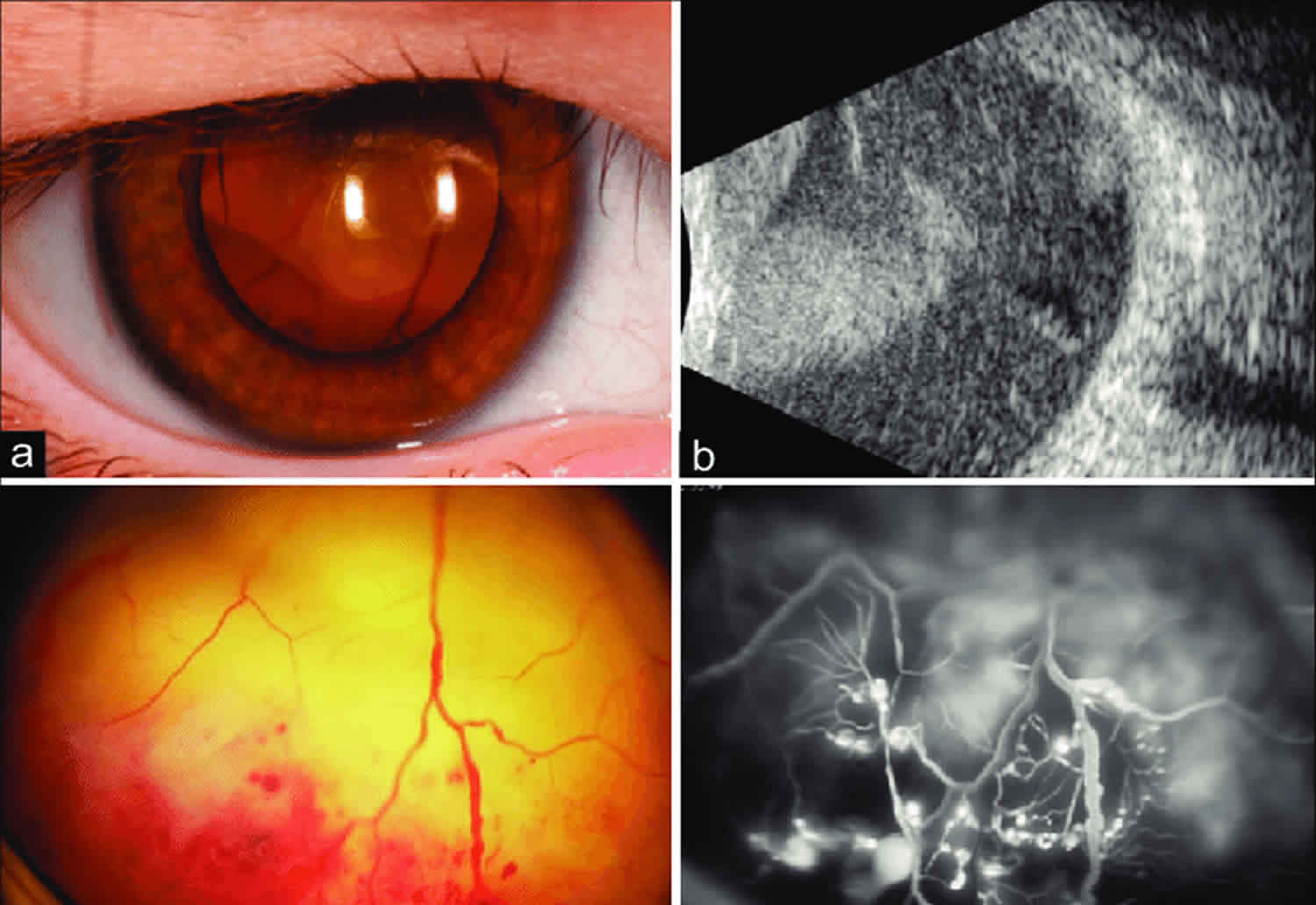
Coats disease
Coats disease is an eye disorder characterized by abnormal development of the blood vessels in the retina (retinal telangiectasia). Coats disease causes blood vessels in the retina to be abnormally enlarged (dilated) and twisted. The abnormal vessels leak fluid, which can eventually cause the layers of the retina to separate (retinal detachment). These eye abnormalities often result in vision loss. Most people with Coats disease begin showing symptoms in childhood. Early signs and symptoms vary but may include vision loss, “crossed eyes” (strabismus), and a white mass in the pupil behind the lens of the eye (leukocoria). Over time, Coats disease may also lead to retinal detachment, glaucoma, and clouding of the lens of the eye (cataracts). In most cases, only one eye is affected. Coats disease is not inherited and the underlying cause is not known. It has been theorized that some cases may be due to somatic mutations (acquired, not inherited) in the NDP gene. Treatment depends on the symptoms present and may include cryotherapy and laser photocoagulation (alone or in combination), steroids for inflammation, and/or surgery for retinal detachment 1.
Coats disease was first described by Dr. George Coats in 1908 2. The definition of the disease has since been refined by Reese in 1956 3 and Shields in 2000 4. Coats disease is characterized by unilateral telangiectatic and aneurysmal retinal vessels with intraretinal fluid, subretinal fluid and lipid exudation. Coats disease is most commonly found in young males although an adult onset form of the disease has been described 5. Affected eyes can develop outer retinal thickening and total or partial exudative retinal detachment. The subretinal fluid is viscous and laden with lipid crystals. Retinal capillary dropout is seen distal to the telangiectasias and can lead to neovascularization of the disc with increasing severity of ischemia. The pathology is present in the retinal vasculature, which acquires numerous aneurysms that rarely bleed. Histopathology of the involved vessels reveals a loss of endothelial cells and pericytes along with adventitial thickening 5. This loss of vascular integrity allows for exudation, resulting in the clinical findings. Research has been undertaken to determine the genetic causes of Coats disease. Several genes such as NDP, CRB1, and PANK2 have been implicated in Coats disease, but none have been definitively related. This has led to the suggestion that a somatic mutation may be the cause 6.
Coats disease key points
- Coats disease is a telangiectatic neovascular disease of the retina of unknown etiology that frequently affects unilateral eyes of young males.
- Young male with painless unilateral decrease in vision
- Commonly presents with leukocoria or strabismus 7
- Incidence: Estimated 0.09 cases per 100,000 8
- Sex: Male predominance (75-85% of cases) 8
- Race: No racial preference 9
- Age (at time of presentation): 1st and 2nd decades of life most common age at presentation (median age 5-8 years old), but can present at any age 9
- Laterality: Unilateral (bilateral cases, while rare, have been noted and generally have minimal findings in the fellow eye) 9
Coats disease stages
The most recently proposed staging classification for Coats disease was developed on based in a large series of affected people. This classification may help in choosing the most appropriate course of treatment as well as predicting the long-term outcome (prognosis) for people with Coats disease 10. A very useful staging is the one developed by Comez-Morales and that of Siegelman.
Gomez- Morales staging
- Stage 1: Focal exudates
- Stage 2: Massive exudation
- Stage 3: Partial exudative retinal detachment
- Stage 4: Total Retinal Detachment
- Stage 5: Complications
Sigelman staging
- Stage 1: Only telangiactasia
- Stage 2: Focal exudates
- Stage 3: Partial exudative retinal detachment
- Stage 4: Total Retinal Detachment
- Stage 5: Complications
Shields’ classification
Shields’ classification (usually followed) 7:
- Stage 1: Retinal telangiectasia only (dilation of capillaries in the retina)
- Stage 2: Telangiectasia and exudation (escape of fluids and material from blood vessels into surrounding tissues)
- A – Extrafoveal exudation (exudation outside of the fovea, which is a small area in the retina responsible for the acute vision)
- B – Foveal exudation (exudation in the fovea)
- Stage 3: Exudative retinal detachment
- A – Subtotal (partial) detachment
- 1. Extrafoveal
- 2. Foveal
- B – Total retinal detachment
- A – Subtotal (partial) detachment
- Stage 4: Total retinal detachment and glaucoma
- Stage 5: Advanced end-stage disease (defined as a blind, non-painful eye with a total retinal detachment, often with cataract and phthisis bulbi)
Coats disease causes
The specific cause of Coats disease is currently unknown. It has been theorized that somatic mutations (acquired, not inherited) in the Norrie disease protein (NDP) gene gene may lead to isolated cases of Coats disease. This gene is an attractive candidate because it has been shown to play a vital role in retinal blood vessel development. One study showed some promise for involvement of the NDP gene in Coats disease, however further studies have not been able to verify this hypothesis. In general, Coats disease is considered a non-genetic, non-heritable condition.
Coats disease has been reported as one feature of several genetic syndromes 11. In these cases, the underlying cause of each syndrome may be responsible for Coats disease. When Coats disease occurs with additional abnormalities that affect the brain, bones, gastrointestinal system, and other parts of the body, it is called Coats plus syndrome and is caused by mutations in the CTC1 gene 12.
Coats disease inheritance pattern
Isolated Coats disease is not an inherited condition 1. Rarely, Coats disease can be a feature of an underlying, inherited genetic syndrome. For example, it is a key feature of a condition called Coats plus syndrome, which is characterized by Coats disease plus abnormalities of the brain, bones, gastrointestinal system, and other parts of the body. Coats plus syndrome is inherited in an autosomal recessive manner 12.
Coats disease symptoms
The signs and symptoms of Coats disease typically begin at an early age (between ages 6 and 8) 10. However, Coats disease may occur at any age, but the majority of people with Coats disease are diagnosed in the first two decades of life. Some people may have few or no symptoms, while others are very severely affected 1. Coats disease is almost always progressive (symptoms get worse over time), although alternating periods of sudden worsening with periods of no apparent progression are common 10.
Early signs and symptoms may include loss of vision, misalignment of the eyes (strabismus), and/or the development of a white mass in the pupil behind the lens of the eye (leukocoria) 1. As the disease progresses, affected people may develop glaucoma, cataracts, reddish discoloration in the iris (rubeosis iridis or neovascular glaucoma), shrinking of the affected eyeball (phthisis bulbi), and/or swelling and irritation of the middle layer of the eye (uveitis). Over time, Coats disease may cause detachment of the retina and substantial loss of vision 1.
When Coats disease occurs with additional abnormalities that affect the brain, bones, gastrointestinal system, and other parts of the body, it is called Coats plus syndrome 12.
In a large study 13 on 150 patients, the first symptom or sign was decreased visual acuity in 68 cases (34%), strabismus in 37 (23%), leukocoria in 31 (20%), and 13 patients (8%) were asymptomatic. Visual acuity at presentation was 20/200 to no light perception in 121 eyes (76%). The anterior segment was normal in 142 eyes (90%). The retinal telangiectasia involved the midperipheral or peripheral fundus in 156 of the 158 eyes (99%) and was restricted to the macular area in two eyes (1%); involved mainly the temporal fundus in 66 eyes (42%), inferior fundus in 41 eyes (26%), and more than one sector in 34 eyes (22%). Retinal exudation was present in all 12 clock hours in 86 eyes (55%) and six or more clock hours in 115 eyes (73%). There was a total retinal detachment in 74 eyes (47%) and neovascular glaucoma in 12 (8%). Retinal macrocysts were present in 18 eyes (11%), a vasoproliferative tumor in nine eyes (6%) and retinal neovascularization in four eyes (3%). Fluorescein angiography in 49 of the 158 eyes (37%) disclosed early hyperfluorescence of the telangiectasias and macular edema in 18 of eyes (37%). Ultrasonography typically showed a retinal detachment but no solid mass’
In mild cases one or two foci of retinal telangiectasia can be found usually on the temporal hemispheres. Microaneurysms, and thickening of retinal vessels are seen. Affected vessels show irregular enlargement with aneurysms which light up like bulb during fluorescein angiogram. The vascular abnormalities are usually prominent near the peripheral capillary dropout. The vascular leakage causes hard exudates which may be peripheral (near the vascular abnormalities) or mid peripheral and central (at macula). In type 1 macular telangiectasia, the telangiectasia and aneurysm with hard exudates (primarily cholesterol) are seen temporal to the fovea. Intraretinal or subretinal fluid/exudate may be noted at the macula.
The exudates vary in size and they have a tendency to occupy the inferior pole, as a result, visual acuity is reduced primarily due to infiltration of the fovea, formation of cystoid macular edema or even exudative retinal detachment. Those exudates ultimately cause discoid glial scarring and subretinal neo-vascularization. Long standing subretinal exudate may cause superficial crystalline deposits. The retinal vessels overlying the subretinal exudates may undego gliotic sheating. Retinal pigment epithelial metaplasia with subretinal fibrosis may be noted. Retinal neovascularization is uncommon, but can occur in late stages.
Anterior segment is usually not involved in early cases, but late cases may cause neovascularization of iris or angle of anterior chamber, ectropion uvae, neovascular glaucoma.
In children, the exudative detachment may be bullous, immobile and touching the posterior capsule of the lens. The irregular caliber of telangiectatic vessels, aneurysms, subretinal exudates and lack of calcification on ultrasound differentiates it from retinoblastoma.
Coats disease complications
Intraretinal/subretinal exudation, retinal detachment (typically worse in younger patients), retinal hemorrhage, vitreous hemorrhage, neovascularization of retina and/or disc, neovascular glaucoma, anterior chamber cholesterolosis, cataracts, uveitis, phthisis.
Coats disease diagnosis
A diagnosis of Coats disease is often suspected based on the presence of signs and symptoms, and findings on a thorough eye examination.
Besides thorough fundoscopy, which ultimately establishes the diagnosis some clinical tests are utilized. The diagnostic tests to confirm the diagnosis and rule out other conditions may include OCT, ultrasound and fluorescence angiography and in some cases, a CT scan of the orbits, and/or MRI.
Fluorescein angiogram
The features include:
- telangiectatic vessels
- aneurysms appearing like light bulb
- early and progressive perivascular leak
- peripheral capillary nonperfusion
- capillary dropout
Coats disease differential diagnosis
The major clinical significance of Coats Disease is to differentiate it from Retinoblastoma since both appear with leukocoria, but calcium seen in CT or ultrasound excludes Coats in favor of Retinoblastoma. Other conditions such as Toxocara infection, retinopathy of prematurity, pars planitis, familial exudative vitreoretinopathy, retinal metastatic lesions, Norrie disease, Eales disease, cavernous retinal hemangioma and leukemia, should be considered. A retinal vasoproliferative tumor is differentiated by presence of feeder and draining vessel, an obvious vascular mass, preretinal fibrosis and extreme peripheral location.
Coats disease treatment
Treatment for Coats disease depends on the severity in each person 1.
The following treatments (used alone or in combination) may be tried:
- Observation– In minimal telangiectatic vessels with little or no exudation and no imminent threat to vision, or in painless and comfortable blind eyes. Clinical observation is recommended for stage 1 and stage 5 disease. Shields et al. 4 showed that Stage 1 disease (telangiectasia with no exudation) that is not threatening vision should simply be observed, as progression is not always imminent. Observation has also been utilized in cases where regaining any vision was highly unlikely. In such cases, enucleation was required in ~20% of cases due to painful secondary glaucoma 4.
- Laser photocoagulation (low power, long duration, large spot burns to the telangiectatic vessels and aneurysms, also laser the peripheral capillary nonperfusion as per scatter laser settings) – in telangiectasia with exudation but no or minimal subretinal fluid. Laser photocoagulation via indirect ophthalmoscopy is the first-line treatment for Coats disease 14. The goal of laser photocoagulation is to cauterize affected retinal vasculature 15. While direct treatment to the telangiectatic vessels is the primary mechanism, additionally treating the areas surrounding exudation to act as a barrier has also been described 14. Argon (most common), Diode, and FD-YAG lasers have all been used 14.
- Cryotherapy (a procedure that uses extreme cold to destroy abnormal blood vessels)- for exudation with subretinal fluid of such thickness that cryo reaction can reach the retina. Historically, cryotherapy was the first-line treatment for Coats disease 4. Now, it’s use is typically limited as an adjuvant to laser photocoagulation in more advanced cases when exudation is too thick for laser penetration 14. When utilized as a treatment modality, a double freeze-thaw method is generally used.
- Anti-vascular endothelial growth factor (anti-VEGF) injections– has been promising but the safety profile for children is as yet unclear. VEGF is increased in the aqueous and vitreous humor of patients with Coats disease 16. Since 2007, there have been numerous case reports of intravitreal injection of anti-VEGF agents decreasing subretinal fluid 17. Vitreoretinal fibrosis and traction retinal detachments have been reported following anti-VEGF therapy, so some authors have recommended that the use of injection therapy should be limited to more severe cases in which a decrease in subretinal fluid would aid laser or cryotherapy 18. Ruling out retinoblastoma is crucial prior to injection as treatment in such an eye could result in cancer spread. Diffuse unilateral retinoblastoma can present with an exudative retinal detachment without a calcified mass, making this differentiation challenging.
- Intravitreal triamcinolone acetonide injections may reduce the exudation, but risks of glaucoma and cataract are there. Intravitreal triamcinolone injection has been shown to reduce subretinal fluid and macular exudates in some studies 19. This can facilitate future treatment with laser photocoagulation or cryotherapy. However, due to the common side effects of cataract formation and ocular hypertension, its use is limited 20. The combination of triamcinolone and cryotherapy has been reported to be associated with inoperable rhegmatogenous retinal detachments and proliferative vitreo-retinopathy 21.
- Vitreoretinal surgery– for advanced stage disease unable to be managed with laser photocoagulation or cryotherapy, surgery may be necessary. Methods such as vitrectomy, posterior retinotomy, intraocular silicone or gas use, scleral buckling, and newer minimally invasive techniques have all been described 22.
- Enucleation– Painful blind eye or when retinoblastoma can not be ruled out by imaging and clinical examination. With advances in treatment and methods to rule out retinoblastoma, enucleation is becoming less common. However, it is still utilized in end stage disease in which secondary glaucoma has resulted in a blind, painful eye.
More advanced disease with extensive retinal detachment may also require surgical interventions such as vitrectomy, scleral buckling to correct a detached retina, and external drainage of fluids 23.
Coats disease prognosis
Coats disease prognosis varies in each case. Factors that effect the prognosis include the stage of disease at the time of diagnosis, the age at diagnosis, the rate of disease progression, and the effectiveness of treatment. Milder Coats disease, older presenting cases, having a better prognosis, of even spontaneous regression, whereas children under the age of three have a poorer prognosis.
In some older children and young adults, spontaneous regression (improvement of symptoms) has been reported. Younger children typically have more aggressive disease and often have a total retinal detachment by the time they are diagnosed; these children usually have a poor visual outcome 24. Some people present with advanced disease which does not benefit from treatment, while others show disease progression despite treatment 23. While most people respond well to treatment, approximately 25% will become worse and require removal of the eye 23. An ophthalmologist with knowledge about Coats disease may be able to make a general prediction about the chances of retaining the eye and preserving vision.
The extent of visual impairment varies considerably from person to person. Favorable visual outcomes are more likely to be achieved with early detection and treatment, with combined therapies as needed. Even when treatment is not able to restore sight, it is beneficial in saving the eye 23.
In the study the Shields and colleagues 7 on 150 patients ‘anatomical improvement or stability was achieved in 76% of eyes, and final visual acuity was 20/50 or better in 17 eyes (14%), 20/60 to 20/100 in eight (6%), 20/200 to finger counting in 30 (24%), and hand motion to no light perception in 49 (40%). Enucleation was ultimately necessary in 20 eyes (16%). Risk factors predictive of poor visual outcome (20/200 or worse) included postequatorial, diffuse or superior location of the telangiectasias and exudation, failed resolution of subretinal fluid after treatment and presence of retinal macrocysts. The main risk factors for enucleation were elevated intraocular pressure (greater than 22 mm Hg) and iris neovascularization.
- Coats disease. https://rarediseases.org/rare-diseases/coats-disease[↩][↩][↩][↩][↩][↩]
- Coats G. Forms of retinal diseases with massive exudation. Graefes Arhiv für Ophthalmologie 1912;17:440-525[↩]
- Reese AB. Telangiectasis of the retina and Coats’ disease: the Eleventh Sanford R. Gifford Lecture. Am J Ophthalmol 1956;42(1):1-8.[↩]
- Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol 2001;131(5):572-583.[↩][↩][↩][↩]
- Grosso A, Pellegrini M, Cereda MG, Panico C, Staurenghi G, Sigler EJ. Pearls and pitfalls in diagnosis and management of coats disease. Retina 2015;35(4):614-623.[↩][↩]
- Sigler EJ, Randolph JC, Calzada JI, Wilson MW, Haik BG. Current management of Coats disease. Surv Ophthalmol 2014;59(1):30-46.[↩]
- Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 Proctor Lecture. Am J Ophthalmol. 2001 May;131(5):572-83.[↩][↩][↩]
- Morris B, Foot B, Mulvihill A. A population-based study of Coats disease in the United Kingdom I: epidemiology and clinical features at diagnosis. Eye (Lond) 2010;24(12):1797-1801.[↩][↩]
- Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol 2001;131(5):561-571.[↩][↩][↩]
- Del Longo A. Coats disease. Orphanet Encyclopedia. September 2004. https://www.orpha.net/data/patho/Pro/en/Coats-FRenPro1645.pdf[↩][↩][↩]
- Del Longo A. Coats disease. Orphanet Encyclopedia. September 2004 http://www.orpha.net/data/patho/Pro/en/Coats-FRenPro1645.pdf[↩]
- Coats Plus Syndrome. http://ghr.nlm.nih.gov/condition/coats-plus-syndrome[↩][↩][↩]
- Shields JA, Shields CL, Honavar SG, Demirci H. Clinical variations and complications of Coats disease in 150 cases: the 2000 Sanford Gifford Memorial Lecture. Am J Ophthalmol. 2001 May;131(5):561-71.[↩]
- Mulvihill A, Morris B. A population-based study of Coats disease in the United Kingdom II: investigation, treatment, and outcomes. Eye (Lond) 2010;24(12):1802-1807.[↩][↩][↩][↩]
- Spitznas M, Joussen F, Wessing A. Treatment of Coats’ disease with photocoagulation. Albrecht Von Graefes Arch Klin Exp Ophthalmol 1976;199(1):31-37.[↩]
- He YG, Wang H, Zhao B, Lee J, Bahl D, McCluskey J. Elevated vascular endothelial growth factor level in Coats’ disease and possible therapeutic role of bevacizumab. Graefes Arch Clin Exp Ophthalmol 2010;248(10):1519-1521.[↩]
- Ghorbanian S, Jaulim A, Chatziralli IP. Diagnosis and treatment of coats’ disease: a review of the literature. Ophthalmologica 2012;227(4):175-182.[↩]
- Ray R, Barañano DE, Hubbard GB. Treatment of Coats’ disease with intravitreal bevacizumab. Br J Ophthalmol 2013;97(3):272-277.[↩]
- Böhm MR, Uhlig CE. Use of intravitreal triamcinolone and bevacizumab in Coats’ disease with central macular edema. Graefes Arch Clin Exp Ophthalmol 2011;249(7):1099-1101.[↩]
- Othman IS, Moussa M, Bouhaimed M. Management of lipid exudates in Coats disease by adjuvant intravitreal triamcinolone: effects and complications. Br J Ophthalmol 2010;94(5):606-610.[↩]
- Bergstrom CS, Hubbard GB, 3rd. Combination intravitreal triamcinolone injection and cryotherapy for exudative retinal detachments in severe Coats disease. Retina 2008;28(3 Suppl):S33-37.[↩]
- Adam RS, Kertes PJ, Lam WC. Observations on the management of Coats’ disease: less is more. Br J Ophthalmol 2007;91(3):303-306.[↩]
- Cebeci Z, Bayraktar S, Yilmaz YC, Tuncer S, Kir N. Evaluation of Follow-Up and Treatment Results in Coats’ Disease. Turk J Ophthalmol. October, 2016; 46(5):226-231. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5200835[↩][↩][↩][↩]
- Shields JA, Shields CL, Honavar SG, Demirci H, Cater J. Classification and management of Coats disease: the 2000 Proctor Lecture. American Journal of Ophthalmology. May 2001; 131(5):572-583.[↩]



{kind=link}