Cornelia de Lange syndrome
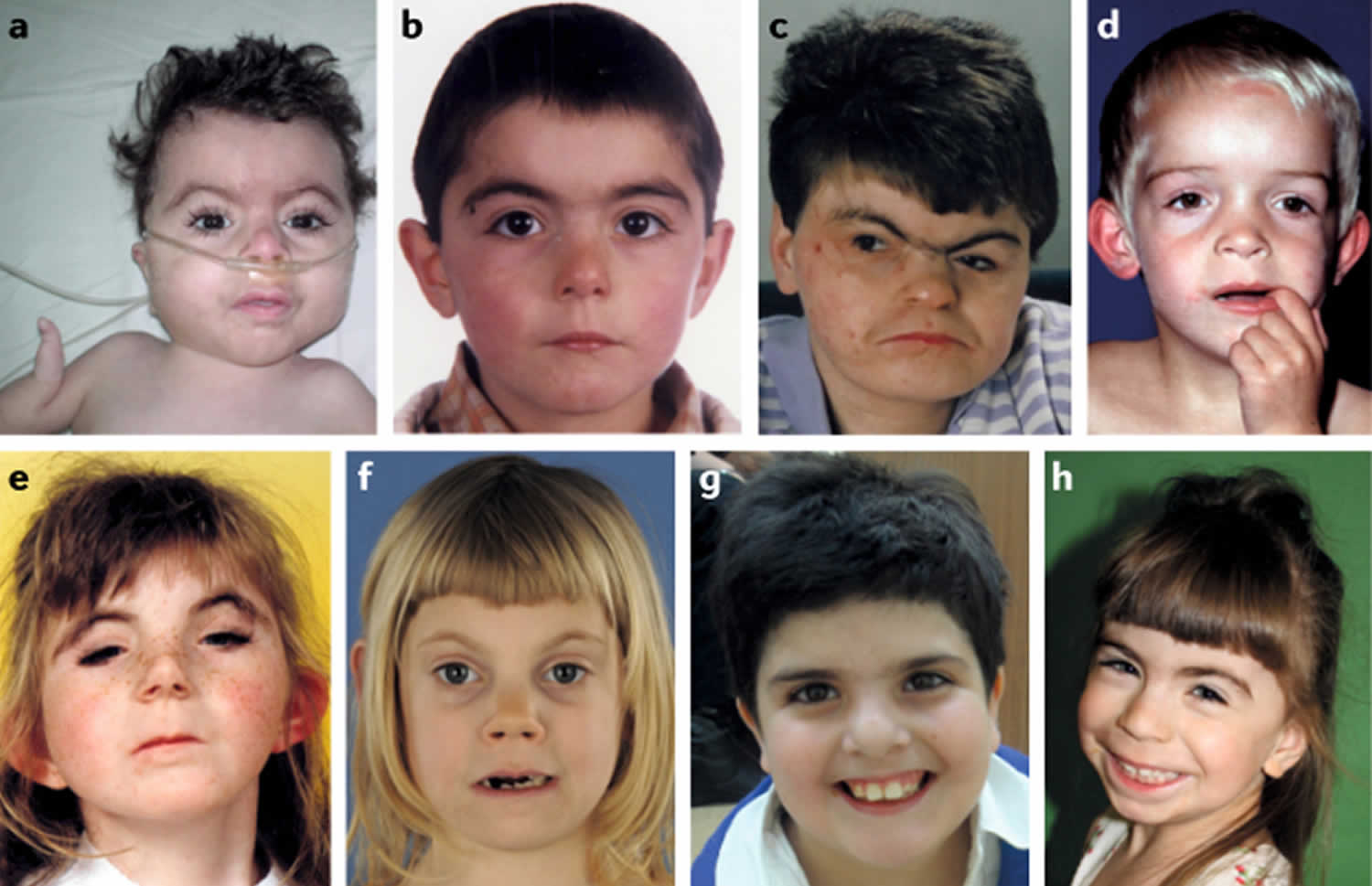
Cornelia de Lange syndrome (CdLS) also known as Brachmann–de Lange Syndrome, de Lange syndrome or Cornelia de Lange Spectrum Disorder, is a genetic developmental disorder that is present from birth (congenital) and it is characterized by congenital head and facial (craniofacial), gastrointestinal, cardiac, musculoskeletal, genitourinary, behavioral and neurodevelopmental abnormalitites, resulting in a distinctive facial appearance, malformations of the hands and arms (upper limbs); and mild to severe intellectual disability 1, 2, 3. The range and severity of associated symptoms and findings may be extremely variable from person to person 4, 5, 6, 7, 8, 9, 10, 11. The most frequent associated congenital malformations of Cornelia de Lange syndrome were limb defects (73.1%), congenital heart defects (45.6%), central nervous system malformations (40.2%), and cleft palate (21.7%) 12. Many infants and children with de Lange syndrome have an unusually small, short head (microbrachycephaly), a prominent vertical groove between the upper lip and nose (philtrum), a depressed nasal bridge, upturned nostrils (anteverted nares) and a small chin (micrognathia) 4. Additional characteristic facial features may include thin, downturned lips, low-set ears, arched, well-defined eyebrows that grow together across the base of the nose (synophrys), an unusually low hairline on the forehead and the back of the neck and curly, unusually long eyelashes 4. Affected individuals may also have distinctive malformations of the limbs, such as unusually small hands and feet, inward deviation (clinodactyly) of the fifth fingers, and webbing (syndactyly) of certain toes. Less commonly, there may be absence of the forearms, hands, and fingers 4. Associated symptoms and findings typically include delays in physical development before and after birth (prenatal and postnatal growth delay) 12. Infants with CdLS may also have feeding and breathing difficulties; an increased susceptibility to respiratory infections; a low-pitched “growling” cry and low voice; heart defects; delayed skeletal maturation; hearing loss; or other physical differences. Cornelia de Lange syndrome is now referred to as Cornelia de Lange Syndrome Spectrum Disorder because of the broad nature of the signs and symptoms. Live born infants with Cornelia de Lange syndrome have a high first week survival.
In 1933, Dutch pediatrician Dr. Cornelia de Lange described two children with similar features 13. The first child had pneumonia and feeding difficulties. She was very small for her age, with a proportionately smaller head circumference. Other unusual facial characteristics were noted by Dr. de Lange. Soon after, she saw a second little girl with common medical problems and physical characteristics. Nowhere was the puzzled physician able to find a similar patient described in the medical literature. Cornelia de Lange is now generally credited with describing the collection of symptoms comprising the syndrome that bears her name.
Cornelia de Lange syndrome is sometimes referred to as Brachmann-de Lange syndrome after Dr. W. Brachmann, who described a similar patient in 1916. Dr. de Lange may have overlooked his report because he concentrated on characteristics of the upper limbs and wrote on the facial symptoms less specifically.
Cornelia de Lange syndrome is a very rare disorder that is apparent at birth (congenital). Cornelia de Lange syndrome (CdLS) occurs in approximately 1.24 per 100,000 live births, but when the mild forms are also taken into account, the prevalence has been reported to be as high as 1 in every 10,000 live births in the United States 12, 14. However, due to problems in the diagnosis of Cornelia de Lange syndrome, particularly for the mild forms because of the lack of objective diagnostic criteria for this subgroup, the exact prevalence is still unknown 1. Males and females appear to be affected in equal numbers. More than 400 cases have been reported in the medical literature, including affected individuals within several families (kindreds). Multiple affected siblings (brothers and sisters) have been reported in some families. The CdLS Foundation serves over 3,800 people with Cornelia de Lange syndrome in the U.S. 15. It is estimated that there is a 1% to 2 % rate of recurrence within affected families 4.
Cornelia de Lange syndrome can be inherited as an autosomal dominant condition or an X-linked condition. However, most cases of Cornelia de Lange syndrome is caused by acquired new gene mutation (de novo mutation) in one of seven important developmental genes at or shortly after conception and occur in people with no family history of the condition. Seven genes have been found to be associated with Cornelia de Lange syndrome including the NIPBL gene on chromosome 5, the SMC1A gene on the X chromosome, the SMC3 gene on chromosome 10, the Rad21 gene on chromosome 8, the HDAC8 gene on the X chromosome, the ANKRD11 on chromosome 16 and the BRD4 gene on chromosome 19 16, 17. Other genes may be found to be associated with Cornelia de Lange syndrome; however, these variants were detected in individuals exhibiting limited clinical CdLS features rather than in individuals fulfilling the clinical diagnostic criteria for CdLS. Approximately 60% of people affected by Cornelia de Lange syndrome affected have a disease-causing change (mutation) in the NIPBL gene, and about 10% of cases are caused by mutations in one of four known genes: SMC1A, SMC3, HDAC8 and RAD21. In the remaining 30% of cases, the underlying genetic cause of de Lange syndrome is unknown 18. Although no specific genotype–phenotype correlations have been firmly established, individuals with missense mutations in NIPBL and SMC1A appear milder than those with other mutations 19.
Most children with Cornelia de Lange syndrome are diagnosed clinically after birth or in childhood based upon a thorough clinical evaluation and identification of characteristic physical findings. A diagnosis of Cornelia de Lange syndrome should be considered in children who exhibit certain distinctive facial features in association with limb anomalies, prenatal and postnatal growth delay and intellectual disability. Diagnosis may be more difficult if symptoms and physical characteristics associated with the disorder are very mild. Molecular genetic testing for mutations in the five genes associated with Cornelia de Lange syndrome is available to confirm the diagnosis and may be particularly helpful when the physical features are mild or unusual. Prenatal diagnosis is available if a specific NIPBL, SMC1A, SMC3, Rad21, HDAC8, ANKRD11 or BRD4 gene mutation has been identified.
Sometimes a diagnosis of Cornelia de Lange syndrome may be suspected before birth (prenatally) through the use of ultrasound imaging. During such testing, reflected sound waves create an image of the fetus that may reveal certain characteristics of Cornelia de Lange syndrome such as delayed growth, limb abnormalities, facial anomalies and/or organ malformations.
Cornelia de Lange syndrome treatment is based on the signs and symptoms present in each person. Treatment may require the efforts of a team of specialists working together to systematically and comprehensively plan an affected child’s treatment. Such specialists may include pediatricians; geneticists; surgeons; specialists who diagnose and treat skeletal disorders (orthopedists); plastic surgeons; orthopedic surgeons; specialists who diagnose and treat abnormalities of the digestive system (gastroenterologists), feeding specialists, disorders of the urinary tract (urologists), and abnormalities of the ears, nose, and throat (otolaryngologists); pediatric heart specialists (cardiologists); dental specialists; speech pathologists; specialists who assess and treat hearing problems (audiologists); eye specialists; physical and occupational therapists and/or other health care professionals. Genetic counseling is recommended for affected individuals and their families.
Figure 1. Facial features of Cornelia de Lange syndrome
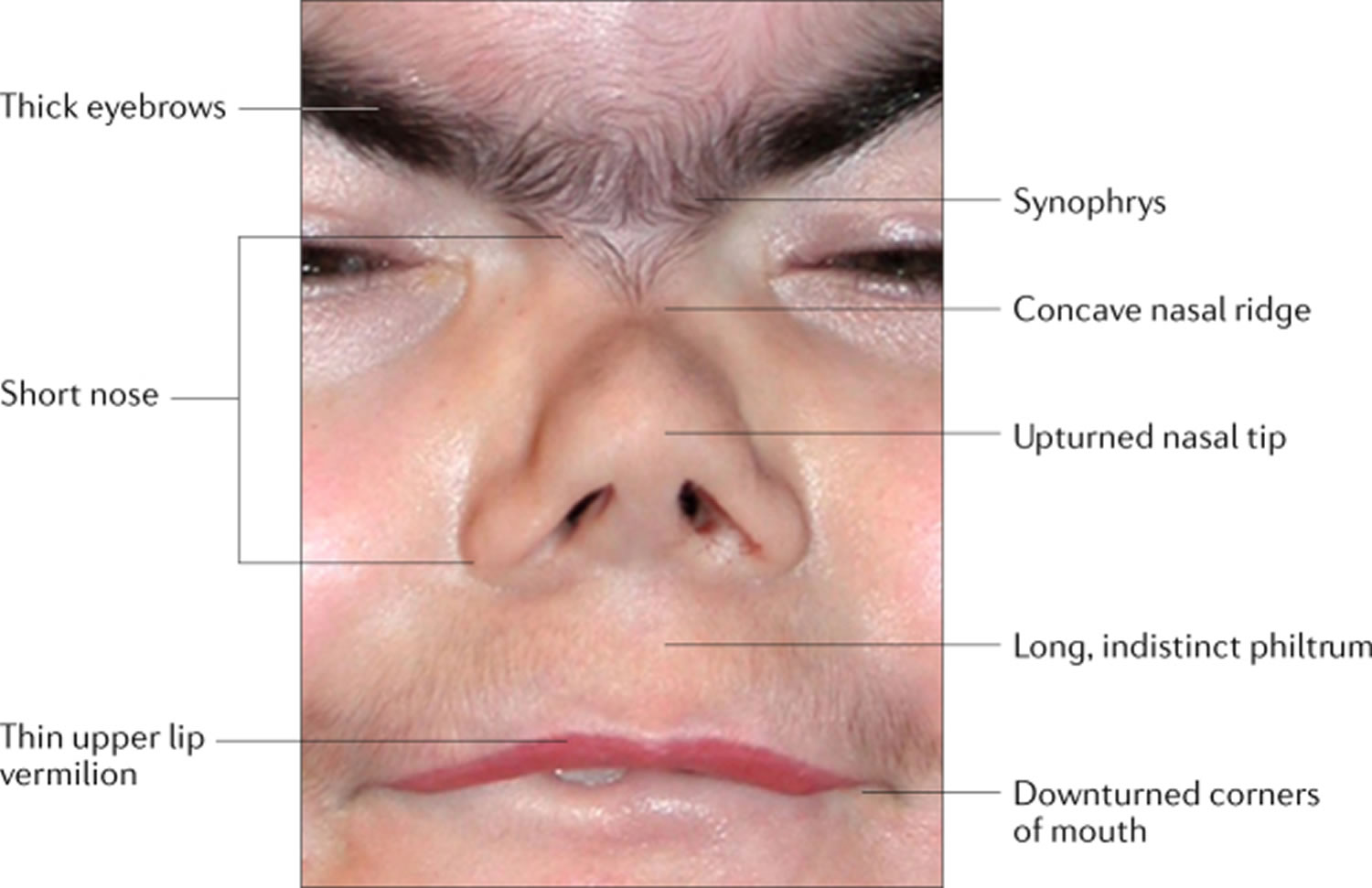
Footnotes: Facial features that are the most characteristic for Cornelia de Lange syndrome (CdLS) include eye manifestations such as synophrys (meeting of the medial eyebrows in the midline) and thick eyebrows, a short nose, concave nasal ridge and upturned nasal tip, a long and smooth philtrum, a thin upper lip vermilion and downturned corners of the mouth. Non-facial features (not shown) that are considered to be cardinal features of CdLS include hand oligodactyly (the congenital absence of one or more fingers), adactyly (the absence of all fingers and/or toes) and congenital diaphragmatic hernia.
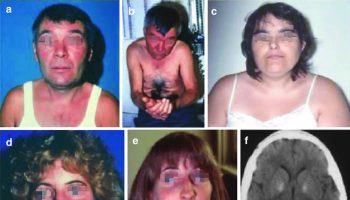
[Source 17 ]Figure 2. Cornelia de Lange syndrome
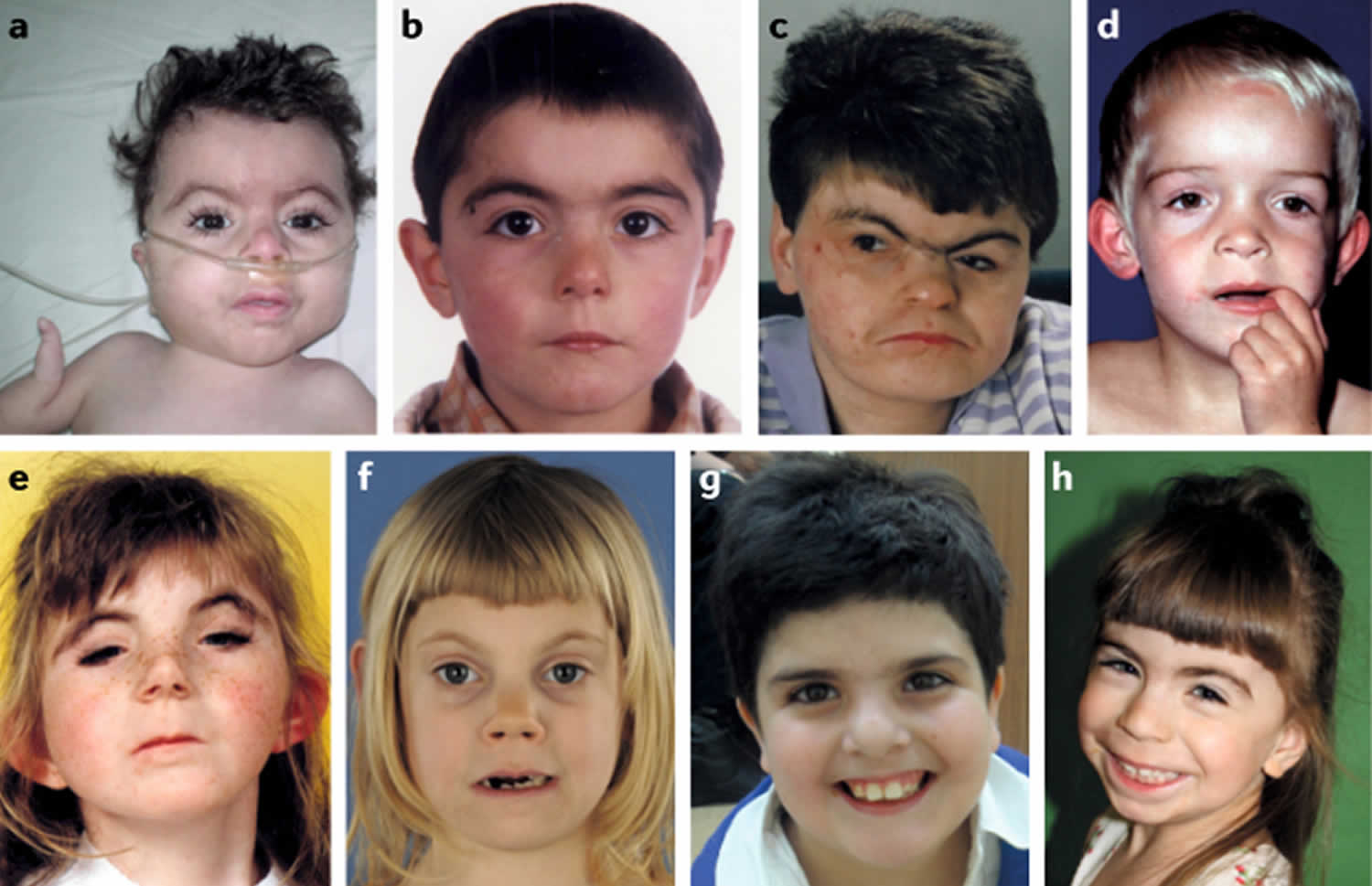
Footnotes: Facial features of individuals with Cornelia de Lange syndrome. (a) Classic Cornelia de Lange syndrome (CdLS) phenotype resulting from an NIPBL gene mutation. (b) Non-classic Cornelia de Lange syndrome (CdLS) facial feature in an individual harbouring an NIPBL variant. (c) Adult with the classic facial feature (NIPBL variant). (d) Non-classic facial feature in individual with an SMC1A variant. (e) Classic facial feature in an individual with an SMC3 variant. (f) Non-classic facial feature in an individual with a RAD21 variant. (g) Non-classic facial feature in an individual with an HDAC8 variant. (h) Non-classic facial feature in an individual with an ANKRD11 variant.
[Source 17 ]Figure 3. Cornelia de Lange syndrome
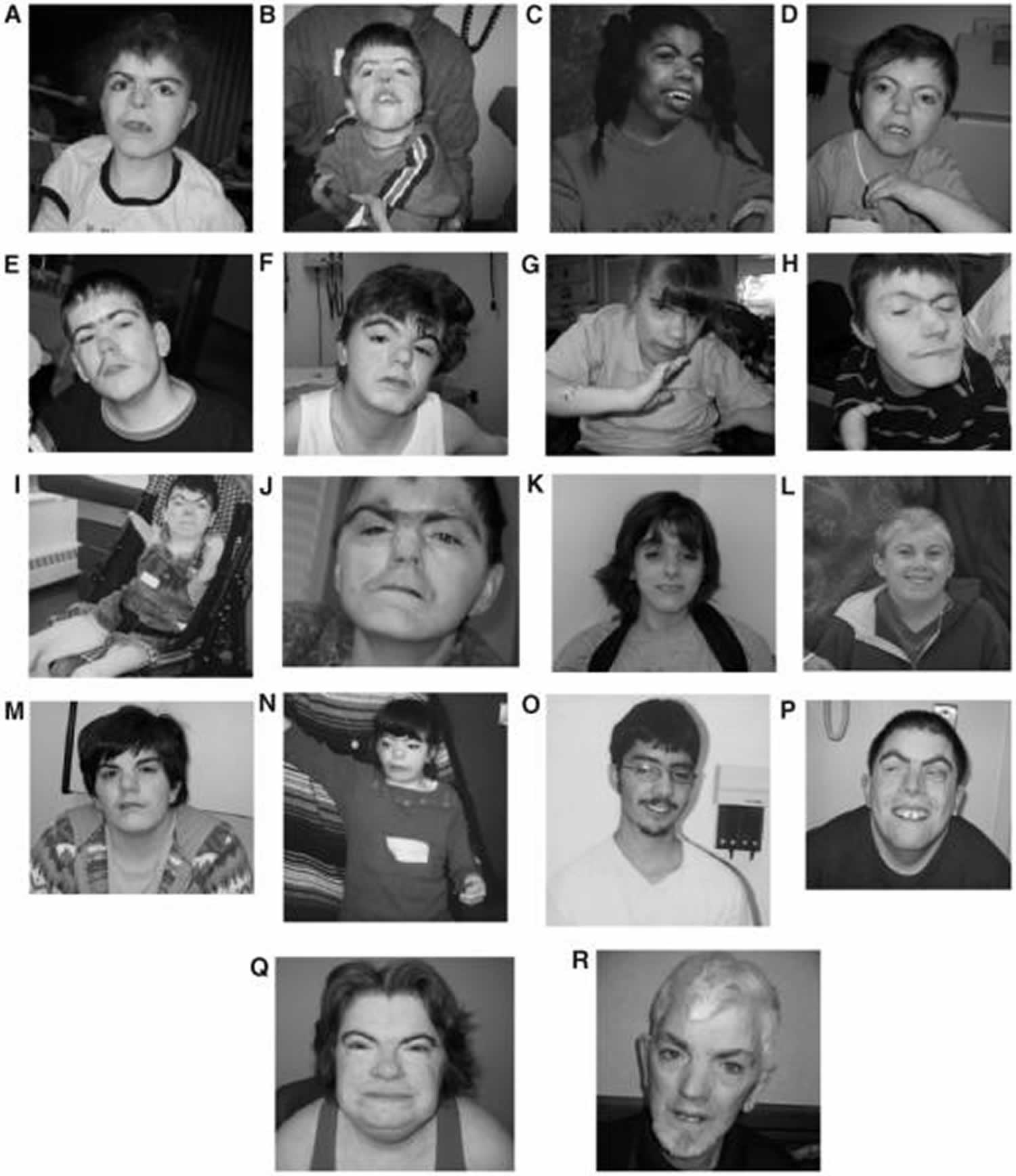
Footnotes: (A to J) Composite of more typical facies from younger to older; (K to R) composite of less typical facies from younger to older. Patients shown in subparts A,D,E,H,I,M,N,P,Q,T are females.
[Source 19 ]Figure 4. Cornelia de Lange syndrome craniofacial features
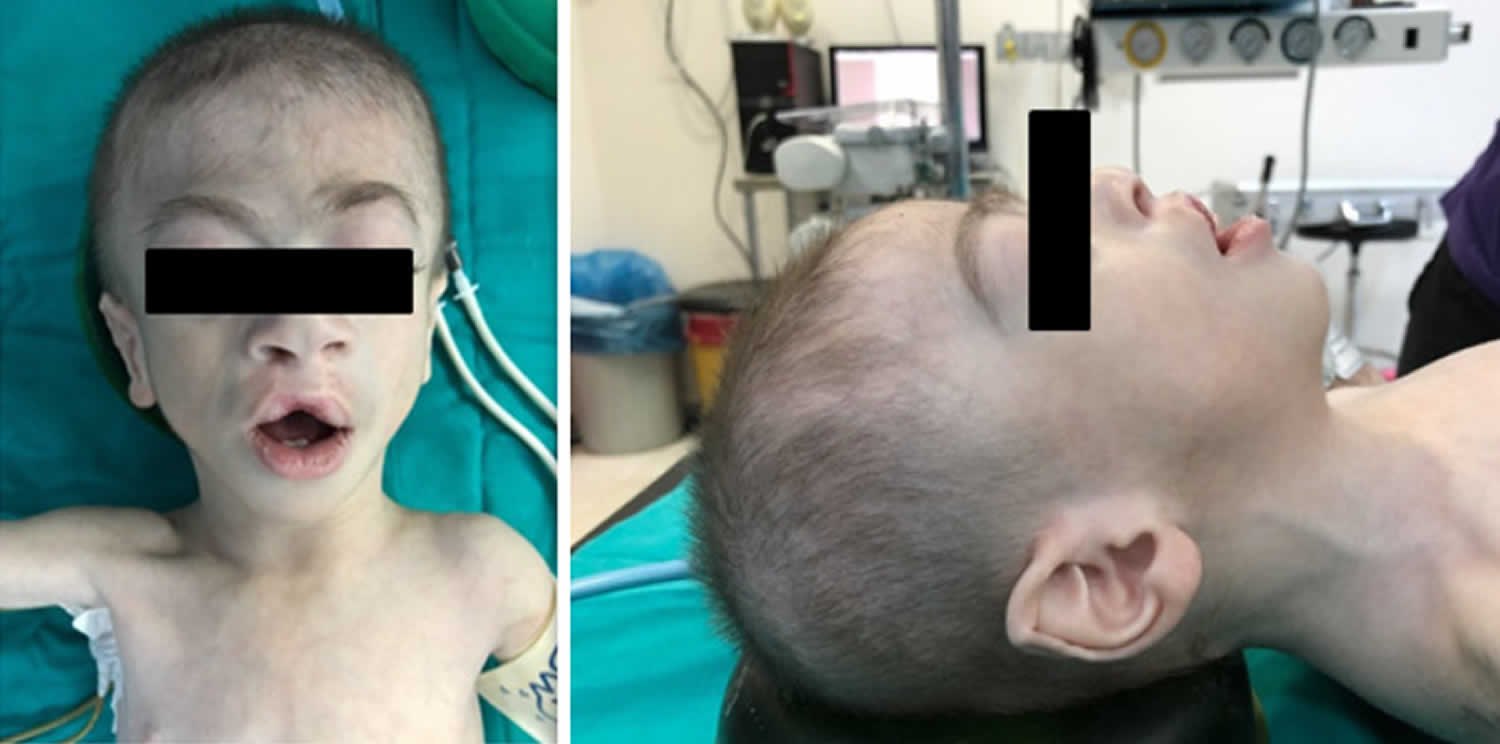
Table 1. Clinical findings of Cornelia de Lange syndrome and recommended interventions in adulthood
| Childhood involvement | Adult involvement | Adult management |
|---|---|---|
| Congenital heart disease | Typically stable | Echocardiogram if never had |
| Primary teeth tend to be retained | Panorex X-ray can be taken | |
| Secondary teeth eruption delayed Sinusitis and/or nasal polyps may produce symptoms | Lingual caries may develop with gastroesophageal reflux disease (GERD) Sinusitis and/or nasal polyps may produce symptoms | Pediatric dental visits every 4–6 months. ENT visits as needed; nasal polypectomy may be helpful |
| Cleft palate—should be repaired Pyloric stenosis requiring surgery Gastroesophageal reflux disease Complications of gastroesophageal reflux disease (GERD) Malrotation may be present | Cleft palate stable—may need repair Constipation may persist Gastroesophageal reflux disease (GERD) may worsen Barrett esophagus may occur Risk for volvulus, intestinal perforation | Craniofacial team visits as needed Diet, medications as needed Regular gastrointestinal follow-up Biopsies every 1–3 years after diagnosis All patients need upper GI series and warning about presenting signs of volvulus (e.g. bilious vomiting) |
| Renal malformation, vesiculo-ureteral reflux (VUR) | Renal malformation, vesiculo-ureteral reflux (VUR) | Renal ultrasound on all patients, as indicated clinically |
| Cryptorchidism in males | Cryptorchidism in males | Orchiopexy and/or orchiectomy in childhood, monitor hormones later |
| Slightly delayed puberty females | Irregular or no menses | Hormonal treatment as needed; routine gynecology care with Pap smears every 3 years |
| Lacrimal duct malformations | Retinal detachment can occur with severe myopia | Regular ophthalmology visits, surgery as needed |
| Blepharitis Hip dislocations | Blepharitis tends to improve Leg length discrepancy, scoliosis, bunions may occur | Baby shampoo rinses Orthopedic visits as needed |
| Seizures may occur or worsen Peripheral neuropathy may produce symptoms | Seizures may occur or worsen Peripheral neuropathy may produce symptoms | Pediatric neurology, medications Medications may be helpful |
| Behavioral issues (self-injurious behavior, anxiety, aggression) may worsen | Behavioral issues (self-injurious behavior, anxiety, aggression) may worsen | Psychiatrist or psychologist intervention may be helpful |
Cornelia de Lange syndrome causes
Cornelia de Lange syndrome can be inherited as an autosomal dominant condition or an X-linked condition 20, 21. However, most cases of Cornelia de Lange syndrome is caused by acquired new gene mutation (de novo mutation) in one of seven important developmental genes at or shortly after conception and occur in people with no family history of the condition. Seven genes have been found to be associated with Cornelia de Lange syndrome including the NIPBL gene on chromosome 5, the SMC1A gene on the X chromosome, the SMC3 gene on chromosome 10, the Rad21 gene on chromosome 8, the HDAC8 gene on the X chromosome, the ANKRD11 gene on chromosome 16 and the BRD4 gene on chromosome 19 16, 17, 22, 23, 24. Other genes may be found to be associated with Cornelia de Lange syndrome; however, these variants were detected in individuals exhibiting limited clinical CdLS features rather than in individuals fulfilling the clinical diagnostic criteria for CdLS. Acquired new gene mutation (de novo mutation) in EP300 were detected in individuals with some features suggestive of Cornelia de Lange syndrome 25 and de novo AFF4 mutations have been reported in three individuals with CHOPS syndrome, which stands for cognitive impairment, coarse facies, heart defects, obesity, pulmonary involvement, short stature and skeletal dysplasia and includes features that overlap with Cornelia de Lange syndrome 26. Gene mutation in NAA10 have been described in a series of individuals with some resemblance to individuals with Cornelia de Lange syndrome that is limited to the periorbital region 27. Finally, recessive TAF6 gene mutations have been reported in two families with children who showed features that overlap with Cornelia de Lange syndrome 28.
Approximately 60% of people affected by Cornelia de Lange syndrome affected have a disease-causing change (mutation) in the NIPBL gene, and about 10% of cases are caused by mutations in one of four known genes: SMC1A, SMC3, HDAC8 and RAD21. In the remaining 30% of cases, the underlying genetic cause of de Lange syndrome is unknown 18. Although no specific genotype–phenotype correlations have been firmly established, individuals with missense mutations in NIPBL and SMC1A appear milder than those with other mutations 19.
Cornelia de Lange syndrome inheritance pattern
Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease (see Figure 5). The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from affected parent to offspring is 50% for each pregnancy. The risk is the same for males and females.
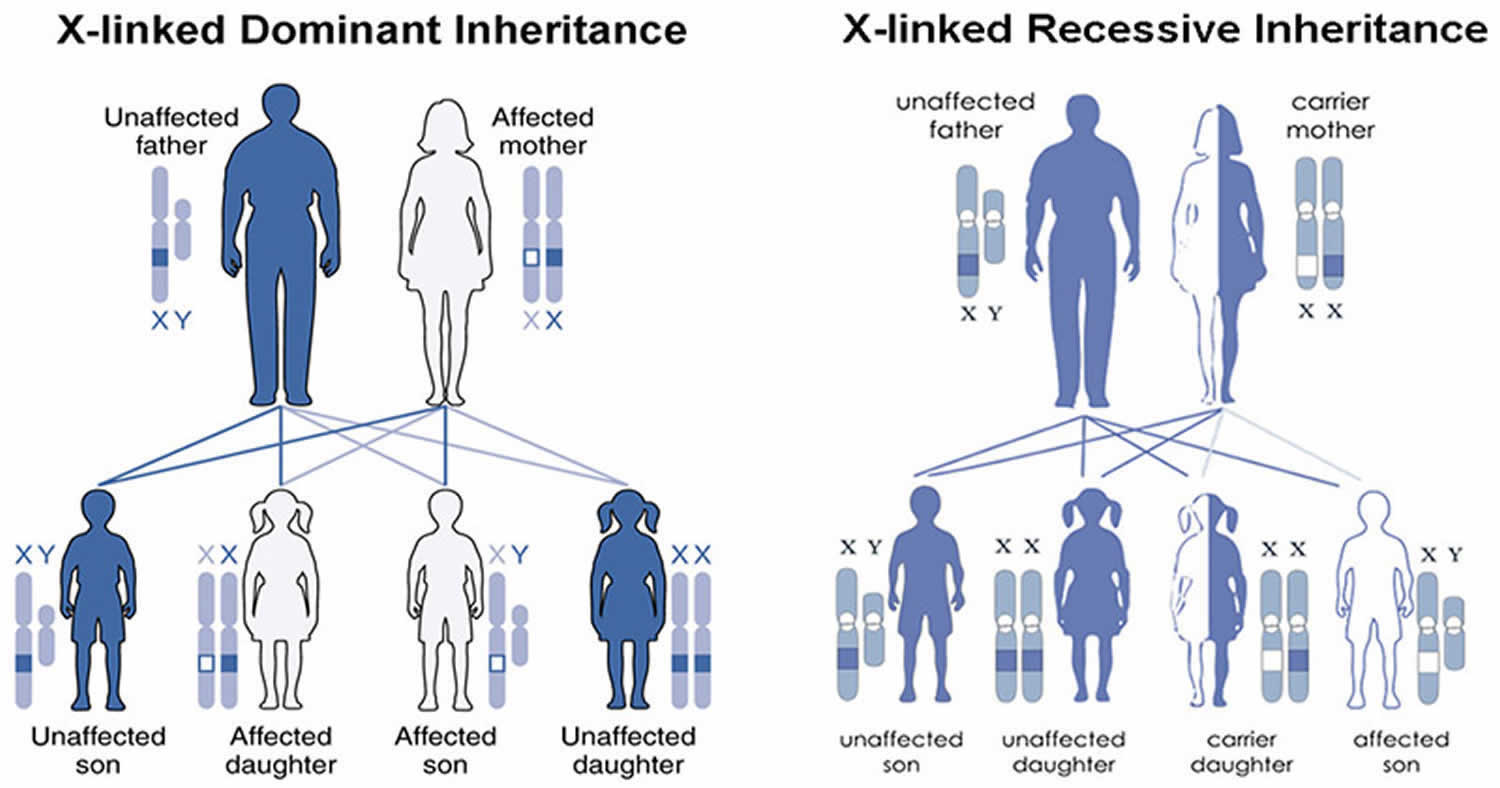
X-linked genetic disorders are conditions caused by an abnormal gene on the X chromosome (sex chromosome) and occur mostly in males (see Figure 6). Humans have two sex chromosomes, X and Y. Females have two X chromosomes in their cells (XX chromosome), while males have one X and one Y (XY chromosome). In the case of an X-linked genetic disorder, it is usually males (XY chromosome) that are affected because they have a single copy of the X chromosome that carries the disease-causing mutation. Females (XX chromosome) that have a disease gene present on one of their X chromosomes are carriers for that disorder. Carrier females usually do not display symptoms because females have two X chromosomes and one is inactivated so that the genes on that chromosome are nonfunctioning. It is often the X chromosome with the abnormal gene that is inactivated. However, in Cornelia de Lange syndrome, because the gene change is likely dominant over the corresponding gene on the X chromosomes, females also often show similar findings as males.
Males (XY chromosome) have one X chromosome that is inherited from their mother and if a male inherits an X chromosome that contains a disease gene he will develop the disease. Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son. Males with X-linked disorders pass the disease gene to all of their daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
Figure 5. Cornelia de Lange syndrome (CdLS) autosomal dominant inheritance pattern
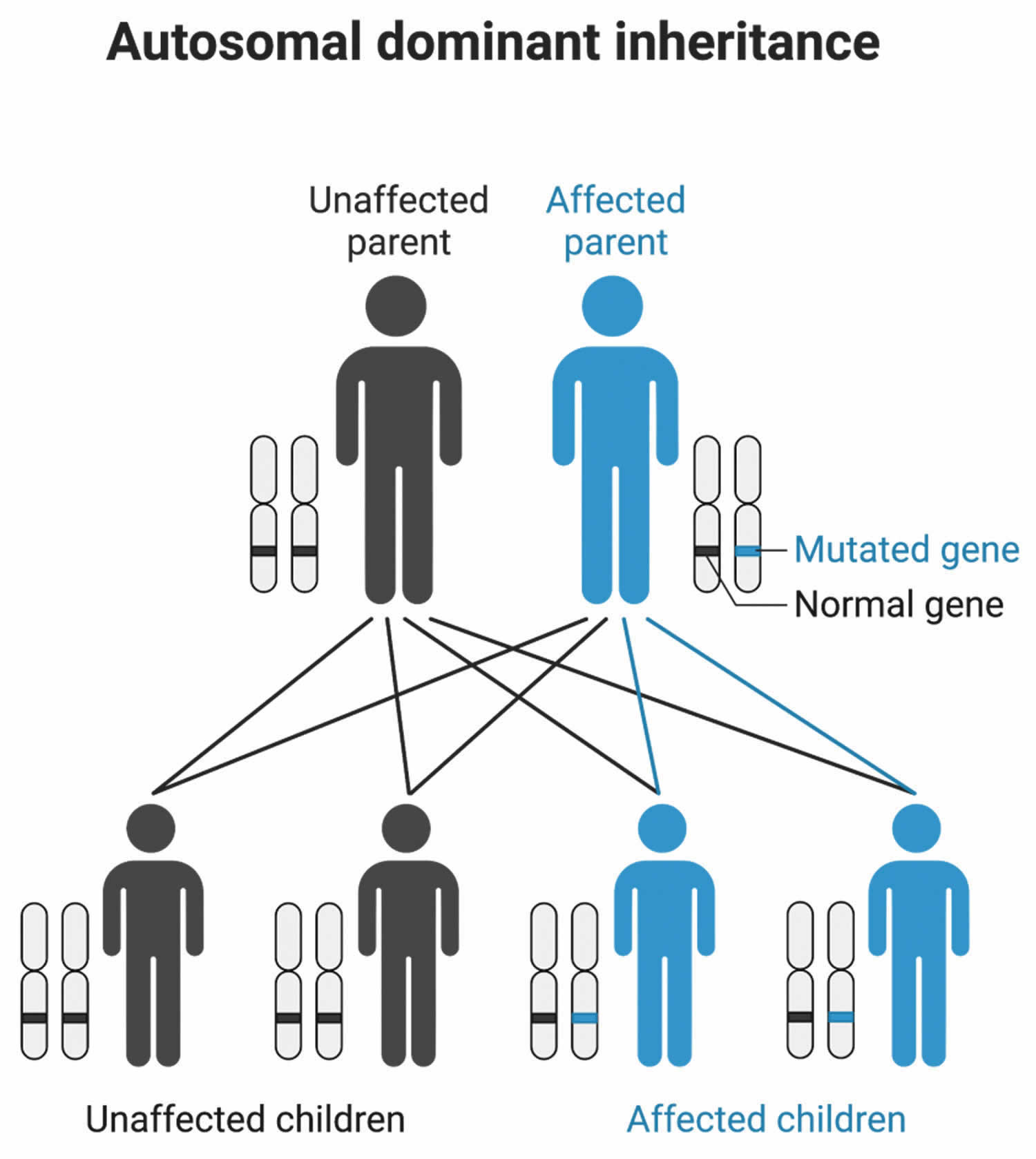
Figure 6. Cornelia de Lange syndrome (CdLS) X-linked inheritance pattern
Cornelia de Lange syndrome signs and symptoms
Cornelia de Lange syndrome severity and the associated signs and symptoms can vary greatly from person to person, but may include distinctive facial characteristics, malformations of the hands, feet, arms, and/or legs (limb anomalies), growth delays, intellectual disability and/or developmental delays 29, 30, 31, 32, 33.
Individuals with Cornelia de Lange syndrome exhibit abnormal growth delays that affect both weight and linear growth before and after birth (prenatal and postnatal growth delay). Most affected infants may have a low birth weight and may fail to gain weight or grow at the expected rate (failure to thrive). Cornelia de Lange syndrome growth charts are available to compare growth to other affected individuals. Individuals may experience feeding, chewing, and swallowing difficulties during the first several months/years of life.
Many affected infants may frequently “spit up” food that has already been swallowed (regurgitation) and may experience episodes of severe, forceful vomiting (projectile vomiting). Infants with Cornelia de Lange syndrome may also demonstrate abnormally increased muscle tone (hypertonicity) and have an unusual, low-pitched, growling cry.
Individuals with Cornelia de Lange syndrome also have distinctive features of the head and facial (craniofacial) area including a small head (microcephaly) that may also be unusually short (brachycephaly); a short, thick neck; low hairline; small, broad, upturned nose with nostrils that tip upwards (anteverted nares); neat, arched eyebrows that grow together (synophrys); long, curly eyelashes and/or excessive hair growth on various areas of the body (hypertrichosis). Additional features may include thin, downturned lips; a long vertical gap between the upper lip (philtrum) and the nose; a small, underdeveloped jaw (micrognathia); late-erupting, widely-spaced, small teeth; and low-set ears. Some affected infants may also have an incomplete closure of the roof of the mouth (cleft palate), a hidden incomplete closure (submucous cleft palate) and/or a highly arched palate.
Most infants with Cornelia de Lange syndrome have hands and feet that are small for their size. In addition, affected individuals may have short fingers that become smaller and thinner toward the ends (tapered fingers), fifth fingers that are permanently curved toward the ring finger (clinodactyly) and/or absence of one or more fingers (oligodactyly). The thumbs may be abnormally positioned (i.e., proximally placed) and the arms may be permanently bent or flexed at the elbows due to bone fusions. Many affected individuals have underdevelopment (hypoplasia) of some of the bones of the fingers and toes, and the second and third toes are often fused or webbed (syndactyly). Some affected infants may also have, in rare cases, missing fingers, hands and forearms. Upper limb differences may involve one side (unilateral) or both sides (bilateral) of the body. If bilateral limb malformations are present, those on one side of the body may be completely different from those on the other side (asymmetrical). Although the feet are small, only in extremely rare cases are there absent bones in the feet or lower legs.
Individuals with Cornelia de Lange syndrome also have delayed bone age. In addition, affected individuals may remain low in weight and have short stature (prenatal and postnatal growth delay), failure to thrive during infancy, delayed bone age and/or other differences. Many individuals with Cornelia de Lange syndrome also exhibit additional skeletal abnormalities. These may include a deformity of the hip (coxa valga), a short breastbone (sternum) and/or abnormally thin ribs.
Many infants and children with Cornelia de Lange syndrome may have delays in the acquisition of skills requiring the coordination of mental and muscular activity (psychomotor delay), mild to severe intellectual disability and/or demonstrate behavioral problems (e.g., episodes of biting, screaming, hitting themselves, etc.). In addition, although affected children may have decreased facial expression based on emotion, they appear to respond positively to certain stimuli (e.g., fast movements). A Cornelia de Lange syndrome developmental chart is available to compare milestones.
Many children with Cornelia de Lange syndrome also have hearing impairment as well as delayed speech development. Middle ear infections (otitis media), which sometimes occur chronically with an accumulation of sticky fluid (otitis media with effusion or glue ear) are common. Younger children may have difficulty speaking (dysphonia and/or apraxia), while older children may have abnormally hoarse speech.
Many individuals with Cornelia de Lange syndrome also exhibit additional physical differences. The skin may appear “marbled” (cutis marmorata) and the skin above the eyes, mouth, and nose may have an unusual bluish tone. Many affected individuals have irregularities in the skin ridge patterns on the palms of the hands (dermatoglyphics). As mentioned earlier, most affected individuals may have excessive hair growth (hypertrichosis) on various areas of the body including the ears. Hair may also tend to appear on the lower back, limbs and/or other areas of the body.
Many individuals with Cornelia de Lange syndrome also have various abnormalities of the gastrointestinal system including gastroesophageal reflux, a condition in which the acidic contents of the stomach flow upward into the lower esophagus; inflammation of the lining of the esophagus (esophagitis) and/or narrowing of the esophagus (esophageal stenosis). In addition, affected individuals are at risk for twisting (malrotation) of the intestines, potentially causing intestinal obstruction (volvulus). In some children, the bands of muscle fibers (pyloric sphincter) at the junction between the stomach and small intestine (pyloric stenosis) may become narrowed (stenosis) in infancy, resulting in obstruction of the normal flow of stomach contents into the small intestine. In addition, some individuals with Cornelia de Lange syndrome may also have protrusion of portions of the large intestine through an opening in musculature lining the abdominal cavity in the area of the groin (inguinal hernia) and/or part of the stomach through an opening where the esophagus passes through the diaphragm (hiatal hernia). Some babies with Cornelia de Lange syndrome are born with diaphragmatic hernia, in which some of the contents of the abdomen have not been separated from the lungs as a fetus; this needs to be repaired for survival. In some individuals with Cornelia de Lange syndrome, certain gastrointestinal abnormalities may lead to intestinal obstruction, potentially causing serious or life-threatening complications if left untreated.
Some individuals with Cornelia de Lange syndrome may also have malformations of the genitourinary tract. In affected males, such abnormalities may include underdevelopment (hypoplasia) of the genitals, failure of one or both of the testes to descend into the scrotum (cryptorchidism) and/or abnormal placement of the urinary opening (urinary meatus) on the underside of the penis (hypospadias). Affected females may have abnormal development of the uterus (e.g., bicornate or septate uterus) and menstruation may be irregular.
Many children with Cornelia de Lange syndrome have additional physical differences including various heart (cardiac) abnormalities. Some affected individuals may also have an increased susceptibility to repeated respiratory infections, eye abnormalities such as nearsightedness (myopia), rapid, involuntary eye movements (nystagmus) and/or abnormal drooping of the upper eyelid(s) (ptosis). Some infants and children with Cornelia de Lange syndrome may also experience episodes of uncontrolled electrical disturbances in the brain (seizures).
Cornelia de Lange syndrome diagnosis
Most children with Cornelia de Lange syndrome are diagnosed clinically after birth or in childhood based upon a thorough clinical evaluation and identification of characteristic physical findings. A diagnosis of Cornelia de Lange syndrome should be considered in children who exhibit certain distinctive facial features in association with limb anomalies, prenatal and postnatal growth delay and intellectual disability. Diagnosis may be more difficult if symptoms and physical characteristics associated with the disorder are very mild. Molecular genetic testing for mutations in the five genes associated with Cornelia de Lange syndrome is available to confirm the diagnosis and may be particularly helpful when the physical features are mild or unusual. Prenatal diagnosis is available if a specific NIPBL, SMC1A, SMC3, Rad21, HDAC8, ANKRD11 or BRD4 gene mutation has been identified.
Sometimes a diagnosis of Cornelia de Lange syndrome may be suspected before birth (prenatally) through the use of ultrasound imaging. During such testing, reflected sound waves create an image of the fetus that may reveal certain characteristics of Cornelia de Lange syndrome such as delayed growth, limb abnormalities, facial anomalies and/or organ malformations.
Cornelia de Lange syndrome genetic testing
The first person to be tested in any family would be the individual with Cornelia de Lange syndrome. Genetic testing for Cornelia de Lange syndrome can be used for future prenatal diagnosis for subsequent pregnancies, confirming a clinical diagnosis in a child who may need surgery or other interventions, or completing a diagnostic odyssey on a child with less clear findings. Testing for changes in Cornelia de Lange syndrome genes is complicated by the fact that the genes are very large. It should be kept in mind that Cornelia de Lange syndrome gene mutations are detectable only in about 80% of affected individuals. For any molecular testing or genetic sequencing, insurance approval should be received prior to sending, since the cost can be very high. Some insurance companies will not approve any molecular or genetic testing, and the family would have to pay out of pocket costs. In that case a clinical diagnosis would have to be enough. Finally, about 20% of individuals with Cornelia de Lange syndrome are mosaic for the gene change, meaning that it would not be detectable in blood but could be present in some other tissue (eg cheek swab or skin sample); in these situations, a portion of the body’s cells are positive for the gene change but not all. Overall, testing should be performed in conjunction with genetic counseling.
Prenatal diagnosis
The major indications for prenatal diagnostics are an earlier child with Cornelia de Lange syndrome, a new pregnancy in a family with a known genetic alteration in a Cornelia de Lange syndrome gene or, as occurs most frequently, no family history but features suggestive of Cornelia de Lange syndrome on fetal ultrasound 17. An ultrasound examination is recommended for all subsequent pregnancies at 18 to 20 weeks, most likely for reassurance. The timing of this scan allows a thorough study of the anatomy of the fetus and accurate dating of the pregnancy. In addition to the routine survey, careful attention should be made to the face, hands, heart, arms, and ventricles of the head. Anatomic abnormalities that have been described on prenatal studies of babies with Cornelia de Lange syndrome include: limb abnormalities (particularly of the upper limbs), abnormal hearts, cleft lip, abnormal facial profile, diaphragmatic hernia, mild enlargement of the ventricles of the head, and gastrointestinal abnormalities.
In 73 published cases involving patients with prenatal findings suggestive of Cornelia de Lange syndrome, symmetric intrauterine growth restriction (IUGR) with onset in the second trimester was noted as the most common finding (80%) 34. Limb anomalies were seen in 66% of fetuses (likely representing a selection bias), and approximately 50% of fetuses had an abnormal facial profile (micrognathia and prominent maxilla) 35. Other reported findings include increased nuchal thickness (51%), diaphragmatic hernia (28%) and cardiac malformation (15%) 34. When considering prenatal investigations, the pros and cons of the prenatal studies need to be discussed with the parents to offer investigations tailored to their wishes and to the technical, medical and legal options available 17.
The most distinctive of these abnormalities are those of the upper limb. Arm bones and fingers should be carefully examined and counted because these may be missing or abnormally short in Cornelia de Lange syndrome. The femur, feet and arm bones should be measured to ensure that their lengths are within the normal range. In the face of a fetus with Cornelia de Lange syndrome, one might find cleft palate, long eyelashes, a small chin, and a small upturned nose. The head tends to be short and small (microbrachycephaly) and should be measured and compared to tables. In many of the children, there are abnormalities of the heart and sometimes a diaphragmatic hernia or abdominal calcification (meconium peritonitis).
Ultrasound is not a perfect tool to diagnose Cornelia de Lange syndrome, nor is it a perfect tool to exclude the possibility of Cornelia de Lange syndrome. Nonetheless, many children with Cornelia de Lange syndrome have fairly severe abnormalities that can be detected prenatally by careful ultrasound with radiologists or obstetricians.
Prenatal molecular testing can be performed on samples obtained from chorionic villous sampling or amniocentesis or by testing embryonic cells obtained through in vitro fertilization 17. Single-gene sequencing with or without deletion or duplication testing is used most frequently, but the advent of panel testing of chorionic villi or amniocytes allows assessment of all known causative genes in a single test in some countries 17.
Non-invasive cell-free fetal DNA multi-gene screening that includes Cornelia de Lange syndrome genes can identify de novo variants in families without a previous child who has Cornelia de Lange syndrome. However, comparison with both biological parental samples is essential to interpret the large number of variants for which pathogenicity may be difficult or impossible to determine, which precludes meaningful use of this approach in routine practice at the present. Owing to the complexity of the molecular findings, prenatal testing for Cornelia de Lange syndrome outside of a known familial pathogenic variant remains challenging 17. Interpretation of novel variants requires caution as pathogenicity may be difficult to determine, and the possibility of undetectable mosaicism often precludes using testing for exclusion purposes. For these reasons, the validity and informative value of prenatal test results, and the ethical issues these may raise for families in deciding whether to continue a pregnancy, must be considered and discussed with couples before sampling 17.
After birth diagnosis
Cornelia de Lange syndrome is a condition with variable severity, signs and symptoms, and therefore diagnosis can be made at many different ages. Sometimes there is a prenatal diagnosis, in which facial features (particularly the profile), malformations (such as absent digits or forearms, diaphragmatic hernia or congenital heart disease) and intrauterine growth retardation become recognizable as Cornelia de Lange syndrome. Often, for the more severely involved individuals, there is a diagnosis made after birth in the newborn period. This is typically based on multiple findings: the small size for gestational age, typical facial features, hirsutism (excessive hair especially on forehead, face and back), small hands or absent digits or forearms, incomplete extension of the elbows, small feet and/or internal organ abnormalities such as diaphragmatic hernia, cleft palate, congenital heart disease, undescended testicles or micropenis in the male, or kidney anomalies. These are all typical, “classical” findings seen in Cornelia de Lange syndrome.
For milder, less severely involved individuals, there are less physical features that are notable, so the diagnosis tends to be later. There continues to be small size, often feeding problems and/or the development of gastroesophageal reflux. There is often developmental delay with early milestones occurring later than average. Even in the most mildly affected children, speech will usually come in later. Facial features are present, but less striking, and there is less excessive hair. Also, the internal organs are usually intact. Behavior can develop into an issue as these children get older, developing ADHD and/or some aggressive or self-injurious behavior. A subset will have autism or autistic-like features. Clinical diagnosis for this group can occur at any age during childhood, often prior to age 8 years, but it can be more challenging. Occasionally, individuals reach adolescence or even early adulthood without a clinical genetic evaluation, and the diagnosis can be missed until that point. Other specialists who may suspect this condition include pediatric gastroenterologists, cardiologist or otolaryngologists.
Cornelia de Lange syndrome treatment
The treatment of Cornelia de Lange syndrome is directed toward the specific symptoms that are apparent in each person. Treatment may require the efforts of a team of specialists working together to systematically and comprehensively plan an affected child’s treatment. Such specialists may include pediatricians; geneticists; surgeons; specialists who diagnose and treat skeletal disorders (orthopedists); plastic surgeons; orthopedic surgeons; specialists who diagnose and treat abnormalities of the digestive system (gastroenterologists), feeding specialists, disorders of the urinary tract (urologists), and abnormalities of the ears, nose, and throat (otolaryngologists); pediatric heart specialists (cardiologists); dental specialists; speech pathologists; specialists who assess and treat hearing problems (audiologists); eye specialists; physical and occupational therapists and/or other health care professionals.
Affected infants and children may be closely monitored for certain abnormalities potentially associated with Cornelia de Lange syndrome (e.g., potential intestinal obstruction due to gastrointestinal abnormalities, cardiac defects, gastroesophageal reflux, glue ear and/or susceptibility to respiratory infections) to ensure early detection and prompt treatment.
Specific therapies for the treatment of Cornelia de Lange syndrome are symptomatic and supportive. In some children, surgery may be performed to help correct cleft palate, cardiac defects and/or diaphragmatic hernias. Plastic surgery may be helpful in reducing excessive hair. Some gastrointestinal, genitourinary and/or cardiac malformations may be treated with certain medications, surgical intervention and/or other techniques. The surgical procedures performed will depend upon the location and severity of the anatomical differences and their associated symptoms. Respiratory infections may be treated with antibiotic drug therapy and/or other medications that may help fight infection.
Various orthopedic techniques may be used to help treat limb differences. Hearing aids may be beneficial in some children. Treatment with anticonvulsant medications may help prevent, reduce or control seizures in some affected children.
Early intervention is important in ensuring that children with Cornelia de Lange syndrome reach their highest potential. Services that may be beneficial include special remedial education, vocational training, speech therapy and/or other medical and/or social services.
Children medical follow-up
Given that Cornelia de Lange syndrome can usually be recognized from birth, the pediatrician has a central role in clinical care 17. Once the clinical diagnosis of Cornelia de Lange syndrome has been confirmed, every infant or child needs to be evaluated for common associated major malformations that require management or surveillance 17. Routine echocardiography and renal ultrasound are indicated in every diagnosed infant and child, given that 25% of individuals with Cornelia de Lange syndrome have a heart anomaly and 10% have a kidney malformation 2. In adolescents, the usefulness of such studies should be guided by symptomatology 17. Central nervous system (brain and spinal cord) imaging is indicated only if neurological symptoms such as seizures present, which is rare 17. Treatment and surveillance of major malformations are the same as for children without Cornelia de Lange syndrome. In 50% of children with Cornelia de Lange syndrome who have undergone intubation, the procedure has been difficult. Moreover, an adverse allergic reaction to midazolam (a short-acting benzodiazepine used for sedation) can occur 36, although complications due to anaesthetic medications are rare 37.
Cornelia de Lange syndrome-specific growth charts are available 38. Weight at birth is usually below the 5th percentile, and height, weight and head circumference all remain below the ranges for the general population 17. The growth charts are derived from clinically diagnosed individuals, and no growth charts subdivided by molecular background are available. Growth is influenced by the nature of the variant and the causative gene and tends to be less compromised in individuals with SMC1A variants compared with those with NIPBL variants 39, 40, 41. If growth velocity is lower than expected, gastrointestinal problems, thyroid dysfunction and growth hormone disturbances should be considered. Growth hormone secretion is normal in most children 42, although a single child with a NIPBL variant with low growth hormone levels and an increase in growth after supplementation has been described 43. The benefits of increased growth by growth hormone supplementation should be weighed against the burden of daily subcutaneous injections and the lack of a positive impact of an increased adult height on the quality of life for most individuals with Cornelia de Lange syndrome 17.
Feeding difficulties are almost universally present in neonates and infants with Cornelia de Lange syndrome and often in children and adults as well. Oral feeding is preferred if it is safe and stress-free and if feeding time does not exceed 3 hours per day, otherwise enteral feeding is recommended57. Involvement of dieticians is essential 17. Gastrostomies are the preferred option if tube feeding is needed for a prolonged period of time. Cleft palate, micrognathia and dental issues may contribute to feeding difficulties 44. Cleft palate, including submucous cleft palate, occurs in 20% of individuals with Cornelia de Lange syndrome. Isolated cleft lip is not related to Cornelia de Lange syndrome. Dental problems consist of delayed secondary tooth eruption, small or absent teeth, malposition, malocclusion, overcrowding of teeth, dental caries on the perilingual maxillary surface (due to gastro-oesophageal reflux disease [GERD]), periodontal disease and bruxism. Dental problems are worsened by poor oral hygiene, especially in those with marked intellectual disability, and owing to limited patient compliance 45, which may lead to early-onset dental decay and periodontal disease 46. Dental treatment by an interdisciplinary health-care team, a healthy diet, topical fluoride application and periodic dental check-ups are crucial in optimal management 47, 48.
Motor development is invariably delayed. Reliable data for a large series of individuals with molecularly confirmed diagnoses are not available. In a small series (n = 51), children with SMC1A variants reached several milestones (sitting, walking and first words) at a younger age than children with NIPBL variants 41. In the latter group, at 5 years of age, 99% were able to sit, 63% could walk independently and 38% had started to speak 17.
Vaccinations should be given according to national schemes 17. Recurrent respiratory infections are common and are thought to be secondary to altered anatomy, hypotonia and coordination of swallowing and coughing. Immunological anomalies occur occasionally; if unusually frequent or severe infections are present, further studies are indicated 49. Thrombocytopenia has been reported but is usually non-progressive and asymptomatic, and specific testing is not needed 50, 51.
Pain can occur in children with Cornelia de Lange syndrome, especially owing to dental problems, bladder and upper respiratory tract (including ears and sinuses) infections, gastro-oesophageal reflux and/or hip anomalies. Limited communicative abilities may hamper the shared recognition of pain and pain can lead to substantial behavioural problems 52. If there is suspicion that a patient with Cornelia de Lange syndrome is in pain, the use of specific tools to identify pain in an individual with intellectual disability, such as the face, legs, activity, cry, consolability (FLACC) assessment tool, is recommended 53.
Most individuals with Cornelia de Lange syndrome will go through puberty. In clinically diagnosed individuals, puberty was mildly delayed (mean age of onset was 15 years for boys and 13 years for girls) 19. That is, on average, menarche is delayed by 1 year compared with the general population; 5% of girls with Cornelia de Lange syndrome will never menstruate. For those who do, the menstrual cycle often remains irregular. A bicornuate uterus is found in 19% of female patients, and approximately 80% of girls develop breast tissue. In boys with Cornelia de Lange syndrome, 80% exhibit cryptorchidism, 37% have a small penis and 9% have hypospadias 19. Surgical correction of cryptorchidism is recommended to reduce the risk of testicular cancer, as in the general population. No lowering of voice in boys at puberty has been reported2. Teenagers with Cornelia de Lange syndrome can become overweight or develop overt obesity, which is often induced by high-calorie food offered by caregivers in combination with limited physical activity 19; regular evaluation of weight is essential.
Preferably, all individuals with Cornelia de Lange syndrome should be followed up by a pediatrician experienced in Cornelia de Lange syndrome 17. Follow-up varies between countries but is frequent in infancy and early childhood, and annually to once every 3–5 years in adolescence and adulthood. In case of problems, the schedule should be adapted to include more frequent follow-up visits 17.
Adult medical follow-up
Care coordination in adults is required, as many medical disciplines are typically involved. Most patients need diagnostic and/or interventional procedures and surgical operations under general anesthesia to survive 1. In several countries, individualized medical alert cards (emergency cards) that report the main clinical data of the patient and the most frequent and potentially life-threatening medical complications of Cornelia de Lange syndrome are used to the satisfaction of families and caregivers alike 17.
A small number of women with Cornelia de Lange syndrome have given birth, and often the diagnosis in the mother has been made only after diagnosis of the child 41, 54, 55, 56. A few men with Cornelia de Lange syndrome are known to have fathered a child, but reliable data on male fertility are not available 57, 58. Sexual education should be offered appropriate to the level of socioemotional and cognitive functioning 59. Contraceptive options are the same as for the general population. For some individuals, suppression of menses is preferred, and several contraceptives can effectively control or suppress menstruation. Hysterectomy is not recommended as a primary method of contraception but is sometimes employed for menorrhagia that does not respond to treatment 60. Premenstrual syndrome and dysmenorrhoea occur in women with Cornelia de Lange syndrome and can be associated with behavioural changes. Treatment options are as in the general population 17.
Several studies 2, 46 indicate that >30% of adults with Cornelia de Lange syndrome are overweight, and at least 50% are considered obese. It remains uncertain whether this percentage is higher than in individuals with the same cognitive level and mobility. Diet and physical activity is indicated 17. Type 2 diabetes mellitus develops in 4% of individuals 19.
Congenital heart anomalies should be detected in infancy or childhood and typically do not cause unexpected complications in adulthood 17. Hypertension and congestive heart failure have been reported in 4–8% and 2–4% of individuals with Cornelia de Lange syndrome, respectively 19, 61. Fatal coronary occlusion and pulmonary artery embolism have been reported once 62. In a retrospective cohort of 97 adults, 2 individuals had a myocardial infarction, and 2 had strokes 61. Renal failure was reported in 1% of individuals but is rarely fatal 61. Structural renal malformations were reported in 30% of adults; of these, 24% had abnormal creatinine clearance rates. It is recommended that renal function be monitored in those with structural renal malformations 51. Prostate enlargement has been found in 10% of men by the age of 41 years, requiring prostate removal in one individual 63. In the general population, benign prostatic hypertrophy is found in 25% of men in their fifties 64 and international management recommendations, which can be followed for men with Cornelia de Lange syndrome as well, start at age 45 years 65.
Cancer of the esophagus has been reported in three individuals with Barrett esophagus. There is no increased risk of cancer at a young age, but reliable data for middle-aged and older individuals are not available. Screening for cervical and breast cancer should be performed according to standard guidelines 66, 67.
Cornelia de Lange syndrome prognosis
Live born infants with Cornelia de Lange syndrome have a high first week survival. Although Cornelia de Lange syndrome patients are expected to have growth retardation, intellectual disabilities and a shorter lifespan due to multiple severe malformations, there have been no previous population-based studies on survival, and some of the patients (particularly with the milder forms) have been reported to reach adulthood 19, 68. Currently, most people with Cornelia de Lange syndrome reach adulthood owing to improved care, especially in the first year of life 17. Individuals with Cornelia de Lange syndrome aged ≥50 years have been described 69, 62. Kline et al. 19 presented the largest group of older patients (over 50 years of age) with Cornelia de Lange syndrome to date. Many of these patients, however, have had inconsistent medical care.
In a study of 295 individuals with Cornelia de Lange syndrome (81 infants, 117 children and 97 adults; 15 with a confirmed NIPBL variant), the most common causes of death in infants were congenital diaphragmatic hernia (17%) and respiratory problems (13%); in children, mortality was greatest owing to complications of congenital heart defects (10%) and respiratory (32%) and gastrointestinal problems (18%) 61. No reliable data are available for the risk of death in infancy or childhood. Causes of death in adults are related to the gastrointestinal, pulmonary and cardiac systems, as well as to infections or to anaesthesia 61, 19, 62, 70.
- Arun O, Oc B, Metin EN, Sert A, Yilmaz R, Oc M. Anesthetic management of a child with Cornelia de Lange Syndrome undergoing open heart surgery: A case report. World J Cardiol. 2022 Jan 26;14(1):54-63. doi: 10.4330/wjc.v14.i1.54[↩][↩][↩][↩]
- Jackson L, Kline AD, Barr MA, Koch S. de Lange syndrome: a clinical review of 310 individuals. Am J Med Genet. 1993 Nov 15;47(7):940-6. doi: 10.1002/ajmg.1320470703[↩][↩][↩]
- Hirai T, Nitahara K, Higa K, Iwakiri S, Shono S, Katori K. [Anesthetic management of an infant with Cornelia de Lange syndrome]. Masui. 2006 Apr;55(4):454-6. Japanese.[↩]
- Cornelia de Lange Syndrome. https://rarediseases.org/rare-diseases/cornelia-de-lange-syndrome[↩][↩][↩][↩][↩]
- Borck G, Zarhrate M, Cluzeau C, Bal E, Bonnefont JP, Munnich A, Cormier-Daire V, Colleaux L. Father-to-daughter transmission of Cornelia de Lange syndrome caused by a mutation in the 5′ untranslated region of the NIPBL Gene. Hum Mutat. 2006 Aug;27(8):731-5. doi: 10.1002/humu.20380[↩]
- Russell KL, Ming JE, Patel K, Jukofsky L, Magnusson M, Krantz ID. Dominant paternal transmission of Cornelia de Lange syndrome: a new case and review of 25 previously reported familial recurrences. Am J Med Genet. 2001 Dec 15;104(4):267-76. doi: 10.1002/ajmg.10066[↩]
- Vuilleumier N, Kövari E, Michon A, Hof PR, Mentenopoulos G, Giannakopoulos P, Bouras C. Neuropathological analysis of an adult case of the Cornelia de Lange syndrome. Acta Neuropathol. 2002 Sep;104(3):327-32. doi: 10.1007/s00401-002-0562-4[↩]
- Ozkinay F, Cogulu O, Gündüz C, Levent E, Ozkinay C. A case of Brachman de Lange syndrome with cerebellar vermis hypoplasia. Clin Dysmorphol. 1998 Oct;7(4):303-5. doi: 10.1097/00019605-199810000-00013[↩]
- Grant RE, Schneider JA, Ferguson EJ, Cummings PB. Total hip reconstruction in a woman with Cornelia de Lange syndrome: a case report. J Natl Med Assoc. 1997 Aug;89(8):530-2. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2568114/pdf/jnma00163-0048.pdf[↩]
- Kim IT, Park JW, Choi WC. A Korean case of Cornelia de Lange syndrome. Korean J Ophthalmol. 2005 Jun;19(2):153-5. doi: 10.3341/kjo.2005.19.2.153[↩]
- Doyle JO, Williams CD, Raymond CA. Hematometra in a patient with Cornelia De Lange syndrome. Obstet Gynecol. 2005 Nov;106(5 Pt 2):1202-4. doi: 10.1097/01.AOG.0000160512.24767.08[↩]
- Barisic, I., Tokic, V., Loane, M., Bianchi, F., Calzolari, E., Garne, E., Wellesley, D., Dolk, H. and (2008), Descriptive epidemiology of Cornelia de Lange syndrome in Europe†. Am. J. Med. Genet., 146A: 51-59. https://doi.org/10.1002/ajmg.a.32016[↩][↩][↩]
- de Lange C. Sur un type nouveau de dégénération (typus Amstelodamensis) Arch Med Enfants. 1933;36:713–719.[↩]
- What is CdLS? https://www.cdlsusa.org/what-is-cdls/[↩]
- Frequently Asked Questions. https://www.cdlsusa.org/faqs/[↩]
- Avagliano L, Parenti I, Grazioli P, et al. Chromatinopathies: A focus on Cornelia de Lange syndrome. Clin Genet. 2020; 97: 3–11. https://doi.org/10.1111/cge.13674[↩][↩]
- Kline AD, Moss JF, Selicorni A, et al. Diagnosis and management of Cornelia de Lange syndrome: first international consensus statement. Nat Rev Genet. 2018 Oct;19(10):649-666. doi: 10.1038/s41576-018-0031-0[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Cornelia de Lange syndrome. https://rarediseases.info.nih.gov/diseases/10109/cornelia-de-lange-syndrome[↩][↩]
- Kline AD, Grados M, Sponseller P, Levy HP, Blagowidow N, Schoedel C, Rampolla J, Clemens DK, Krantz I, Kimball A, Pichard C, Tuchman D. Natural history of aging in Cornelia de Lange syndrome. Am J Med Genet C Semin Med Genet. 2007 Aug 15;145C(3):248-60. doi: 10.1002/ajmg.c.30137[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- McConnell V, Brown T, Morrison PJ. An Irish three-generation family of Cornelia de Lange syndrome displaying autosomal dominant inheritance. Clin Dysmorphol. 2003 Oct;12(4):241-4. doi: 10.1097/00019605-200310000-00006[↩]
- de Die-Smulders C, Theunissen P, Schrander-Stumpel C, Frijns JP. On the variable expression of the Brachmann-de Lange syndrome. Clin Genet. 1992 Jan;41(1):42-5. doi: 10.1111/j.1399-0004.1992.tb03628.x[↩]
- Krantz ID, McCallum J, DeScipio C, Kaur M, Gillis LA, Yaeger D, Jukofsky L, Wasserman N, Bottani A, Morris CA, Nowaczyk MJ, Toriello H, Bamshad MJ, Carey JC, Rappaport E, Kawauchi S, Lander AD, Calof AL, Li HH, Devoto M, Jackson LG. Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nat Genet. 2004 Jun;36(6):631-5. doi: 10.1038/ng1364[↩]
- Tonkin ET, Wang TJ, Lisgo S, Bamshad MJ, Strachan T. NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nat Genet. 2004 Jun;36(6):636-41. doi: 10.1038/ng1363[↩]
- Musio A, Selicorni A, Focarelli ML, Gervasini C, Milani D, Russo S, Vezzoni P, Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat Genet. 2006 May;38(5):528-30. doi: 10.1038/ng1779[↩]
- Woods SA, Robinson HB, Kohler LJ, Agamanolis D, Sterbenz G, Khalifa M. Exome sequencing identifies a novel EP300 frame shift mutation in a patient with features that overlap Cornelia de Lange syndrome. Am J Med Genet A. 2014 Jan;164A(1):251-8. doi: 10.1002/ajmg.a.36237[↩]
- Izumi K, Nakato R, Zhang Z, Edmondson AC, Noon S, Dulik MC, Rajagopalan R, Venditti CP, Gripp K, Samanich J, Zackai EH, Deardorff MA, Clark D, Allen JL, Dorsett D, Misulovin Z, Komata M, Bando M, Kaur M, Katou Y, Shirahige K, Krantz ID. Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet. 2015 Apr;47(4):338-44. doi: 10.1038/ng.3229[↩]
- Saunier C, Støve SI, Popp B, Gérard B, Blenski M, AhMew N, de Bie C, Goldenberg P, Isidor B, Keren B, Leheup B, Lampert L, Mignot C, Tezcan K, Mancini GM, Nava C, Wasserstein M, Bruel AL, Thevenon J, Masurel A, Duffourd Y, Kuentz P, Huet F, Rivière JB, van Slegtenhorst M, Faivre L, Piton A, Reis A, Arnesen T, Thauvin-Robinet C, Zweier C. Expanding the Phenotype Associated with NAA10-Related N-Terminal Acetylation Deficiency. Hum Mutat. 2016 Aug;37(8):755-64. doi: 10.1002/humu.23001[↩]
- Yuan B, Pehlivan D, Karaca E, Patel N, Charng WL, Gambin T, Gonzaga-Jauregui C, Sutton VR, Yesil G, Bozdogan ST, Tos T, Koparir A, Koparir E, Beck CR, Gu S, Aslan H, Yuregir OO, Al Rubeaan K, Alnaqeb D, Alshammari MJ, Bayram Y, Atik MM, Aydin H, Geckinli BB, Seven M, Ulucan H, Fenercioglu E, Ozen M, Jhangiani S, Muzny DM, Boerwinkle E, Tuysuz B, Alkuraya FS, Gibbs RA, Lupski JR. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J Clin Invest. 2015 Feb;125(2):636-51. doi: 10.1172/JCI77435[↩]
- Pashayan H, Whelan D, Guttman S, Fraser FC. Variability of the de Lange syndrome: report of 3 cases and genetic analysis of 54 families. J Pediatr. 1969 Nov;75(5):853-8. doi: 10.1016/s0022-3476(69)80310-0[↩]
- Leroy JG, Persijn J, Van de Weghe V, Van Hecke R, Oostra A, De Bie S, Craen M. On the variability of the Brachmann-de Lange syndrome in seven patients. Am J Med Genet. 1993 Nov 15;47(7):983-91. doi: 10.1002/ajmg.1320470709[↩]
- Selicorni A, Lalatta F, Livini E, Briscioli V, Piguzzi T, Bagozzi DC, Mastroiacovo P, Zampino G, Gaeta G, Pugliese A, et al. Variability of the Brachmann-de Lange syndrome. Am J Med Genet. 1993 Nov 15;47(7):977-82. doi: 10.1002/ajmg.1320470708[↩]
- Van Allen MI, Filippi G, Siegel-Bartelt J, Yong SL, McGillivray B, Zuker RM, Smith CR, Magee JF, Ritchie S, Toi A, et al. Clinical variability within Brachmann-de Lange syndrome: a proposed classification system. Am J Med Genet. 1993 Nov 15;47(7):947-58. doi: 10.1002/ajmg.1320470704[↩]
- Allanson JE, Hennekam RC, Ireland M. De Lange syndrome: subjective and objective comparison of the classical and mild phenotypes. J Med Genet. 1997 Aug;34(8):645-50. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1051026/pdf/jmedgene00250-0029.pdf[↩]
- Dempsey MA, Knight Johnson AE, Swope BS, Moldenhauer JS, Sroka H, Chong K, Chitayat D, Briere L, Lyon H, Palmer N, Gopalani S, Siebert JR, Lévesque S, Leblanc J, Menzies D, Haverfield E, Das S. Molecular confirmation of nine cases of Cornelia de Lange syndrome diagnosed prenatally. Prenat Diagn. 2014 Feb;34(2):163-7. doi: 10.1002/pd.4279[↩][↩]
- Clark DM, Sherer I, Deardorff MA, Byrne JL, Loomes KM, Nowaczyk MJ, Jackson LG, Krantz ID. Identification of a prenatal profile of Cornelia de Lange syndrome (CdLS): a review of 53 CdLS pregnancies. Am J Med Genet A. 2012 Aug;158A(8):1848-56. doi: 10.1002/ajmg.a.35410[↩]
- Stevic M, Milojevic I, Bokun Z, Simic D. Unpredictable drug reaction in a child with Cornelia de Lange syndrome. Int J Clin Pharm. 2015 Feb;37(1):1-3. doi: 10.1007/s11096-014-0050-7[↩]
- Moretto A, Scaravilli V, Ciceri V, Bosatra M, Giannatelli F, Ateniese B, Mariani M, Cereda A, Sosio S, Zanella A, Pesenti A, Selicorni A. Sedation and general anesthesia for patients with Cornelia De Lange syndrome: A case series. Am J Med Genet C Semin Med Genet. 2016 Jun;172(2):222-8. doi: 10.1002/ajmg.c.31493[↩]
- Kline AD, Barr M, Jackson LG. Growth manifestations in the Brachmann-de Lange syndrome. Am J Med Genet. 1993 Nov 15;47(7):1042-9. doi: 10.1002/ajmg.1320470722[↩]
- Bhuiyan ZA, Klein M, Hammond P, van Haeringen A, Mannens MM, Van Berckelaer-Onnes I, Hennekam RC. Genotype-phenotype correlations of 39 patients with Cornelia De Lange syndrome: the Dutch experience. J Med Genet. 2006 Jul;43(7):568-75. doi: 10.1136/jmg.2005.038240[↩]
- Huisman SA, Redeker EJ, Maas SM, Mannens MM, Hennekam RC. High rate of mosaicism in individuals with Cornelia de Lange syndrome. J Med Genet. 2013 May;50(5):339-44. doi: 10.1136/jmedgenet-2012-101477[↩]
- Huisman S, Mulder PA, Redeker E, et al. Phenotypes and genotypes in individuals with SMC1A variants. Am J Med Genet A. 2017 Aug;173(8):2108-2125. doi: 10.1002/ajmg.a.38279[↩][↩][↩]
- Kousseff BG, Thomson-Meares J, Newkirk P, Root AW. Physical growth in Brachmann-de Lange syndrome. Am J Med Genet. 1993 Nov 15;47(7):1050-2. doi: 10.1002/ajmg.1320470723[↩]
- de Graaf M, Kant SG, Wit JM, Willem Redeker EJ, Eduard Santen GW, Henriëtta Verkerk AJM, Uitterlinden AG, Losekoot M, Oostdijk W. Successful Growth Hormone Therapy in Cornelia de Lange Syndrome. J Clin Res Pediatr Endocrinol. 2017 Dec 15;9(4):366-370. doi: 10.4274/jcrpe.4349[↩]
- Romano C, van Wynckel M, Hulst J, Broekaert I, Bronsky J, Dall’Oglio L, Mis NF, Hojsak I, Orel R, Papadopoulou A, Schaeppi M, Thapar N, Wilschanski M, Sullivan P, Gottrand F. European Society for Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for the Evaluation and Treatment of Gastrointestinal and Nutritional Complications in Children With Neurological Impairment. J Pediatr Gastroenterol Nutr. 2017 Aug;65(2):242-264. doi: 10.1097/MPG.0000000000001646[↩]
- Toker AS, Ay S, Yeler H, Sezgin I. Dental findings in Cornelia de Lange syndrome. Yonsei Med J. 2009 Apr 30;50(2):289-92. doi: 10.3349/ymj.2009.50.2.289[↩]
- Mariani M, Decimi V, Bettini LR, Maitz S, Gervasini C, Masciadri M, Ajmone P, Kullman G, Dinelli M, Panceri R, Cereda A, Selicorni A. Adolescents and adults affected by Cornelia de Lange syndrome: A report of 73 Italian patients. Am J Med Genet C Semin Med Genet. 2016 Jun;172(2):206-13. doi: 10.1002/ajmg.c.31502[↩][↩]
- Mouradian WE. The face of a child: children’s oral health and dental education. J Dent Educ. 2001 Sep;65(9):821-31.[↩]
- Scarpelli AC, Pordeus IA, Resende VL, Castilho LS, Marques LS, Paiva SM. Cornelia De Lange syndrome: a case report of a Brazilian boy. Cleft Palate Craniofac J. 2011 Jul;48(4):490-3. doi: 10.1597/10-025[↩]
- Jyonouchi S, Orange J, Sullivan KE, Krantz I, Deardorff M. Immunologic features of Cornelia de Lange syndrome. Pediatrics. 2013 Aug;132(2):e484-9. doi: 10.1542/peds.2012-3815[↩]
- Cavalleri V, Bettini LR, Barboni C, Cereda A, Mariani M, Spinelli M, Gervasini C, Russo S, Biondi A, Jankovic M, Selicorni A. Thrombocytopenia and Cornelia de Lange syndrome: Still an enigma? Am J Med Genet A. 2016 Jan;170A(1):130-4. doi: 10.1002/ajmg.a.37390[↩]
- Lambert MP, Jackson LG, Clark D, Kaur M, Krantz ID, Deardorff MA. The incidence of thrombocytopenia in children with Cornelia de Lange syndrome. Am J Med Genet A. 2011 Jan;155A(1):33-7. doi: 10.1002/ajmg.a.33631[↩][↩]
- Collis L, Moss J, Jutley J, Cornish K, Oliver C. Facial expression of affect in children with Cornelia de Lange syndrome. J Intellect Disabil Res. 2008 Mar;52(Pt 3):207-15. doi: 10.1111/j.1365-2788.2007.01004.x[↩]
- Malviya S, Voepel-Lewis T, Burke C, Merkel S, Tait AR. The revised FLACC observational pain tool: improved reliability and validity for pain assessment in children with cognitive impairment. Paediatr Anaesth. 2006 Mar;16(3):258-65. doi: 10.1111/j.1460-9592.2005.01773.x[↩]
- Bonora E, Bianco F, Cordeddu L, Bamshad M, Francescatto L, Dowless D, Stanghellini V, Cogliandro RF, Lindberg G, Mungan Z, Cefle K, Ozcelik T, Palanduz S, Ozturk S, Gedikbasi A, Gori A, Pippucci T, Graziano C, Volta U, Caio G, Barbara G, D’Amato M, Seri M, Katsanis N, Romeo G, De Giorgio R. Mutations in RAD21 disrupt regulation of APOB in patients with chronic intestinal pseudo-obstruction. Gastroenterology. 2015 Apr;148(4):771-782.e11. doi: 10.1053/j.gastro.2014.12.034[↩]
- Kaiser FJ, Ansari M, Braunholz D, et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum Mol Genet. 2014 Jun 1;23(11):2888-900. doi: 10.1093/hmg/ddu002[↩]
- Feingold M, Lin AE. Familial Brachmann-de Lange syndrome: further evidence for autosomal dominant inheritance and review of the literature. Am J Med Genet. 1993 Nov 15;47(7):1064-7. doi: 10.1002/ajmg.1320470726[↩]
- Chodirker BN, Chudley AE. Male-to-male transmission of mild Brachmann-de Lange syndrome. Am J Med Genet. 1994 Sep 1;52(3):331-3. doi: 10.1002/ajmg.1320520315[↩]
- McKenney RR, Elder FF, Garcia J, Northrup H. Brachmann-de Lange syndrome: autosomal dominant inheritance and male-to-male transmission. Am J Med Genet. 1996 Dec 30;66(4):449-52. doi: 10.1002/(SICI)1096-8628(19961230)66:4<449::AID-AJMG13>3.0.CO;2-U[↩]
- Quint EH. Menstrual and reproductive issues in adolescents with physical and developmental disabilities. Obstet Gynecol. 2014 Aug;124(2 Pt 1):367-375. doi: 10.1097/AOG.0000000000000387[↩]
- American College of Obstetricians and Gynecologists. Menstrual manipulation for adolescents with physical and developmental disabilities. ACOG Committee Opinion 668, August 2016 (ACOG, 2016).[↩]
- Schrier SA, Sherer I, Deardorff MA, Clark D, Audette L, Gillis L, Kline AD, Ernst L, Loomes K, Krantz ID, Jackson LG. Causes of death and autopsy findings in a large study cohort of individuals with Cornelia de Lange syndrome and review of the literature. Am J Med Genet A. 2011 Dec;155A(12):3007-24. doi: 10.1002/ajmg.a.34329[↩][↩][↩][↩][↩]
- Beck B, Fenger K. Mortality, pathological findings and causes of death in the de Lange syndrome. Acta Paediatr Scand. 1985 Sep;74(5):765-9. doi: 10.1111/j.1651-2227.1985.tb10028.x[↩][↩][↩]
- Oliver C, Bedeschi MF, Blagowidow N, Carrico CS, Cereda A, Fitzpatrick DR, Gervasini C, Griffith GM, Kline AD, Marchisio P, Moss J, Ramos FJ, Selicorni A, Tunnicliffe P, Wierzba J, Hennekam RC. Cornelia de Lange syndrome: extending the physical and psychological phenotype. Am J Med Genet A. 2010 May;152A(5):1127-35. doi: 10.1002/ajmg.a.33363[↩]
- McVary KT. BPH: epidemiology and comorbidities. Am J Manag Care. 2006 Apr;12(5 Suppl):S122-8.[↩]
- McVary KT, Roehrborn CG, Avins AL, Barry MJ, Bruskewitz RC, Donnell RF, Foster HE Jr, Gonzalez CM, Kaplan SA, Penson DF, Ulchaker JC, Wei JT. Update on AUA guideline on the management of benign prostatic hyperplasia. J Urol. 2011 May;185(5):1793-803. doi: 10.1016/j.juro.2011.01.074[↩]
- WHO Guidelines for Screening and Treatment of Precancerous Lesions for Cervical Cancer Prevention. Geneva: World Health Organization; 2013. Available from: https://www.ncbi.nlm.nih.gov/books/NBK195239[↩]
- WHO Position Paper on Mammography Screening. Geneva: World Health Organization; 2014. Available from: https://www.ncbi.nlm.nih.gov/books/NBK269545[↩]
- Torres MD, Calvo E, Fernández Esplá F, Gilsanz F. Anesthetic management of an adult patient with Cornelia de Lange Syndrome. Minerva Anestesiol. 2010 Mar;76(3):229-31.[↩]
- Russo S, Masciadri M, Gervasini C, Azzollini J, Cereda A, Zampino G, Haas O, Scarano G, Di Rocco M, Finelli P, Tenconi R, Selicorni A, Larizza L. Intragenic and large NIPBL rearrangements revealed by MLPA in Cornelia de Lange patients. Eur J Hum Genet. 2012 Jul;20(7):734-41. doi: 10.1038/ejhg.2012.7[↩]
- Filippi G. The de Lange syndrome. Report of 15 cases. Clin Genet. 1989 May;35(5):343-63. doi: 10.1111/j.1399-0004.1989.tb02955.x[↩]

{kind=link}