Congenital hepatic fibrosis
Congenital hepatic fibrosis is an inherited disease of the liver that is present from birth. Congenital hepatic fibrosis is characterized by malformation of the bile ducts and the blood vessels of the hepatic portal system. Congenital hepatic fibrosis may occur alone, in which case it is called isolated congenital hepatic fibrosis. More frequently, congenital hepatic fibrosis can cause the liver, kidneys and bile ducts to form abnormally before birth and may also cause fibrosis and scarring on the liver. occurs as a feature of genetic syndromes that also affect the kidneys, such as autosomal recessive polycystic kidney disease (ARPKD) a severe form of polycystic kidney disease (PKD) 1 and Caroli syndrome (Caroli disease). Congenital hepatic fibrosis can cause the liver, kidneys and bile ducts to form abnormally before birth and may also cause fibrosis and scarring on the liver.
Because of the variable clinical presentations, congenital hepatic fibrosis is believed to represent a broad spectrum of hepatic and renal lesions rather than a single clinical entity 2. Symptoms, which may be early or late, are mostly characterized by hepatosplenomegaly and portal hypertension 3.
Isolated congenital hepatic fibrosis is rare, but is often concomitant with a wide range of disorders caused by various gene mutations like autosomal recessive polycystic kidney disease (ARPKD) and Caroli syndrome. Its prevalence is unknown. The total prevalence of syndromes that include congenital hepatic fibrosis as a feature is estimated to be 1 in 10,000 to 20,000 individuals 4.
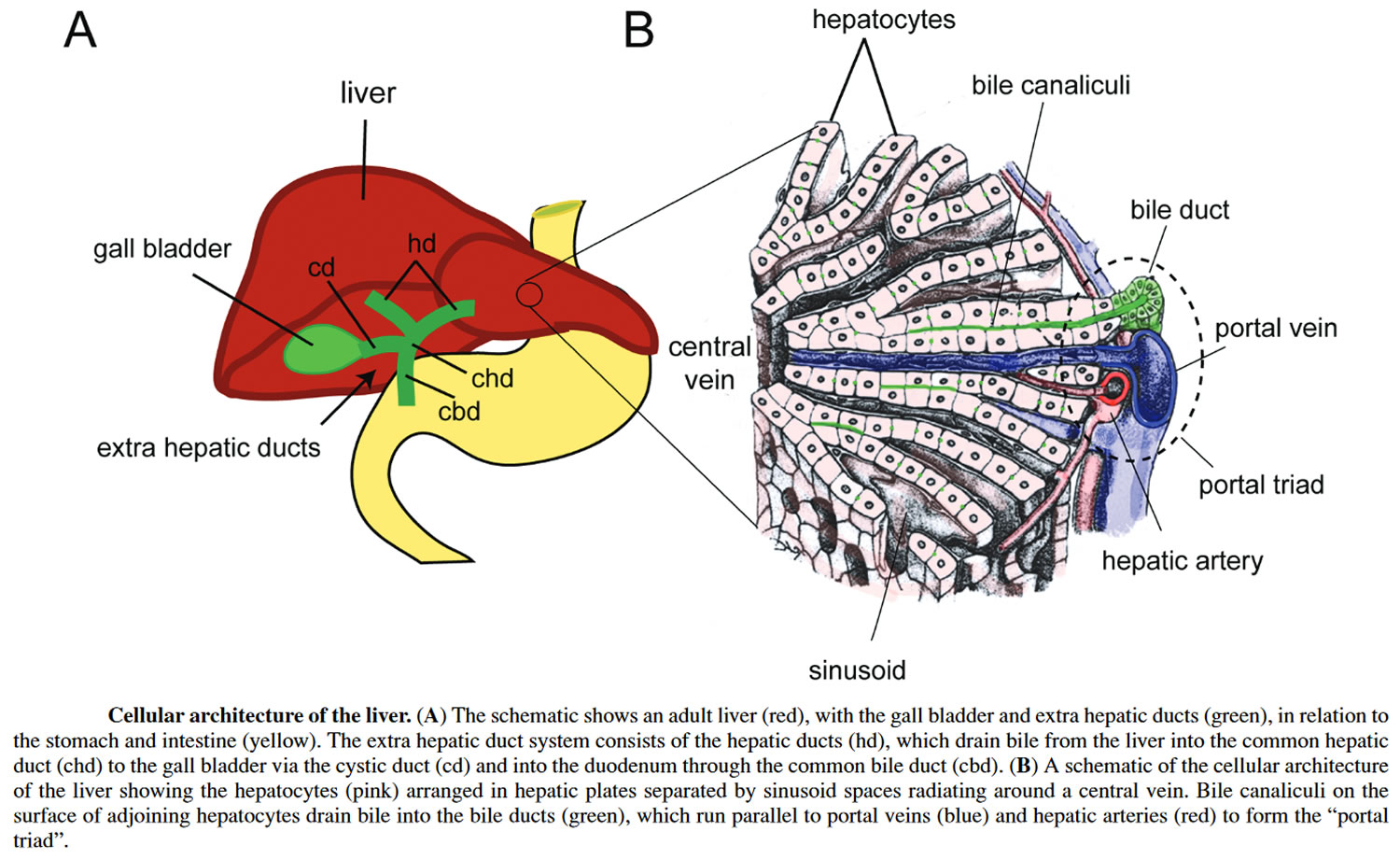
The liver has many important functions, including producing various substances needed by the body and breaking down other substances into smaller parts to be used or eliminated. Bile ducts carry bile (a fluid that helps to digest fats) from the liver to the gallbladder and small intestine. The hepatic portal system is a branching network of veins (portal veins) that carry blood from the gastrointestinal tract to the liver for processing.
A buildup of scar tissue (fibrosis) in the portal tracts also occurs in this disorder. Portal tracts are structures in the liver that bundle the vessels through which blood, lymph, and bile flow. Lymph is a fluid that helps exchange immune cells, proteins, and other substances between the blood and tissues. Fibrosis in the portal tracts can restrict the normal movement of fluids in these vessels.
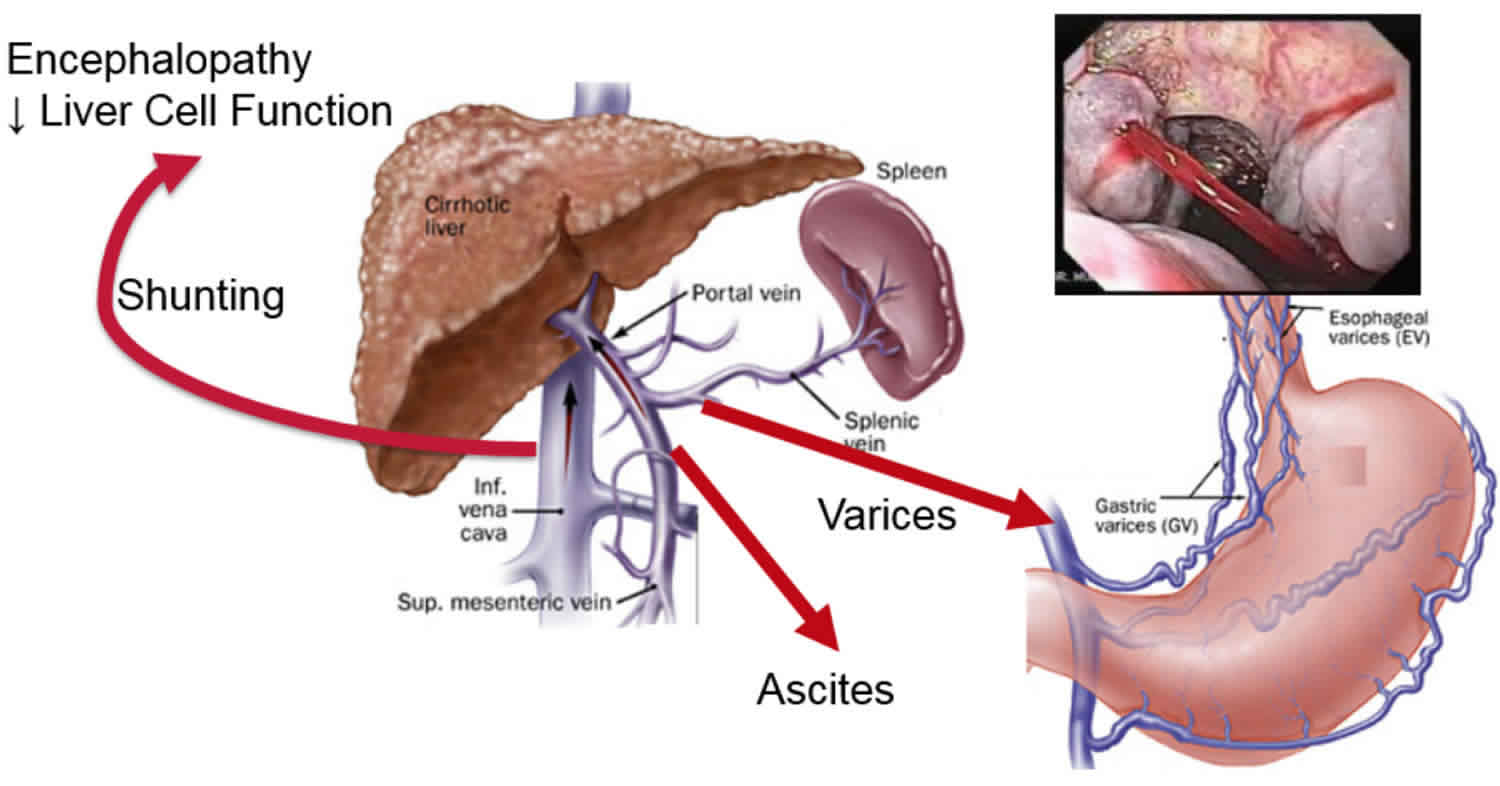
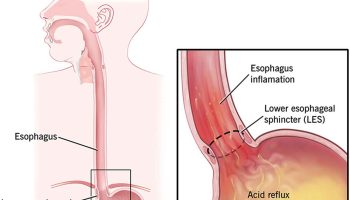
Constriction of the portal veins due to malformation and portal tract fibrosis results in high blood pressure in the hepatic portal system (portal hypertension). Portal hypertension impairs the flow of blood from the gastrointestinal tract, causing an increase in pressure in the veins of the esophagus, stomach, and intestines. These veins may stretch and their walls may become thin, leading to a risk of abnormal bleeding.
People with congenital hepatic fibrosis have an enlarged liver and spleen (hepatosplenomegaly). The liver is also abnormally shaped. Affected individuals also have an increased risk of infection of the bile ducts (cholangitis), hard deposits in the gallbladder or bile ducts (gallstones), and cancer of the liver or gallbladder.
Figure 1. Liver location
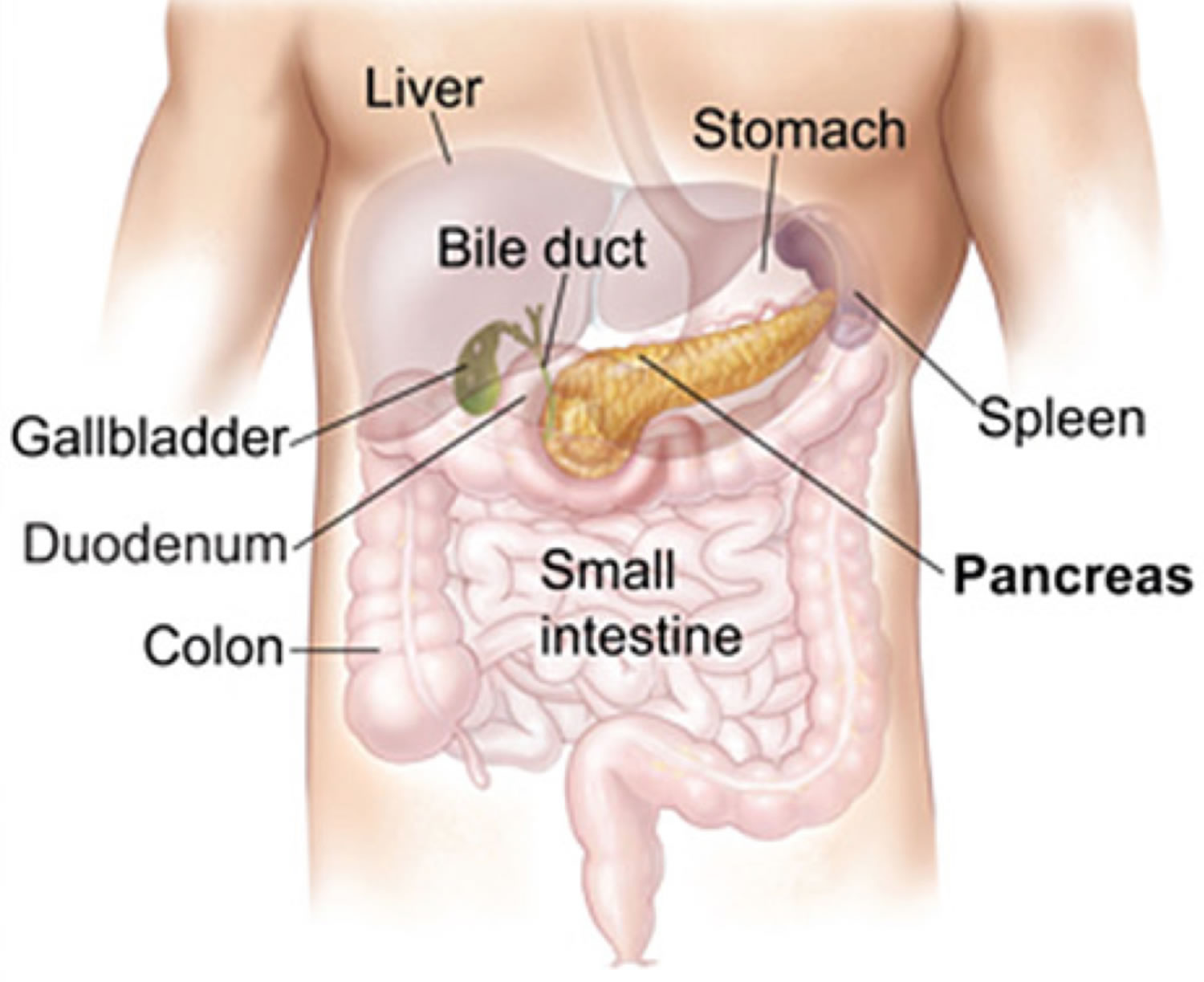
Figure 2. Liver anatomy
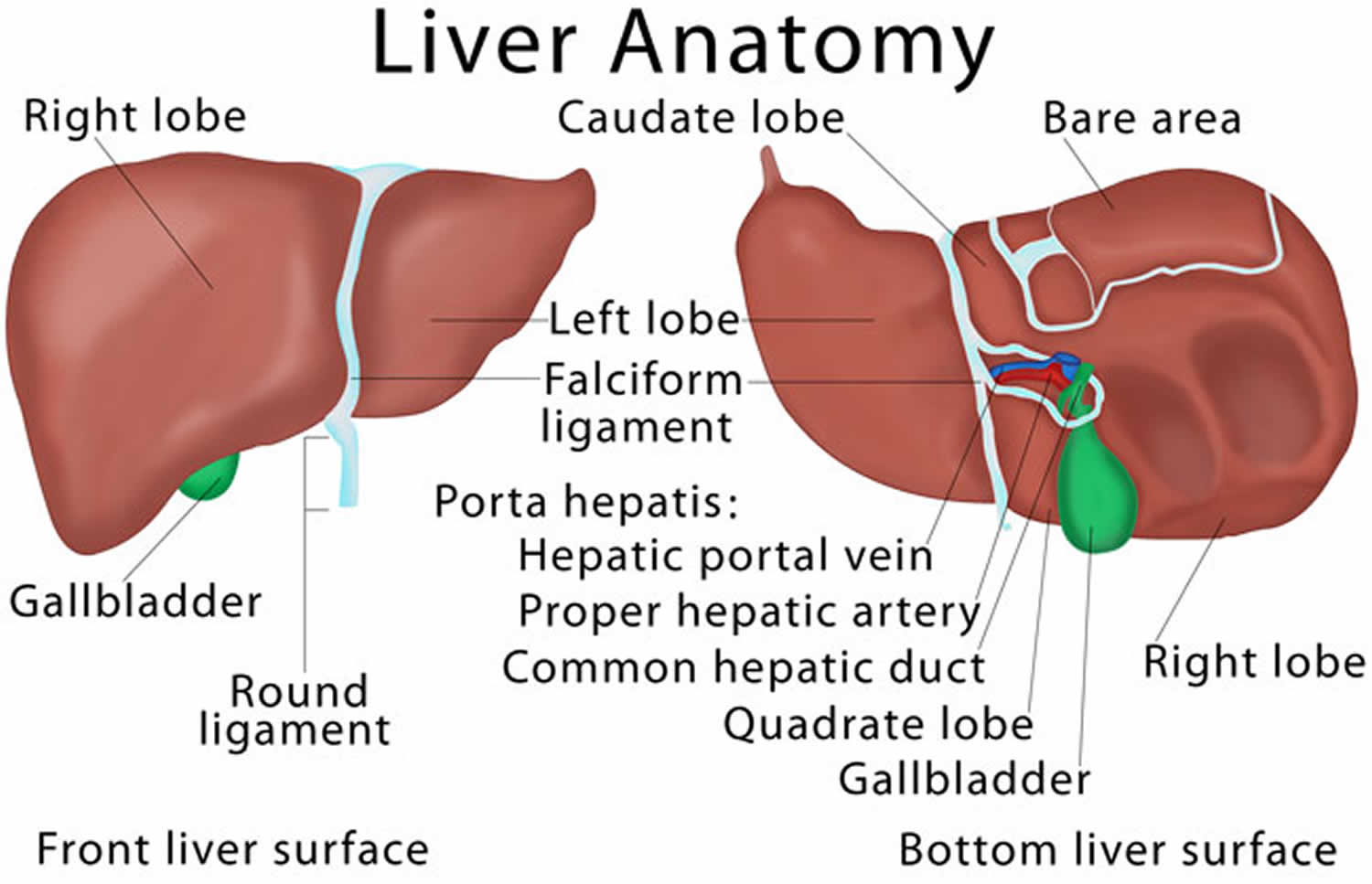
Figure 3. Liver cellular architecture
Congenital hepatic fibrosis causes
No specific genes have been associated with isolated congenital hepatic fibrosis 5. Isolated congenital hepatic fibrosis is rare 4. Congenital hepatic fibrosis is usually associated with conditions known as hepatorenal fibrocystic diseases (FCD) that can also affect the kidneys. Examples of fibrocystic diseases include polycystic kidney disease (PKD) and nephronophthisis (NPHP), chronic tubulointerstitial disease and others. Fibrocystic diseases are caused by defects in proteins on the primary (immotile) cilia that interfere with receiving signals from other cells or fluids nearby. Fibrocystic diseases can be inherited as autosomal recessive , autosomal dominant , or X-linked recessive disorders 5.
Congenital hepatic fibrosis is caused by problems in the development of the portal veins and bile ducts before birth. These problems include malformation of embryonic structures called the ductal plates (the embryological precursor of the biliary system), secondary biliary strictures, and periportal fibrosis 6. The ductal plate is the cylindrical layer of cells surrounding branches of the portal veins. The ductal plate is a precursor of the intrahepatic bile ducts. The ductal plates normally develop into the network of bile ducts. Ductal plates arise around the smaller portal vein branches at a distance from the hilum. Progressive remodeling starts at 12 weeks’ gestation. Both interlobular and intralobular bile ductules develop from the ductal plate. The lack of remodeling of the ductal plate results in persistence of an excess of embryonic duct structures. This abnormality has been termed the ductal plate malformation 7 and consists of persistence of the ductal plate with an increase in duct elements and an increase in portal fibrous tissue.
In congenital hepatic fibrosis, the development of the ductal plates does not proceed normally, and the bile ducts remain immature. Branching of the portal vein network also proceeds abnormally, and excess fibrous tissue develops in the portal tracts. The malformation of the portal veins and bile ducts disrupts the normal flow of blood and bile, which leads to the progressive signs and symptoms of congenital hepatic fibrosis resulting in the development of portal hypertension.
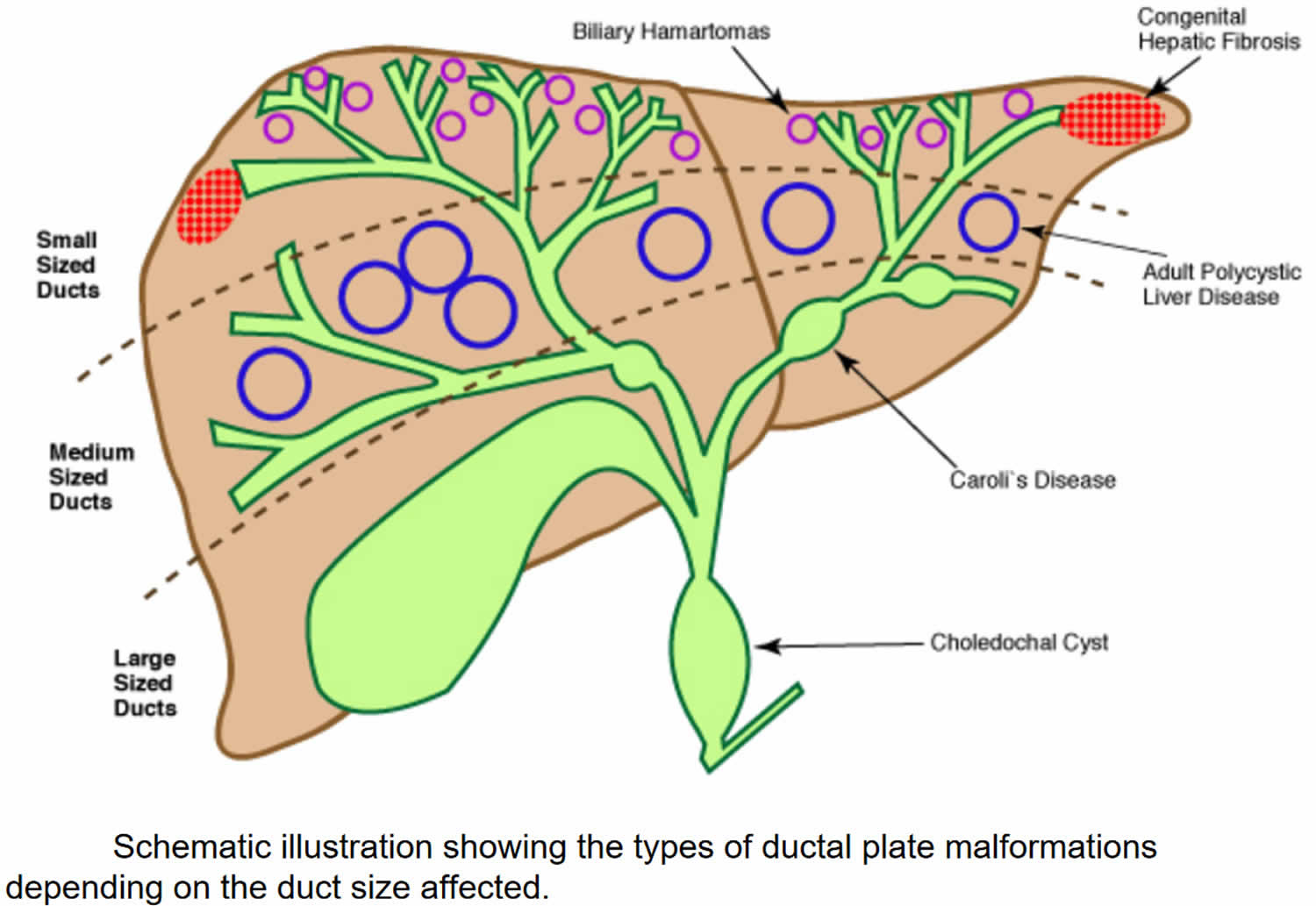
The family of fibropolycystic diseases are characterized by varying degrees of persistent bile duct structures, fibrosis, and duct dilatation. They are all developmental anomalies of the duct plate and occurred at various stages of remodeling. Congenital hepatic fibrosis is a ductal plate malformation of the small interlobular bile ducts, whereas Caroli disease involves the large intrahepatic bile ducts (see Figure 4).
Congenital hepatic fibrosis can manifest with hepatic fibrosis alone or it can be an important component of various fibrocystic diseases involving multiple systems 8. Also, congenital hepatic fibrosis is observed in X-linked and autosomal dominant inherited disease like oral-facial-digital syndrome type 1 and autosomal dominant polycystic kidney disease (ADPKD), respectively, and is reported in Prader-Willi syndrome, abernethy malformation, and different types of hepatic nodules (e.g., hepatocellular adenoma and hepatoblastoma) in individual case reports 9. In some cases, it was found that congenital hepatic fibrosis could coexist with congenital biliary atresia and ABCB4 mutation, but their association with congenital hepatic fibrosis has not been verified 10. Also, image analysis suggested Caroli syndrome or polycystic kidney disease despite the lack of genetic testing, which was often categorized into the “hepatorenal fibrocystic disease” family 11.
The classic renal lesion associated with congenital hepatic fibrosis is autosomal recessive polycystic kidney disease (ARPKD), which results in an impairment of renal functions. Its association with autosomal dominant polycystic kidney disease (ADPKD) is also recognized, especially among adults. The relationship of autosomal recessive polycystic kidney disease (ARPKD) to congenital hepatic fibrosis remains a controversial issue. The 2 conditions may actually be one disorder with different clinico-pathological presentations.
Autosomal recessive polycystic kidney disease (ARPKD) is caused by mutations in the polycystic kidney and hepatic disease 1 (PKHD1) gene 12, which consists of 86 exons that are variably assembled into numerous alternatively spliced transcripts 13. Most cases of autosomal recessive polycystic kidney disease (ARPKD) and congenital hepatic fibrosis are genetically homogeneous. However, the exact pathogenesis of association between congenital hepatic fibrosis and autosomal dominant polycystic kidney disease (ADPKD) still requires further research and study.
In all cases of congenital hepatic fibrosis and autosomal recessive polycystic kidney disease (ARPKD), a hepatic lesion of ductal plate malformation of the interlobular bile ducts is found; the difference in its presentation is primarily age dependent. Gradual disappearance of bile duct profiles associated with increased periportal fibrosis results from a progressive destructive cholangiopathy that involves the immature bile duct structures.
The hepatic disease progresses to develop portal hypertension associated with splenomegaly and esophageal varices. Congenital hepatic fibrosis is characterized by the intrahepatic form of portal hypertension, which is caused by the intrahepatic obstruction that affects the blood supply to the liver and subsequently leads to the development of cavernous transformations of the portal vein with a rise in portal venous pressure.
Congenital hepatic fibrosis is also associated with cholangitis. The presence of cholangitis or its repeated occurrence may influence the status of the hepatic lesion and the prognosis of the disease. Commonly, the hepatic lesion is associated with renal involvement characterized by cystic tubular dilatations, which affect both the cortical and medullary portions of the kidney. The longer the patient survives, the less characteristic the renal pathology becomes.
Figure 4. Congenital hepatic fibrosis malformation of the ductal plate
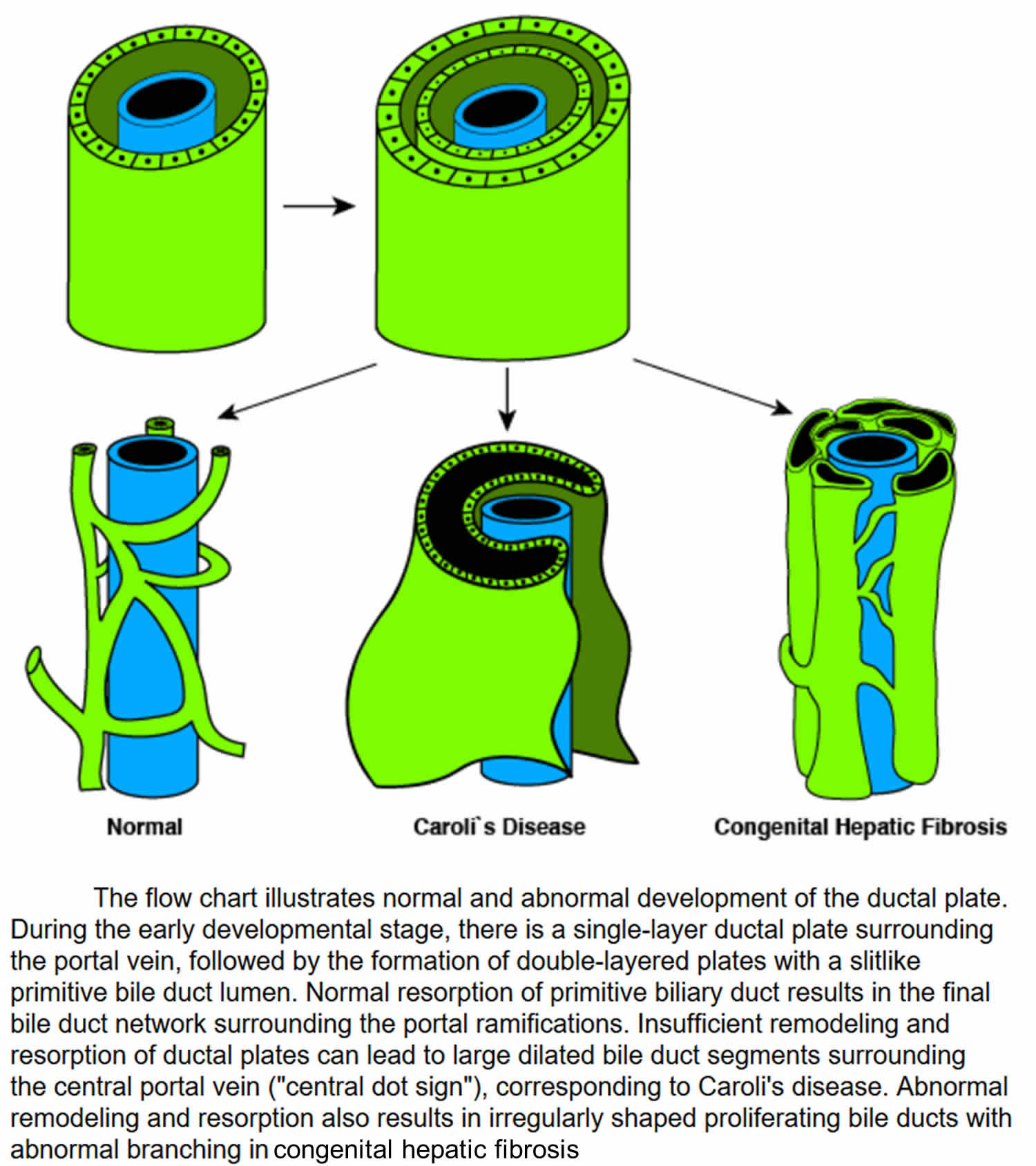
Figure 5. Ductal plate malformations
Congenital hepatic fibrosis inheritance pattern
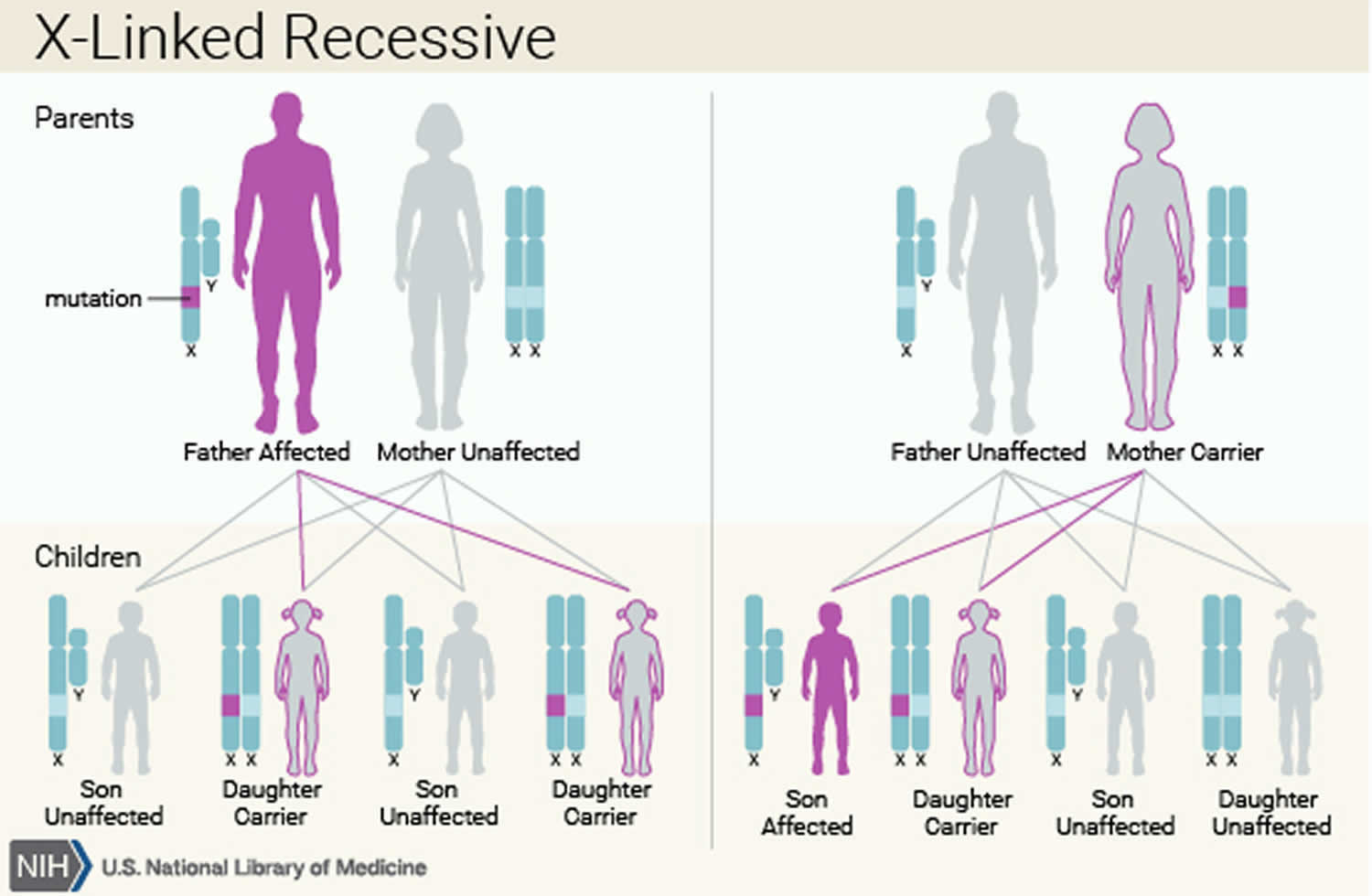
The various syndromes that include congenital hepatic fibrosis can have different inheritance patterns. Most of these disorders are inherited in an autosomal recessive pattern, which means both copies of the associated gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. Rare syndromes involving congenital hepatic fibrosis may be inherited in an X-linked recessive pattern, in which the gene associated with the syndrome is located on the X chromosome, which is one of the two sex chromosomes.
In isolated congenital hepatic fibrosis, the inheritance pattern is unknown.
Figure 6. Congenital hepatic fibrosis autosomal recessive inheritance pattern
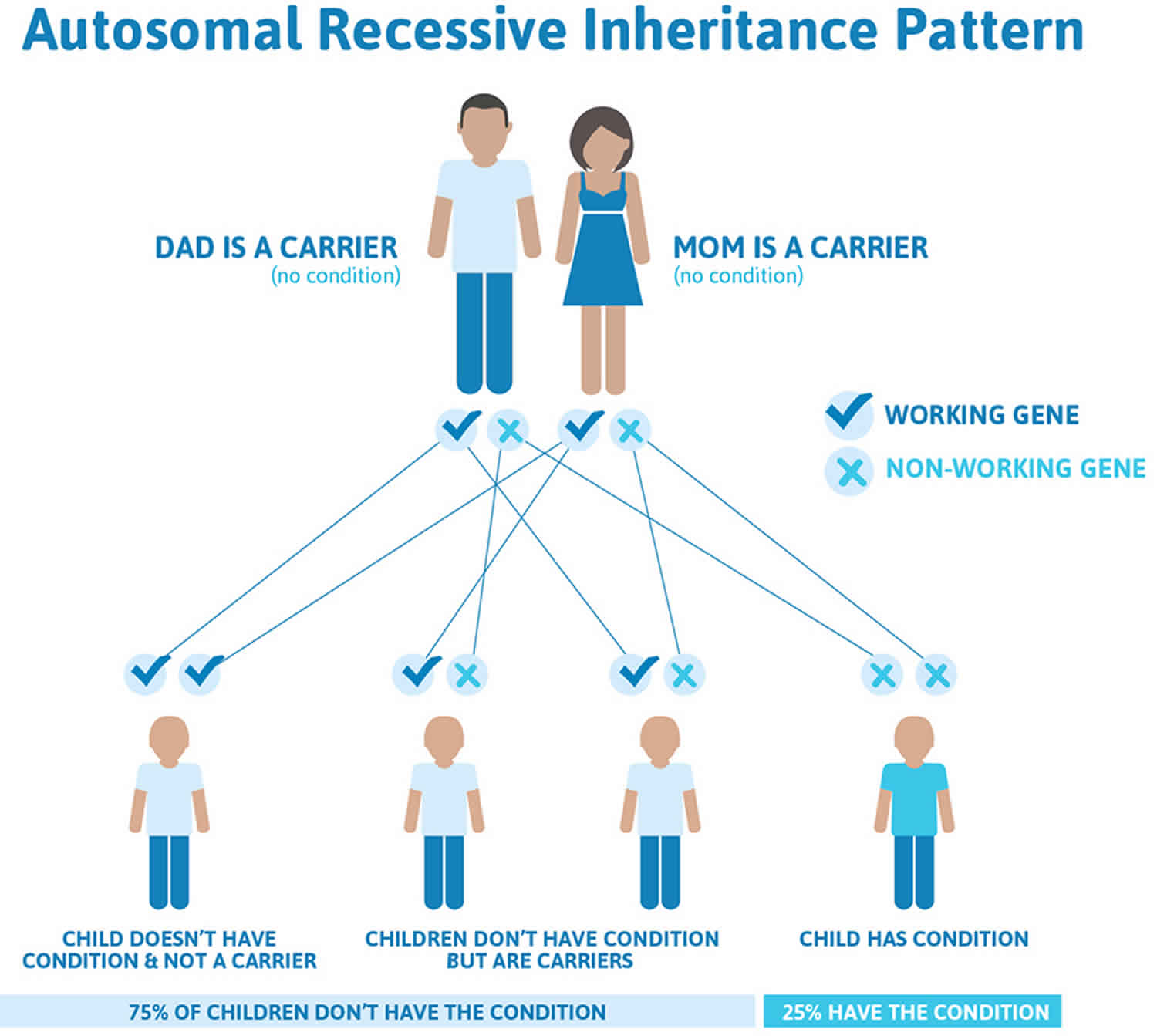
Figure 7. Congenital hepatic fibrosis X-linked recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Congenital hepatic fibrosis symptoms
The more obvious symptoms are a swollen abdomen, a firm, slightly enlarged and abnormally shaped liver and vomiting blood (hematemesis) due to bleeding from the enlarged blood vessels (varices) under the inner lining of the esophagus, stomach, and intestines. There is an increased risk for inflamed bile ducts (cholangitis) as well. The main findings in this disorder are identified through diagnostic testing. Many of the following signs are present in affected individuals with this disorder:
Portal hypertension
Increased pressure in the venous system that carries blood from multiple organs to the liver (portal system). Portal hypertension is strictly defined as an increase in the portal venous pressure leading to clinical sequelae, is the predominant manifestation of congenital hepatic fibrosis 14. This increased blood pressure is caused by the probable congenital abnormality of the portal vein as well as blockage of the portal blood supply to the liver due to excess connective tissue growth (fibrosis) in the liver. Portal hypertension can cause enlargement of the spleen and swollen or dilated veins of the esophagus.
In general, as hepatic fibrosis increases and portal hypertension worsens, the spleen increases in size, platelets and white blood cells decrease in number (hypersplenism), and porto-systemic vascular collaterals develop, including esophageal and gastric varices. As varices enlarge, the risk for bleeding increases 15. Variceal bleeding can occur at any age starting from infancy; however, significant portal hypertension takes time to develop and most commonly occurs in older children and adults.
Pulmonary hypertension and vascular shunts in the pulmonary parenchyma (hepatopulmonary syndrome) are complications of portal hypertension that can also be rarely seen in congenital hepatic fibrosis 5.
Other complications of portal hypertension including ascites and encephalopathy are less common in congenital hepatic fibrosis than in cirrhosis. Furthermore, individuals with congenital hepatic fibrosis rarely manifest other systemic features associated with chronic liver disease, such as gynecomastia and enlarged parotid gland, with the exception of spider angiomata 15.
Hepatic fibrosis: a fiber-like connective tissue that spreads through the liver.
Splenomegaly: an enlarged spleen. Liver function tests are usually normal in people with congenital hepatic fibrosis.
Nephromegaly: enlarged kidney.
Gastrointestinal bleeding: bleeding from the esophagus, and/or stomach and intestines that may cause the affected individual to vomit red blood or have dark black stools.
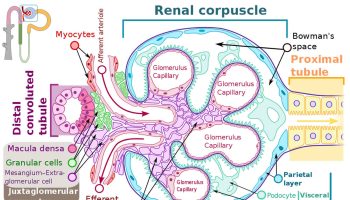
Polycystic kidney disease
Polycystic kidney disease is an inherited disorder in which cysts invade both kidneys. This causes enlargement in the size of the kidney while at the same time, reducing the amount of functional kidney tissue by compression.
Congenital hepatic fibrosis diagnosis
Congenital hepatic fibrosis is diagnosed by ultrasound exam and magnetic resonance imaging of the liver and kidneys, and rarely, by liver biopsy. congenital hepatic fibrosis and Caroli’s syndrome are often associated with cystic disease of the kidneys. Family history, physical exam, and various tests including kidney ultrasound exam, kidney function tests, X-rays, eye exam, brain MRI, and molecular genetic testing can help to determine the underlying fibrocystic disease syndrome.
Congenital hepatic fibrosis pathology
Liver histology, as revealed through the biopsy, reveals extensive hepatic fibrosis. The widened fibrous bands in the portal tract contain an increased number of ectatic and dysplastic branches of the interlobular bile ducts. The irregularly shaped proliferating bile ducts are lined by normal cuboidal epithelium.
The hepatic lobules are usually normal.
Cholestasis is observed in association with cholangitis. Other findings include portal vein branch hypoplasia and degeneration of the bile duct epithelium. Hypoplasia of the portal vein branches in association with supernumerous hepatic artery branches is also observed.
Liver biopsy reveals the following characteristic abnormalities associated with ductal plate malformation:
- Abundant, abnormally formed bile ducts in the portal tracts caused by an excess of embryonic bile duct structures remaining in their primitive ductal plate configuration 16 (often incorrectly described as “bile duct proliferation”)
- Abnormal branching of the portal vein
- Periportal fibrosis without inflammation
- Portal-portal bridging fibrosis (i.e., not the portal tract to central vein bridging that is typical of cirrhosis)
- Multiple bile duct hamartomas (von Meyenburg complexes) within dense fibrous stroma
- Inspissated bile in the lumen of some ducts
Note: The hepatic parenchyma is normal without intrahepatic cholestasis or disruption of the hepatocellular plates.
Histopathologic findings on liver biopsy are the gold standard for diagnosis of congenital hepatic fibrosis. However, liver biopsy is not required in most individuals (especially those with fibrocystic renal disease) because the diagnosis can be established on clinical findings alone 17.
Congenital hepatic fibrosis treatment
Treatment of congenital hepatic fibrosis is symptomatic and supportive. Complications of congenital hepatic fibrosis including gastrointestinal bleeding, hypersplenism and cholangitis can be routinely treated. Treatment is not available to correct the developmental abnormalities in the portal veins and bile ducts or reverse the fibrosis.
Affected individuals should avoid alcohol, medications known to impair liver function, and nonsteroidal anti-inflammatory drugs (NSAIDS).
Portal hypertension with secondary esophageal varices also requires treatment. Some episodes of variceal bleeding may spontaneously resolve. However, persistent hemorrhage that lasts longer than 12 hours or requires blood transfusion warrants the consideration of medical therapy, surgical therapy, or both.
Acute management includes intravenous fluid administration, nasogastric tube placement, and, once the patient is stable, an endoscopy. An initial pharmacologic approach with vasopressin, somatostatin, or other vasoconstricting medications is preferred in pediatrics. Each is discussed more thoroughly in the Medication section.
In cases of uncontrolled hemorrhage, one may resort to other interventions, including endoscopic sclerotherapy or band ligation, transjugular intrahepatic portosystemic shunting, or surgical shunting. Moreover, spontaneous extrahepatic portosystemic shunting has been reported in congenital hepatic fibrosis 18.
Genetic counseling is recommended for affected individuals and their families.
Surgical care
Portosystemic shunt surgery is the treatment of choice for these patients because the risk of postoperative hepatic encephalopathy is low. Patients also have a patent portal vein and preserved liver function. External or internal drainage may be required to resolve the refractory hepatobiliary infection.
Sclerotherapy is indicated for the treatment of acute hemorrhage from esophageal varices and as a primary therapy for management of recurrent or chronic variceal bleeding. Relative contraindications to the procedure include uncorrectable severe coagulopathy, fever, or compromise of respiratory status. Complications of sclerotherapy include ulcers, strictures, rebleeding, perforations, and bacteremia.
A Sengstaken-Blakemore tube may be required in some patients to control massive life-threatening bleeding. However, its current use is very much limited to patients who fail to respond to endoscopic sclerotherapy and in whom band ligation is not possible.
Endoscopic variceal ligation is an effective and safe method for early variceal obliteration in children. It is effective in controlling active bleeding and preventing recurrences. Types of surgical shunt include nonselective total portosystemic shunts, nonselective partial portosystemic shunts that maintain some antegrade blood flow to the liver, and selective portosystemic shunts, which decompress the gastroesophageal junction and the spleen through the splenic vein to the left renal vein.
Transjugular intrahepatic portosystemic shunts are considered for patients not amenable to sclerotherapy. It is particularly valuable in treating patients with refractory bleeding before liver transplantation.
Early shunt surgery with splenorenal or portacaval shunting may be required if repeated endoscopic sclerotherapy fails to arrest the variceal bleeding. Select the type of shunt carefully so that renal or hepatic transplantation remains a future option, with minimal limitations and complications 19.
Liver transplantation is also considered in the management of congenital hepatic fibrosis complicated by recurrent cholangitis or failure to respond to various medical and surgical therapeutic modalities resulting in progressive hepatic dysfunction 20.
Congenital hepatic fibrosis prognosis
The severity and prognosis of congenital hepatic fibrosis differ according to onset ages, and the therapeutic regimens vary accordingly 10. Although the liver disease in individuals with congenital hepatic fibrosis or Caroli syndrome is usually asymptomatic, the risk is increased for cholangitis and, less commonly, biliary stone formation and cholangiocarcinoma, which can develop at a relatively young age 15.
No prospective studies of the natural history of congenital hepatic fibrosis have been published. A recent thorough review of the literature evaluated a total of 1230 individuals with congenital hepatic fibrosis who had been published in 155 articles 21. A majority (64%) of the cases were classified as autosomal recessive polycystic kidney disease (ARPKD)-congenital hepatic fibrosis and 9.5% were diagnosed as isolated congenital hepatic fibrosis. Follow-up time ranged from 0 to 38 years with a mean of 7.5 years. Most affected individuals were diagnosed in childhood and sequelae of portal hypertension was the predominant reported finding (409 affected individuals). Of those with portal hypertension 164 had varices, 74 of whom had bled and 81 of whom had portosystemic shunting performed. Cholangitis was reported in 152 affected individuals and was fatal in three out of 23 persons who had undergone renal transplantation. Twenty-one affected individuals were reported to have had hepatobiliary cancers, 19 of whom had cholangiocarcinoma. The youngest reported individual with hepatobiliary cancer was age 33 years. The mean age of diagnosis for cholangiocarcinoma was 60.1 years.
Most other reports on the natural history of congenital hepatic fibrosis include small numbers of individuals with advanced congenital hepatic fibrosis and an undetermined type of renal disease. Whether the manifestations or rate of progression of congenital hepatic fibrosis differ according to the associated genetic disorder has not been determined. Although the manifestations of non-cirrhotic portal hypertension and the complications of the bile duct abnormalities associated with autosomal recessive polycystic kidney disease (ARPKD)-congenital hepatic fibrosis are well recognized, the variability in progression of congenital hepatic fibrosis, even within the same family, makes prognostication difficult 22.
Factors that could exacerbate or accelerate fibrosis include excessive alcohol intake, steatohepatitis, hepatotoxic medicines, and viral hepatitis.
- Lipschitz B, Berdon WE, Defelice AR, Levy J. Association of congenital hepatic fibrosis with autosomal dominant polycystic kidney disease. Report of a family with review of literature. Pediatr Radiol. 1993. 23(2):131-3.[↩]
- Congenital Hepatic Fibrosis. https://emedicine.medscape.com/article/927984-overview[↩]
- Hoyer PF. Clinical manifestations of autosomal recessive polycystic kidney disease. Curr Opin Pediatr. 2015 Apr. 27 (2):186-92.[↩]
- Congenital hepatic fibrosis. https://ghr.nlm.nih.gov/condition/congenital-hepatic-fibrosis[↩][↩]
- Gunay-Aygun M, Gahl WA, Heller T. Congenital Hepatic Fibrosis Overview ─ ARCHIVED CHAPTER, FOR HISTORICAL REFERENCE ONLY. 2008 Dec 9 [Updated 2014 Apr 24]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK2701[↩][↩][↩]
- Akhan O, Karaosmanoglu AD, Ergen B. Imaging findings in congenital hepatic fibrosis. Eur J Radiol. 2007 Jan. 61(1):18-24.[↩]
- Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology. 1992 Oct. 16(4):1069-83.[↩]
- Janowski K., Goliszek M., Cielecka-Kuszyk J., Jankowska I., Pawlowska J. Congenital hepatic fibrosis in a 9-year-old female patient – a case report. Clinical and Experimental Hepatology. 2017;3(3):176–179. doi: 10.5114/ceh.2017.70299[↩]
- Kadakia N., Lobritto S. J., Ovchinsky N., et al. A challenging case of hepatoblastoma concomitant with autosomal recessive polycystic kidney disease and Caroli syndrome-review of the literature. Frontiers in Pediatrics. 2017;5:p. 114. doi: 10.3389/fped.2017.00114[↩]
- Zhu B, Du Z, Wang Z, Li Y, Zhang J, Zhu H. Congenital Hepatic Fibrosis in Children and Adults: Clinical Manifestations, Management, and Outcome-Case Series and Literature Review. Gastroenterol Res Pract. 2020;2020:8284274. Published 2020 Apr 21. doi:10.1155/2020/8284274 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7191434[↩][↩]
- Shenoy P., Zaki S. A., Shanbag P., Bhongade S. Caroli’s syndrome with autosomal recessive polycystic kidney disease. Saudi Journal of Kidney Diseases and Transplantation. 2014;25(4):840–843. doi: 10.4103/1319-2442.135176[↩]
- Harris PC, Rossetti S. Molecular genetics of autosomal recessive polycystic kidney disease. Mol Genet Metab. 2004 Feb. 81(2):75-85.[↩]
- Losekoot M, Haarloo C, Ruivenkamp C, et al. Analysis of missense variants in the PKHD1-gene in patients with autosomal recessive polycystic kidney disease (ARPKD). Hum Genet. 2005 Nov. 118(2):185-206.[↩]
- Summerfield JA, Nagafuchi Y, Sherlock S, Canafalch J, Scheuer PJ. Hepatobiliary fibropolycystic disease: a clinical and histological review of 51 patients. J Hepatol. 1986;2:141–56.[↩]
- Fonck C, Chauveau D, Gagnadoux MF, Pirson Y, Grunfeld JP. Autosomal recessive polycystic kidney disease in adulthood. Nephrol Dial Transplant. 2001;16:1648–52.[↩][↩][↩]
- Desmet VJ. Ludwig symposium on biliary disorders–part I. Pathogenesis of ductal plate abnormalities. Mayo Clin Proc. 1998;73:80–9.[↩]
- Gunay-Aygun M, Font-Montgomery E, Lukose L, Tuchman Gerstein M, Piwnica-Worms K, Choyke P, Daryanani K, Turkbey B, Fischer R, Bernardini I, Sincan M, Zhao X, Sandler N, Roque A, Douek D, Graf J, Huizing M, Bryant J, Mohan P, Gahl W, Heller T. Characteristics of congenital hepatic fibrosis in a large cohort of patients with autosomal recessive polycystic kidney disease. Gastroenterology. 2013;144:112–21.e2.[↩]
- Rossi G, di Chio T, Nastasio S, Maggiore G. Spontaneous Extra-hepatic Portosystemic Shunt in Congenital Hepatic Fibrosis. J Pediatr Gastroenterol Nutr. 2016 Feb 4.[↩]
- Al-Lawati TT. Fibropolycystic disease of the liver and kidney in Oman. Arab J Gastroenterol. 2013 Dec. 14 (4):173-5.[↩]
- Chandar J, Garcia J, Jorge L, Tekin A. Transplantation in autosomal recessive polycystic kidney disease: liver and/or kidney?. Pediatr Nephrol. 2015 Aug. 30 (8):1233-42.[↩]
- Srinath A, Shneider BL. Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease. J Pediatr Gastroenterol Nutr. 2012;54(5):580-587. doi:10.1097/MPG.0b013e31824711b7 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4369775[↩]
- Adeva M, El-Youssef M, Rossetti S, Kamath PS, Kubly V, Consugar MB, Milliner DM, King BF, Torres VE, Harris PC. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 2006;85:1–21.[↩]


{kind=link}