Costello syndrome
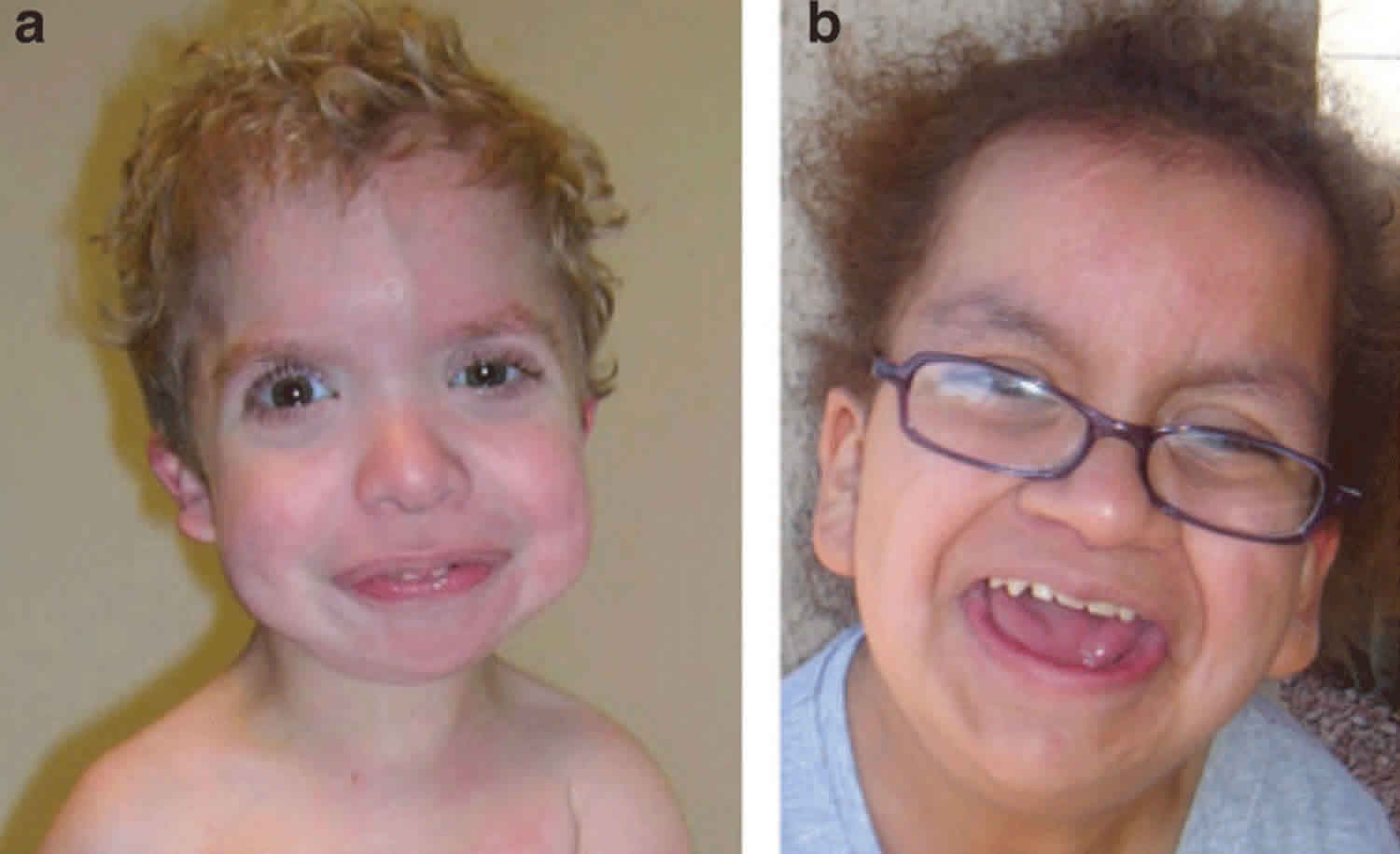
Costello syndrome is an extremely rare disorder that affects many parts of the body. Costello syndrome is characterized by delayed development and intellectual disability, loose folds of skin which are especially noticeable on the hands and feet, unusually flexible joints, and distinctive facial features including a large mouth with full lips. Heart problems are common, including an abnormal heartbeat (arrhythmia), structural heart defects, and a type of heart disease that enlarges and weakens the heart muscle (hypertrophic cardiomyopathy). Infants with Costello syndrome may be larger than average at birth, but most have difficulty feeding and grow more slowly than other children. People with this condition have relatively short stature and may have reduced growth hormone levels. Other signs and symptoms of Costello syndrome can include tight Achilles tendons (which connect the calf muscles to the heel), weak muscle tone (hypotonia), a structural abnormality of the brain called a Chiari 1 malformation, skeletal abnormalities, dental problems, and problems with vision.
Beginning in early childhood, people with Costello syndrome are at an increased risk of developing certain cancerous and noncancerous tumors. The most common noncancerous tumors associated with this condition are papillomas, which are small, wart-like growths that usually develop around the nose and mouth or near the anus. The most common cancerous tumor associated with Costello syndrome is a childhood cancer called rhabdomyosarcoma, which begins in muscle tissue. Neuroblastoma, a tumor that arises in developing nerve cells, also has been reported in children and adolescents with this syndrome. In addition, some teenagers with Costello syndrome have developed transitional cell carcinoma, a form of bladder cancer that is usually seen in older adults.
Costello syndrome belongs to a group of related conditions called the RASopathies. The signs and symptoms of Costello syndrome overlap significantly with those of two other genetic conditions, cardiofaciocutaneous syndrome (CFC syndrome) and Noonan syndrome. In affected infants, it can be difficult to tell the three conditions apart based on their physical features. However, the conditions can be distinguished by their genetic cause and by specific patterns of signs and symptoms that develop later in childhood.
Costello syndrome is very rare; it probably affects 200 to 300 people worldwide. Reported estimates of Costello syndrome prevalence range from 1 in 300,000 to 1 in 1.25 million people 1.
Costello syndrome is caused by changes (mutations) in the HRAS gene. Costello syndrome is considered an autosomal dominant condition, but almost all cases are the result of de novo gene mutations and occur in people with no family history of the condition 2.
The treatment of Costello syndrome is directed toward the specific signs and symptoms that are apparent in each person 3. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, physicians who diagnose and treat abnormalities of the heart (cardiologists), physicians who diagnose and treat skeletal abnormalities (orthopedists), orthopedic surgeons, specialists who diagnose and treat abnormalities of the skin (dermatologists), speech pathologists, dietitians, and other health care professionals may need to systematically and comprehensively plan an affected child’s treatment.
Individuals with cardiac abnormalities, such as hypertrophic cardiomyopathy, may be treated with certain medications (e.g., beta-blockers or calcium channel blockers, antiarrhythmic medications), surgical intervention, and/or other measures may be necessary. The specific surgical procedures performed will depend upon the severity and location of the anatomical abnormalities, their associated symptoms, and other factors.
Bracing, occupational and physical therapy may be used to treat ulnar deviation of the wrists. Surgery may be used to lengthen Achilles tendons. Facial papillomata can be removed with dry ice.
Early intervention is important to ensure that children with Costello syndrome reach their potential. Services that may be beneficial include special remedial education, speech therapy, special social support, and other medical, social, and/or vocational services.
Genetic counseling is recommended for affected individuals and their families. Other treatment for this disorder is symptomatic and supportive.
Costello syndrome quick facts
- Costello syndrome is a rare, genetic disorder that affects many parts of the body. There is no cure.
- Costello syndrome causes neurocognitive delays and impaired learning.
- Many children struggle to walk, talk, and feed themselves.
- Although infants with Costello syndrome may be large at birth, they have difficulty feeding and grow more slowly than other infants. They may require a feeding tube as infants but are often able to eat on their own as they grow.
- Many people with Costello syndrome have gastroesophageal reflux disease (GERD).
- Problems with the heart are common. About 85% of those with Costello syndrome have at least one issue with their heart.
- Beginning in early childhood, people with Costello syndrome have a greater risk of developing cancerous (malignant) and noncancerous (benign) tumors. Individuals with Costello syndrome have a 15% lifetime risk of developing malignant tumors, with the highest risk occurring during childhood. Noncancerous (benign) papillomas and cancerous (malignant) tumors may occur in a muscle, nerve cells, or bladder. Rhabdomyosarcoma, neuroblastoma, and bladder carcinoma are the most common malignant tumors seen in people with Costello syndrome.
- Most people with Costello syndrome will have skeletal and orthopedic problems.
Costello syndrome causes
Mutations in the HRAS gene cause Costello syndrome. This gene provides instructions for making a protein called H-Ras, which is part of a pathway that helps control cell growth and division. Mutations that cause Costello syndrome lead to the production of an H-Ras protein that is abnormally turned on (active). The overactive protein directs cells to grow and divide constantly, which can lead to the development of cancerous and noncancerous tumors. It is unclear how mutations in the HRAS gene cause the other features of Costello syndrome, but many of the signs and symptoms probably result from cell overgrowth and abnormal cell division.
Some people with signs and symptoms like those of Costello syndrome do not have an identified mutation in the HRAS gene. These individuals may actually have CFC syndrome or Noonan syndrome, which are caused by mutations in related genes. The proteins produced from these genes interact with one another and with the H-Ras protein as part of the same cell growth and division pathway. These interactions help explain why mutations in different genes can cause conditions with overlapping signs and symptoms.
Costello syndrome inheritance pattern
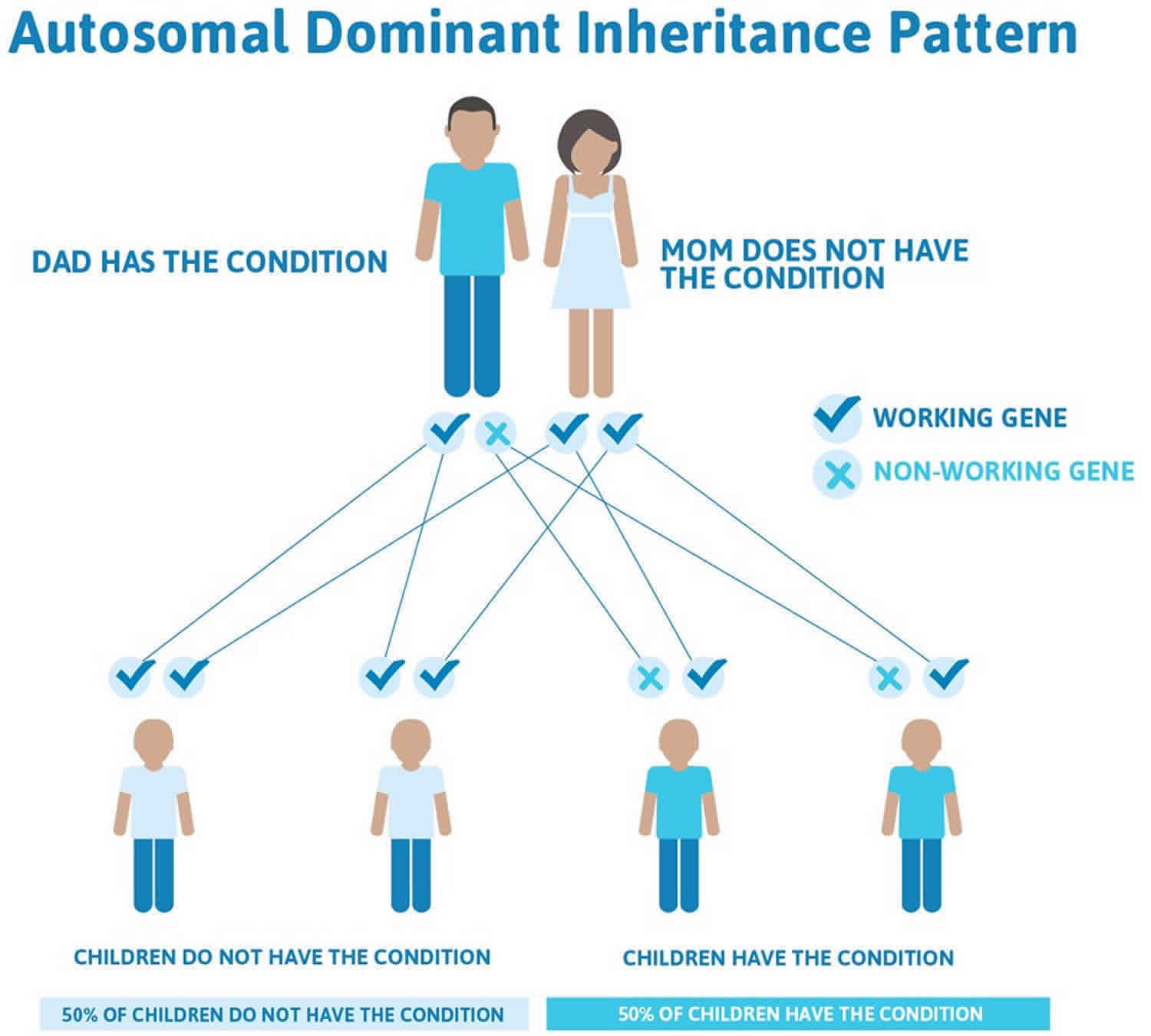
Costello syndrome is considered to be an autosomal dominant condition, which means one copy of the altered gene in each cell is sufficient to cause the disorder. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females. However, almost all reported cases have resulted from new gene mutations and have occurred in people with no history of the disorder in their family.
There have been a few reports of more than one sibling with Costello syndrome in a family. This is most likely due to germ cell mosaicism in which some of the parent’s reproductive cells (germ cells) carry the HRAS gene mutation whereas others contain a normal gene. As a result, one or more of the parent’s children may inherit the gene mutation, leading to manifestation of the autosomal dominant disorder, but the parent may have no apparent symptoms.
Figure 1. Costello syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Costello syndrome symptoms
Infants with Costello syndrome typically have a normal or high birth weight, but show poor sucking ability, have swallowing difficulties, and fail to grow and gain weight at the expected rate (failure to thrive). Growth delay after birth typically results in short stature during childhood and adulthood. Affected children may have developmental delay or mild to moderate intellectual disability. In some individuals, speech development and/or the ability to walk is significantly delayed. Children with Costello syndrome generally have warm, sociable personalities.
Individuals with Costello syndrome typically have loose skin (cutis laxa) on the neck, palms, fingers, and soles. The skin in these areas may lack elasticity and hang loosely; in addition, the skin may appear wrinkled and thickened. In some cases, certain areas of the skin may become unusually dark (hyperpigmentation). In addition, most patients with this disorder develop dry hardened patches of skin (hyperkeratosis) with unusually deep creases on the palms and soles. Some affected individuals may also have skeletal abnormalities such as dislocated hips, abnormally flexible (hyperextensible) joints of the fingers, wrists bent toward the little finger (ulnar deviation) and/or unusual tightening of the fibrous cords on the back of the heels (Achilles tendon). Additional skeletal abnormalities include side-to-side curvature of the spine (scoliosis), front-to-back curvature of the spine (kyphosis), and reduced range of motion in the shoulder and elbows.
Children with Costello syndrome usually devlelop papillomata around the mouth and nostrils. Papillomata may develop as early as two years of age or at older ages. In some cases, these wart-like (verrucal) lesions may be found near the anus. Papillomata usually become more apparent with age. Other benign tumors have also been reported.
Children with Costello syndrome have a distinctive facial appearance. Characteristic facial features may include an abnormally large head (macrocephaly); low-set ears with large, thick lobes; unusually thick lips; a large, depressed nasal bridge; abnormally wide nostrils (nares); and a coarse facial appearance. In addition, affected children may have unusually curly hair and/or sparse, thin hair on the front (anterior) of the head. Some children have folds of skin over the inner corners of the eyes (epicanthal folds).
In early childhood relative overgrowth of the hindbrain compared to the space available in the posterior fossa of skull cavity can result in crowding and neurologic problems. Because severe crowding requires surgical intervention, screening with brain and cervical spine MRI has been suggested.
Eye and vision changes are common and include nystagmus (rapid eye movements) in younger individuals, strabismus and rarely in older individuals keratoconus (abnormal thickening of the cornea).
Children with Costello syndrome often have certain heart abnormalities. These may include structural malformations of the heart that are present at birth (congenital heart defects); abnormal thickening of the muscular walls of the left lower chamber of the heart (hypertrophic cardiomyopathy); leakage of the valve between the left upper (atrial) and lower (ventricular) heart chambers (mitral valve prolapse); and/or other cardiac defects. Associated symptoms and findings may include abnormal heart sounds (heart murmurs) that may be detected by a physician through use of a stethoscope; shortness of breath, particularly upon exertion; faintness; chest pain; abnormal heart rhythms (arrhythmias); and/or other findings that may potentially lead to life-threatening complications without appropriate treatment.
Affected individuals have an approximately 15% lifetime risk to develop malignant tumors such as a cancer of the muscle tissue (rhabdomyosarcoma), a cancer of the nerve cells (neuroblastoma), and transitional cell carcinoma of the bladder.
In some cases, the symptoms and findings of Costello syndrome overlap with two similar disorders known as Noonan syndrome and cardiofaciocutaneous syndrome which are caused by mutations in different genes.
Clinical description
Costello syndrome affects multiple organ systems. Its typical presentation is characterized by diffuse hypotonia and severe feeding difficulties in infancy; short stature; developmental delay or intellectual disability; characteristic facial features; curly or sparse, fine hair; loose, soft skin with deep palmar and plantar creases; papillomata of the face and perianal region; joint laxity with ulnar deviation of the wrists and fingers; tight Achilles tendons; and cardiac involvement (hypertrophic cardiomyopathy, congenital heart defect, and arrhythmia). Postnatal cerebellar overgrowth can result in Chiari I malformation with associated hydrocephalus or syringomyelia. An approximately 15% lifetime risk for malignant tumors includes rhabdomyosarcoma and neuroblastoma in young children and transitional cell carcinoma of the bladder in adolescents and young adults. Females and males are equally affected.
In rare instances, related to the underlying HRAS pathogenic variant, the presentation is more severe with intrauterine hydrops, postnatal pulmonary effusions with respiratory compromise, and severe progressive hypertrophic cardiomyopathy, resulting in early lethality 4. Other rare variants are associated with a milder or attenuated phenotype, encompassing milder developmental delay, less striking facial features resembling Noonan syndrome, and a lower risk for malignancies 5.
Growth
Increased birth weight and head circumference (often >50th centile) for gestational age can lead to the categorization of Costello syndrome as macrosomia, which is misleading. Short stature is universal, delayed bone age is common 6 and testing may show partial or complete growth hormone deficiency.
Normative growth charts, derived from measurements of individuals who had not used growth hormone, document the very slow weight gain in early infancy, as well as the short stature with the 95th centile for individuals with Costello syndrome falling into the low normal range of typical age-matched individuals 7. The reported adult height range is 135-150 cm 8.
Failure to thrive and severe feeding difficulties are almost universal and typically necessitate the placement of a gastric feeding tube. Anecdotally, affected children have very high caloric needs. Even after nutrition is improved through supplemental feeding, growth retardation persists; therefore, aggressive feeding therapy is not effective.
Children are able to take oral feeds beginning between ages two and four years.
The first acceptable tastes are often strong (e.g., ketchup).
Neurologic
Most infants show hypotonia, irritability, developmental delay, and nystagmus.
Hypotonia may be severe with low muscle mass and a skeletal myopathy 9.
Progressive postnatal cerebellar overgrowth may result in development of a Chiari 1 malformation, syringomyelia, and hydrocephalus 10. Cerebellar abnormalities include tonsillar ectopia or Chiari malformation, occasionally associated with syringomyelia 11.
EEG abnormalities are seen in approximately one third of individuals; between 20% and 50% have seizures 12.
Cardiac
Cardiac abnormalities, which typically present in infancy or early childhood, may be recognized at any age. In 146 individuals with molecularly confirmed Costello syndrome, 87% had some type of cardiovascular abnormality. A congenital heart defect was present in 44%, with non-progressive valvar pulmonic stenosis being the most common finding. Rarely, atrial septal defects are seen. hypertrophic cardiomyopathy comprising typical subaortic septal hypertrophy was noted in 61% and pathologic myocardial disarray was seen in 70% of those studied.
A few neonates can present with very severe hypertrophic cardiomyopathy that is lethal. In other infants, progressively severe hypertrophic cardiomyopathy and/or severe multifocal atrial tachycardia can lead to death in the first two years of life. Multifocal atrial tachycardia and other atrial tachycardia may be very concerning but are usually self-limited with aggressive treatment.
Pulmonic valve stenosis is usually mild to moderate, and infrequently requires surgery or interventional catheterization.
Most children with hypertrophic cardiomyopathy have either mild or moderate involvement. Of great interest are the few with moderate-to-severe involvement who appear to have “remodeling” over many years which gives the impression of disappearance of (or marked decrease in) left ventricular obstruction. Only a small number of these individuals are being followed, and their long-term natural history is incomplete 13. In addition to the rare severe lethal form, hypertrophic cardiomyopathy can be chronic (persistence of a gradient) or progressive (increase in gradient severity; 14/37 [37%]), stabilizing (without further increase in severity; 10/37 [27%]), or decreasing (resolving; 5/37 [14%]). Outcome was unavailable in 8/37 (22%) 13, necessitating prudent surveillance.
Non-reentrant atrial tachycardias are generally self-limited, but may persist or worsen in approximately one fourth of affected individuals. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy 14.
Older individuals (ages 16 to 40 years) with moderate hypertrophic cardiomyopathy or new-onset arrhythmia (both atrial and ventricular) represent the greatest challenge and do not constitute a predictable outcome “phenotype” until more information is obtained. Hypertension is not uncommon.
Mild-moderate aortic dilation not associated with bicuspid aortic valve is a recent cardiovascular finding 13 that occurs in approximately 5% of affected individuals.
Primary vascular disease has rarely been reported. In one individual with early lethal Costello syndrome due to the rare p.Gly12Glu variant, pulmonary vascular dysplasia affecting small arteries and veins with abnormal elastin distribution was seen in the absence of significant hypertrophic cardiomyopathy 15.
Developmental delay and intellectual disability
Developmental delay or intellectual disability is present in all individuals 16.
Recognition memory in verbal memory functioning is relatively preserved compared to other cognitive tasks 17.
The onset of speech frequently coincides with the willingness to feed orally.
Separation anxiety, seen in 39% of individuals with Costello syndrome, is more common in males than in females 16.
Many children younger than age four years meet criteria on a screening tool for autism spectrum disorder. There is a positive correlation with the need for gastrostomy tube and inability to walk independently. In contrast, none of the children older than age four years met criteria for autism, suggesting that early signs consistent with autism spectrum disorder tend to resolve by age four years 18.
Limited detailed information is available on the quality of life in older individuals with Costello syndrome. Quality of life in individuals age 16-34 years is compromised by four factors 19: limited relationships outside of the immediate circle of friends and family, lack of independence, male gender, and the presence of major medical issues 19. Functional limitations from orthopedic problems regarding mobility, as well as limitations in the social and cognitive domains, were documented using normative scales 20.
Dermatologic
Papillomata, absent in infancy, appear in young children, usually in the perinasal region and less commonly in the perianal region, torso, and extremities. While papillomata are mostly of cosmetic concern, they can become noticeable and at times bothersome.
Palmoplantar keratoderma is common and can affect function in severe cases 21. Additional findings include acanthosis nigricans and thick toenails.
Musculoskeletal
Individuals with Costello syndrome have very loose joints, particularly involving the fingers. Ulnar deviation of the wrists and fingers is also common. Developmental hip dysplasia may result in severe pain and prevent ambulation. Tight Achilles tendons may develop.
More than half of a cohort of 43 individuals examined by an orthopedic surgeon with review of radiographs as available showed ligamentous laxity, scoliosis, kyphosis, characteristic hand and wrist deformities, shoulder and elbow contractures, tight Achilles tendons, and flat feet 22. Hip dysplasia, seen in 45%, was not universally congenital but acquired in some.
Osteoporosis is common in young adults with Costello syndrome 22. In adults ranging in age from 16 to 40 years, all eight individuals who had a bone density measurement had abnormal results that suggested osteoporosis or osteopenia; three had bone pain, vertebral fractures, and height loss 23. In a study of nine individuals with Costello syndrome who had dual-energy X-ray absorptiometry, all showed significantly decreased bone mineral density compared to age-matched controls 24.
Respiratory
Seven of ten individuals ages three to 29 years undergoing polysomnography in the sleep laboratory had obstructive events 25. A literature review showed respiratory complications in 78% of neonates, with the majority resolving and more severe complications only in those with rare HRAS pathogenic variants associated with the severe phenotype 26.
Upper-airway obstruction was seen more often in older children and young adults 26.
Endocrine
Neonatal hyperinsulinism has been reported 27 and, in one case, was correlated to focal uniparental disomy for 11p within the pancreatic nodule 28.
In older individuals, hypoglycemia may be related to growth hormone deficiency. Growth hormone deficiency is common (30%-50%) 10.
Several patients have been diagnosed with hypothyroidism requiring treatment with hormone replacement.
Other endocrine issues may include delayed or dysregulated puberty including precocious puberty.
Solid tumors
Benign and malignant solid tumors occur with far greater frequency in individuals with Costello syndrome than in the general population. The overall tumor incidence is approximately 15% over the lifetime of individuals with an identified HRAS pathogenic variant 29. Kratz et al 30 reviewed published cases and confirmed the 15% cumulative incidence of cancer in individuals with Costello syndrome by age 20 years. Rhabdomyosarcoma occurs most frequently, followed by neuroblastoma, transitional cell carcinoma of the bladder, and other solid tumors 31.
Rhabdomyosarcoma and neuroblastoma, tumors of early childhood, present in Costello syndrome at ages comparable to the general population. In contrast, transitional cell carcinoma of the bladder, which occurs in older adults (70% age >65 years) in the general population, may be found in adolescents with Costello syndrome. The ages at presentation in the three individuals with Costello syndrome with transitional cell carcinoma of the bladder were ten, 11, and 16 years.
Other
- Pyloric stenosis occurs more commonly than in the general population 32.
- Adult-onset gastroesophageal reflux was present in four individuals in the series of White et al 23; additional cases are known.
- Dental abnormalities, including enamel defects, occur frequently. Malocclusion with maxillary first molars positioned posteriorly to the mandibular first molars is common and may contribute to obstructive sleep apnea 33. Excessive secretions are often noted 6.
- In addition to the common vision disturbance and nystagmus, less common eye abnormalities include retinal dystrophy 34 and keratoconus 35.
- Adolescents may appear older than their chronologic age because of worsening kyphoscoliosis, sparse hair, and prematurely aged skin.
Costello syndrome diagnosis
Costello syndrome is diagnosed by clinical examination and specific diagnostic criteria have been developed. Molecular genetic testing for mutations in the HRAS gene is available to confirm the diagnosis. Most clinically affected individuals have an identifiable HRAS mutation. Experts in the field suggested that individuals without an identifiable HRAS mutation should not be diagnosed with Costello syndrome, as they most likely have a related condition such as Noonan or cardiofaciocutaneous syndrome. The specific mutation in the HRAS gene, often referred to by the resulting amino acid change, is important to identify. While most individuals with Costello syndrome share a mutation that results in a change of the amino acid glycine in position 12 to serine, a number of changes have been seen. The prognosis for a patient is affected by the specific mutation, as some present with more severe medical problems than others.
Costello syndrome treatment
Failure to thrive is the most common and challenging clinical problem; most infants require nasogastric or gastrostomy feeding; many require Nissen fundoplication. Treatment of cardiac manifestations and malignancy is routine. Ulnar deviation of the wrists and fingers often requires early bracing and occupational and/or physical therapy; tight Achilles tendons may require surgical tendon lengthening. Developmental disability requires early-intervention programs and individualized education strategies. Recurrent facial papillomata may require routine removal with dry ice. Hemodynamically significant valvar stenoses require antibiotic prophylaxis for subacute bacterial endocarditis; anesthesia may pose a risk to those with hypertrophic cardiomyopathy or those predisposed to some types of atrial tachycardia.
Table 1. Recommended evaluations following initial diagnosis in individuals with Costello syndrome
| System/Concern | Evaluation | Comment |
|---|---|---|
| Constitutional | Measurement of height, weight, head circumference | Failure to thrive is common & persists in spite of adequate caloric intake. |
| Gastrointestinal/ Feeding | Asses nutritional status, feeding, gastroesophageal reflux disease | Failure to thrive is typical; a feeding tube is typically necessary. |
| Neurologic | Examination by neurologist for clinical signs of Chiari I malformation &/or sryingomyelia |
|
| EEG if seizures are a concern | ||
| Development | Developmental assessment |
|
| Cardiac | Evaluation by cardiologist for congenital heart defects, hypertrophic cardiomyopathy, arrhythmia | Standard evaluation |
| Respiratory | Refer as needed to a pulmonologist | Upper- & lower-airway issues may occur; hypertrophy of tonsils & adenoids may contribute to upper-airway obstruction. |
| Musculoskeletal | Evaluation by pediatric orthopedist | Assess spine & extremities w/attention to hip joint abnormalities & range of motion |
| Genitourinary | Assess males for cryptorchidism | |
| Eyes | Ophthalmology evaluation |
|
| Skin | Papillomata and hyperkeratosis require referral to a dermatologist | Treat symptomatically |
| Dental | Evaluation by a pediatric dentist recommended | Enamel defects & malocclusion are common |
| Endocrine | Pediatric endocrinology evaluation for hypoglycemia, growth hormone deficiency | May require additional evaluation for dysregulated puberty |
| Psychiatric/ Behavioral | Neuropsychiatric evaluation as needed | In individuals age >4 yrs: screen for behavior problems incl sleep disturbances, attention-deficient and hyperactivity disorder, anxiety, &/or traits suggestive of autism spectrum disorder. |
| Miscellaneous/ Other | Consultation w/clinical geneticist &/or genetic counselor | To include genetic counseling |
Growth hormone treatment
If treatment with growth hormone is contemplated, its unproven benefit and potential risks should be thoroughly discussed in view of the established risks of cardiomyopathy and malignancy in individuals with Costello syndrome. Growth hormone replacement has not been shown to increase these risks.
- Unproven benefit. Individuals with Costello syndrome frequently have low growth hormone levels:
- True growth hormone deficiency requires growth hormone replacement. Three individuals with growth hormone deficiency showed increased growth velocity without adverse effects after three to seven years of replacement therapy, but two continued to have short stature 36.
- It is unclear from the literature if the use of growth hormone is beneficial in individuals with Costello syndrome with partial growth hormone deficiency. An abnormal growth hormone response on testing and a good initial growth response have been reported 37.
- Cardiac hypertrophy. Whether the anabolic actions of growth hormone accelerate pre-existing cardiac hypertrophy is not known, but early descriptive studies do not suggest a clear association 13. In rare cases, cardiomyopathy has progressed after initiation of growth hormone treatment; whether the relationship was causal or coincidental is unknown 38.
- Malignancy. The effect of growth hormone on tumor predisposition has not been determined. Two reports have raised the possibility of an association:
Developmental Delay and Intellectual Disability Management Issues
The following information represents typical management recommendations for individuals with developmental delay / intellectual disability in the United States; standard recommendations may vary from country to country.
- Ages 0-3 years. Referral to an early intervention program is recommended for access to occupational, physical, speech, and feeding therapy. In the US, early intervention is a federally funded program available in all states.
- Ages 3-5 years. In the US, developmental preschool through the local public school district is recommended. Before placement, an evaluation is made to determine needed services and therapies and an individualized education plan (Individual Education Plan) is developed.
- All ages. Consultation with a developmental pediatrician is recommended to ensure the involvement of appropriate community, state, and educational agencies and to support parents in maximizing quality of life. Some issues to consider:
Individual Education Plan services:
- An Individual Education Plan provides specially designed instruction and related services to children who qualify.
- Individual Education Plan services will be reviewed annually to determine if any changes are needed.
- As required by special education law, children should be in the least restrictive environment feasible at school and included in general education as much as possible and when appropriate.
- Vision and hearing consultants should be a part of the child’s Individual Education Plan team to support access to academic material.
PT, OT, and speech services will be provided in the Individual Education Plan to the extent that the need affects the child’s access to academic material. Beyond that, private supportive therapies based on the affected individual’s needs may be considered. Specific recommendations regarding type of therapy can be made by a developmental pediatrician. - As a child enters teen years, a transition plan should be discussed and incorporated in the Individual Education Plan. For those receiving Individual Education Plan services, the public school district is required to provide services until age 21.
- A 504 plan (Section 504: a US federal statute that prohibits discrimination based on disability) can be considered for those who require accommodations or modifications such as front-of-class seating, assistive technology devices, classroom scribes, extra time between classes, modified assignments, and enlarged text.
- Developmental Disabilities Administration (DDA) enrollment is recommended. DDA is a US public agency that provides services and support to qualified individuals. Eligibility differs by state but is typically determined by diagnosis and/or associated cognitive/adaptive disabilities.
- Families with limited income and resources may also qualify for supplemental security income (SSI) for their child with a disability.
Motor Dysfunction
Gross motor dysfunction
- Physical therapy is recommended to maximize mobility.
- Consider use of durable medical equipment as needed (e.g., wheelchairs, walkers, bath chairs, orthotics, adaptive strollers).
Fine motor dysfunction. Occupational therapy is recommended for difficulty with fine motor skills that affect adaptive function such as feeding, grooming, dressing, and writing.
Oral motor dysfunction. Assuming that the individual is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended for affected individuals who have difficulty feeding due to poor oral motor control.
Oral motor dysfunction should be assessed at each visit and clinical feeding evaluations and/or radiographic swallowing studies should be obtained for choking/gagging during feeds, poor weight gain, frequent respiratory illnesses or feeding refusal that is not otherwise explained. Assuming that the child is safe to eat by mouth, feeding therapy (typically from an occupational or speech therapist) is recommended to help improve coordination or sensory-related feeding issues. Feeds can be thickened or chilled for safety. Severe feeding dysfunction typically requires an NG-tube or G-tube.
Communication issues. Consider evaluation for alternative means of communication (e.g., Augmentative and Alternative Communication) for individuals who have expressive language difficulties. An Augmentative and Alternative Communication evaluation can be completed by a speech-language pathologist who has expertise in the area. The evaluation will consider cognitive abilities and sensory impairments to determine the most appropriate form of communication. AAC devices can range from low-tech, such as picture exchange communication, to high-tech, such as voice-generating devices. Contrary to popular belief, Augmentative and Alternative Communication devices do not hinder verbal development of speech and in many cases, can improve it.
Social and Behavioral Concerns
Children may qualify for and benefit from interventions used in treatment of autism spectrum disorder, including applied behavior analysis (ABA). Applied behavior analysis therapy is targeted to the individual child’s behavioral, social, and adaptive strengths and weaknesses and typically performed one on one with a board-certified behavior analyst.
Consultation with a developmental pediatrician may be helpful in guiding parents through appropriate behavior management strategies or providing prescription medications, such as medication used to treat attention-deficit/hyperactivity disorder, when necessary.
Concerns about serious aggressive or destructive behavior can be addressed by a pediatric psychiatrist.
Surveillance
Monitoring for neonatal hypoglycemia; echocardiography with electrocardiogram at diagnosis with subsequent follow up by a cardiologist who is aware of the spectrum of cardiac disease and its natural history; abdominal and pelvic ultrasound examinations to screen for rhabdomyosarcoma and neuroblastoma every three to six months until age eight to ten years may be considered; annual urinalysis for evidence of hematuria to screen for bladder cancer beginning at age ten years.
Costello syndrome lifespan
A formal epidemiologic study of Costello syndrome, with lifespan analysis, survival and cumulative mortality has not been performed 40. Lin et al. 13 studied 146 mutation-confirmed patients, with at least another 85 clinically defined cases from the literature. The total is at least 220, and with additional unreported patients around the world, it is likely the figure approaches 300. As part of the recent cardiovascular study 41, deaths were reported in 10% and in the study and 20% of literature patients, providing preliminary descriptive data. Causes of death reported in 10% of individuals included in an analysis of cardiovascular findings 13 and in 20% of affected individuals described in the literature were: hypertrophic cardiomyopathy in all 11 (48%) accompanied by neoplasia, coronary artery fibromuscular dysplasia, and multifocal tachycardia in four. Neoplasia was noted at the cause of death in five (22%), pulmonary cause in two (9%), and multiorgan failure in five (four with hypertrophic cardiomyopathy) (22%).
- Costello syndrome. https://ghr.nlm.nih.gov/condition/costello-syndrome[↩]
- Costello syndrome. https://rarediseases.org/rare-diseases/costello-syndrome[↩]
- Gripp KW, Rauen KA. Costello Syndrome. 2006 Aug 29 [Updated 2019 Aug 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1507[↩]
- Lo IF, Brewer C, Shannon N, Shorto J, Tang B, Black G, Soo MT, Ng DK, Lam ST, Kerr B. Severe neonatal manifestations of Costello syndrome. J Med Genet. 2008;45:167–71.[↩]
- Bertola D, Buscarilli M, Stabley DL, Baker L, Doyle D, Bartholomew DW, Sol-Church K, Gripp KW. Phenotypic spectrum of Costello syndrome individuals harboring the rare HRAS mutation p.Gly13Asp. Am J Med Genet. 2017;173:1309–18.[↩]
- Johnson JP, Golabi M, Norton ME, Rosenblatt RM, Feldman GM, Yang SP, Hall BD, Fries MH, Carey JC. Costello syndrome: phenotype, natural history, differential diagnosis, and possible cause. J Pediatr. 1998;133:441–8.[↩][↩]
- Sammon MR, Doyle D, Hopkins E, Sol-Church K, Stabley DL, McGready J, Schulze K, Alade Y, Hoover-Fong J, Gripp KW. Normative growth charts for individuals with Costello syndrome. Am J Med Genet. 2012;158A:2692–9.[↩]
- Hennekam RC. Costello syndrome: an overview. Am J Med Genet Part C Semin Med Genet. 2003;117C:42–8.[↩]
- Tidyman WE, Lee HS, Rauen KA. Skeletal muscle pathology in Costello and cardio-facio-cutaneous syndrome: developmental consequences of germline Ras/MAPK activation on myogenesis. Am J Med Genet C Semin Med Genet. 2011;157C:104–14.[↩]
- Gripp KW, Hopkins E, Doyle D, Dobyns WB. High incidence of progressive postnatal cerebellar enlargement in Costello syndrome: brain overgrowth associated with HRAS mutations as the likely cause of structural brain and spinal cord abnormalities. Am J Med Genet A. 2010;152A:1161–8.[↩][↩]
- Calandrelli R, D’Apolito G, Marco P, Zampino G, Tartaglione T, Colosimo C. Costello syndrome: Analysis of the posterior cranial fossa in children with posterior fossa crowding. Neuroradiol J. 2015;28:254–8.[↩]
- Kawame H, Matsui M, Kurosawa K, Matsuo M, Masuno M, Ohashi H, Fueki N, Aoyama K, Miyatsuka Y, Suzuki K, Akatsuka A, Ochiai Y, Fukushima Y. Further delineation of the behavioral and neurologic features in Costello syndrome. Am J Med Genet A. 2003;118A:8–14.[↩]
- Lin AE, Alexander ME, Colan SD, Kerr B, Rauen KA, Noonan J, Baffa J, Hopkins E, Sol-Church K, Limongelli G, Digilio MC, Marino B, Ines AM, Aoki Y, Silberbach M, Del-Rue MA, While SM, Hamilton RM, O’Connor W, Grossfeld PD, Smoot LB, Padera RF, Gripp KW. Clinical, pathological and molecular analyses of cardiovascular abnormalities in Costello syndrome: A Ras/MAPK Pathway syndrome. Am J Med Genet A. 2011;155A:486–507.[↩][↩][↩][↩][↩][↩]
- Levin MD, Saitta SC, Gripp KW, Wenger TL, Ganesh J, Kalish JM, Epstein MR, Smith R, Czosek RJ, Ware SM, Goldenberg P, Myers A, Chatfield KC, Gillespie MJ, Zackai EH, Lin AE. Nonreentrant atrial tachycardia occurs independently of hypertrophic cardiomyopathy in RASopathy patients. Am J Med Genet. 2018;176:1711–22.[↩]
- Weaver KN, Wang D, Cnota J, Gardner N, Stabley D, Sol-Church K, Gripp KW, Witte DP, Bove KE, Hopkin RJ. Early-lethal Costello syndrome due to rare HRAS tandem base substitution (c.35_36GC>AA; p.G12E) associated pulmonary vascular disease. Pediatr Dev Pathol. 2014;17:421–430.[↩]
- Axelrad ME, Schwartz DD, Katzenstein JM, Hopkins E, Gripp KW. Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am J Med Genet C Semin Med Genet. 2011;157C:115–22.[↩][↩]
- Schwartz DD, Katzenstein JM, Hopkins E, Stabley DL, Sol-Church K, Gripp KW, Axelrad ME. Verbal memory functioning in adolescents and young adults with Costello syndrome: evidence for relative preservation in recognition memory. Am J Med Genet. 2013;161A:2258–65.[↩]
- Schwartz DD, Katzenstein JM, Highley EJ, Stabley DL, Sol-Church K, Gripp KW, Axelrad ME. Age-related differences in prevalence of autism spectrum disorder symptoms in children and adolescents with Costello syndrome. Am J Med Genet. 2017;173:1294–1300.[↩]
- Hopkins E, Lin AE, Krepkovich KE, Axelrad ME, Sol-Church K, Stabley DL, Hossain J, Gripp KW. Living with Costello syndrome: Quality of life issues in older individuals. Am J Med Genet A. 2010;152A:84–90.[↩][↩]
- Johnson B, Goldberg-Strassler D, Gripp K, Thacker M, Leoni C, Stevenson D. Function and disability in children with Costello syndrome and Cardiofaciocutaneous syndrome. Am J Med Genet. 2015;167A:40–4.[↩]
- Marukian NV, Levinsohn JL, Craiglow BG, Milstone LM, Choate KA. Palmoplantar Keratoderma in Costello Syndrome Responsive to Acitretin. Pediatr Dermatol. 2017;34:160–162.[↩]
- Detweiler S, Thacker MM, Hopkins E, Conway L, Gripp KW. Orthopedic manifestations and implications for individuals with Costello syndrome. Am J Med Genet. 2013;161A:1940–9.[↩][↩]
- White SM, Graham JM Jr, Kerr B, Gripp K, Weksberg R, Cytrynbaum C, Reeder JL, Stewart FJ, Edwards M, Wilson M, Bankier A. The adult phenotype in Costello syndrome. Am J Med Genet A. 2005;136:128–35.[↩][↩]
- Leoni C, Stevenson DA, Martini L, De Sanctis R, Mascolo G, Pantaleoni F, De Santis S, La Torraca I, Persichilli S, Caradonna P, Tartaglia M, Zampino G. Decreased bone mineral density in Costello syndrome. Mol Genet Metab. 2014;111:41–5.[↩]
- Della Marca G, Vasta I, Scarano E, Rigante M, De Feo E, Mariotti P, Rubino M, Vollono C, Mennuni GF, Tonali P, Zampino G. Obstructive sleep apnea in Costello syndrome. Am J Med Genet A. 2006;140:257–62.[↩]
- Gomez-Ospina N, Kuo C, Ananth AL, Myers A, Brennan ML, Stevenson DA, Bernstein JA, Hudgins L. Respiratory system involvement in Costello syndrome. Am J Med Genet. 2016;170:1849–57.[↩][↩]
- Sheffield BS, Yip S, Ruchelli ED, Dunham CP, Sherwin E, Brooks PA, Sur A, Singh A, Human DG, Patel MS, Lee A. Fatal congenital hypertrophic cardiomyopathy and a pancreatic nodule morphologically identical to focal lesion of congenital hyperinsulinism in an infant with Costello syndrome. case report and review of the literature. Pediatr Dev Pathol. 2015;18:237–44.[↩]
- Gripp KW, Robbins KM, Sheffield BS, Lee AF, Patel MS, Yip S, Doyle D, Stabley D, Sol-Church K. Paternal uniparental disomy 11p15.5 in the pancreatic nodule of an infant with Costello syndrome: Shared mechanism for hyperinsulinemic hypoglycemia in neonates with Costello and Beckwith-Wiedemann syndrome and somatic loss of heterozygosity in Costello syndrome driving clonal expansion. Am J Med Genet. 2016;170:559–64.[↩]
- Gripp KW, Lin AE, Stabley DL, Nicholson L, Scott CI Jr, Doyle D, Aoki Y, Matsubara Y, Zackai EH, Lapunzina P, Gonzalez-Meneses A, Holbrook J, Agresta CA, Gonzalez IL, Sol-Church K. HRAS mutation analysis in Costello syndrome: genotype and phenotype correlation. Am J Med Genet A. 2006a;140:1–7.[↩]
- Kratz CP, Rapisuwon S, Reed H, Hasle H, Rosenberg PS. Cancer in Noonan, Costello, cardiofaciocutaneous and LEOPARD syndromes. Am J Med Genet C Semin Med Genet. 2011;157C:83–9.[↩]
- Gripp KW. Tumor predisposition in Costello syndrome. Am J Med Genet C Semin Med Genet. 2005;137C:72–7.[↩]
- Gripp KW, Innes AM, Axelrad ME, Gillan TL, Parboosingh JS, Davies C, Leonard NJ, Lapointe M, Doyle D, Catalano S, Nicholson L, Stabley DL, Sol-Church K. Costello syndrome associated with novel germline HRAS mutations: an attenuated phenotype? Am J Med Genet A. 2008;146A:683–90.[↩]
- Goodwin AF, Oberoi S, Landan M, Charles C, Massie JC, Fairley C, Rauen KA, Klein OD. Craniofacial and dental development in Costello syndrome. Am J Med Genet. 2014;164A:1425–30.[↩]
- Pierpont ME, Richards M, Engel WK, Mendelsohn NJ, Summers CG. Retinal dystrophy in two boys with Costello syndrome due to the HRAS p.Gly13Cys mutation. Am J Med Genet. 2017;173:1342–7.[↩]
- Gripp KW, Demmer LA. Keratoconus in Costello syndrome. Am J Med Genet. 2013;161A:1132–6.[↩]
- Stein RI, Legault L, Daneman D, Weksberg R, Hamilton J. Growth hormone deficiency in Costello syndrome. Am J Med Genet A. 2004;129A:166–70.[↩]
- Legault L, Gagnon C, Lapointe N. Growth hormone deficiency in Costello syndrome: a possible explanation for the short stature. J Pediatr. 2001;138:151–2.[↩]
- Kerr B, Einaudi MA, Clayton P, Gladman G, Eden T, Saunier P, Genevieve D, Philip N. Is growth hormone treatment beneficial or harmful in Costello syndrome? J Med Genet. 2003;40:e74[↩][↩]
- Gripp KW, Scott CI Jr, Nicholson L, Figueroa TE. Second case of bladder carcinoma in a patient with Costello syndrome. Am J Med Genet. 2000;90:256–9.[↩]
- Gripp, K., Lin, A. Costello syndrome: a Ras/mitogen activated protein kinase pathway syndrome (rasopathy) resulting from HRAS germline mutations. Genet Med 14, 285–292 (2012). https://doi.org/10.1038/gim.0b013e31822dd91f[↩]
- Lin AE, Alexander ME, Colan SD,et al. Clinical, pathological, and molecular analyses of cardiovascular abnormalities in Costello syndrome: a Ras/MAPK pathway syndrome. Am J Med Genet A 2011;155A:486–507.[↩]


{kind=link}