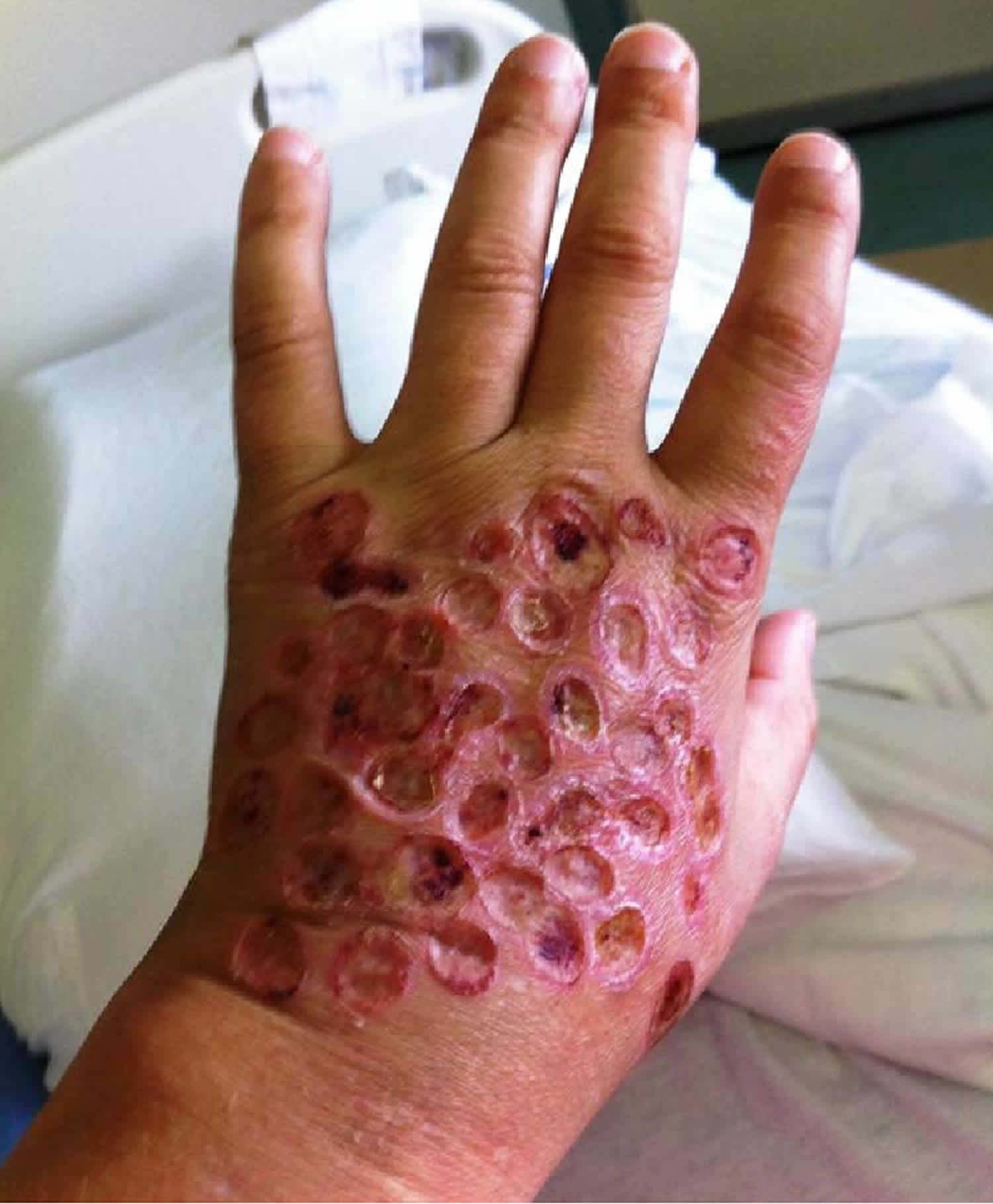
What is Degos disease
Degos disease also known as malignant atrophic papulosis is a very rare disease that affects a person’s small blood vessels. The blood vessels affected include those supplying the skin, gastrointestinal tract and central nervous system. When cells in the linings of veins and arteries under the skin become inflamed and swell, this restricts blood flow, resulting in spots or lesions that appear on the skin. The spots on the skin first appear small, red rimmed with a white center, and raised, but over time flatten and become depressed. The blood vessels affected include those that supply the skin, eyes, gastrointestinal tract, heart, bladder, and central nervous system.
Degos disease or malignant atrophic papulosis generally occurs in young Caucasian adults but has been reported in other races and at all ages. However, one study found that women developed the benign cutaneous form of Degos disease more often than men. Approximately 200 cases of Degos disease have been reported in the medical literature. The exact incidence of the disorder is unknown 1.
There is a spectrum of Degos disease progression. Degos disease starts on the skin and for an estimated two thirds of patients does not go beyond a cutaneous form. Approximately one third of Degos disease cases become systemic, meaning that patients develop internal manifestations of the lesions, which can block blood vessels and result in a host of life threatening complications, such as bowel perforation and stroke.
Degos disease was first described in the early 1940s by a French dermatologist, but the cause is still not known. Treatments for Degos disease (malignant atrophic papulosis) are limited, with no FDA-approved medications available for this indication.
Degos disease cause
The underlying cause of the occlusion of the blood vessels in Degos disease (malignant atrophic papulosis) is at present unknown. Some have classified Degos disease as a vasculitis, a mucinosis, or a thrombotic disorder 2. It is likely due to a genetic mutation of complement or some other clotting factor.
Many theories have been proposed as to what is the underlying cause of Degos disease. Three main theories include (1) viral infection, (2) defects in the body’s blood clotting ability (primary disorder of coagulation) and (3) autoimmunity, whereby the body’s immune system mistakenly attacks healthy tissue.
Some cases of Degos disease have run in families suggesting a genetic predisposition to the disorder may exist in these cases. The mode of inheritance of this familial variant of Degos disease is unknown. Interestingly, this form of Degos disease is usually limited to the skin (benign cutaneous Degos disease).
The disease process causes the cells lining the arteries to multiply, which contributes to the narrowing or blocking of arteries (arterial occlusion). Areas of severely damaged tissue (necrosis) may appear when narrowed or blocked arteries restrict blood flow (occlusive arteriopathy). The effects of Degos disease depend upon the location of the blocked arteries and necrotic lesions.
Degos disease symptoms
In most cases, the initial symptoms of Degos disease are distinct skin lesions or a rash. Affected individuals develop small elevated bumps or spots (papules) of varying shape, usually on the trunk and upper arms and upper legs. Initially, only a few lesions may be apparent. Eventually, 10-40 lesions may slowly develop and, in some cases, hundreds may develop. The palms, soles, and face are usually spared. The lesions may sometimes itch (pruritis). The lesions start as reddish or pink bumps and eventually the center degenerates (atrophies) such that older lesions have a reddish border with porcelain-white, atrophic centers.
In certain individuals, blood vessels in areas other than the skin eventually become involved which can result in serious complications. The specific organ systems affected and the severity of associated symptoms can vary greatly. Individuals who develop systemic Degos disease will not develop all of the symptoms discussed below.
The most common area affected by Degos disease outside of the skin is the gastrointestinal tract. Gastrointestinal involvement can occur anywhere from a few weeks to a few years after the skin lesions develop. In extremely rare cases, gastrointestinal involvement may precede the development of skin lesions.
When lesions form in the small intestines, they may cause abdominal pain, cramping, nausea, vomiting, diarrhea, bloating, constipation, and the passing of blood with bowel movements or vomiting up of blood. Some affected individuals may experience weakness, fatigue and weight loss from malabsorption. The intestinal lesions can tear or rupture (perforate) causing the contents of the intestines to leak into the abdominal cavity. This results in inflammation of the membranes lining the abdominal cavity (peritonitis), a life-threatening complication.
Some individuals with systemic Degos disease experience involvement of the central nervous system (CNS). Symptoms of CNS involvement vary depending upon the specific areas affected, but may include headaches, dizziness, seizures, paralysis (palsy) of cranial nerves, weakness of one side of the body (hemiparesis), strokes, and damage to small areas of cells in the brain due to blocked arteries (cerebral infarcts.) Nonspecific neurological symptoms such as memory loss or altered sensations may also occur.
Additional organ systems that can become involved include the eyes, heart, lungs, and kidneys. When the eyes are affected, individuals may develop double vision (diplopia), drooping of the eyelids (ptosis), clouding of the lenses of the eyes (cataracts), atrophy of the optic nerve, swelling of the optic nerve (papilledema), and loss of part of the field of vision (visual-field defects).
When the heart is affected, individuals may develop weakness, shortness of breath upon exertion, and chest pain. In some cases, inflammation of the sac-like membrane surrounding the heart (pericardium) may occur. This may develop into permanent thickening, with resulting scarring and contracture of the pericardium (constrictive pericarditis).
Inflammation of the membranes lining the lungs (pleuritis) and fluid collection around the lungs (pleural effusion) has also been reported.
Skin
Skin lesions are the characteristic feature of Degos disease (malignant atrophic papulosis). The lesions are usually multiple and occur predominantly on the trunk and arms. They usually start as small red raised spots of 2–5 mm in diameter. After a few days, they enlarge and develop a central white spot that is depressed in comparison to the red skin around it. They heal leaving depressed porcelain-white scars.
Gastrointestinal system
Gastrointestinal complications of Degos disease (malignant atrophic papulosis) in 50% of patients with systemic disease result from lack of blood supply to the lining of the gut. They may lead to serious complications including perforation of the bowel. The usual symptoms are sudden onset of abdominal pain or gastrointestinal bleeding (vomiting blood or passing blood with the bowel motion).
Nervous system
Occlusion of blood vessels in the brain in Degos disease (malignant atrophic papulosis) in 20% of patients can lead to strokes, headaches, epilepsy or non-specific neurological symptoms, such as memory loss or altered sensation.
Other organs
Other organs sometimes affected by Degos disease (malignant atrophic papulosis) include eyes, kidneys, heart and liver.
Degos disease diagnosis
Degos disease (malignant atrophic papulosis) is diagnosed clinically and by skin biopsy revealing occlusion of small blood vessels.
There are no specific blood tests, and most results are normal. Further investigations including imaging will depend on systemic symptoms and signs.
Degos disease treatment
No specific therapy has been identified for Degos disease. Treatment is directed toward the specific symptoms that are apparent in each individual.
Anti-platelet drugs like aspirin and dipyridamole may reduce the number of new lesions in some patients with only skin involvement. A combination of two of these drugs (i.e., aspirin and dipyridamole) has been reported to improve skin and eye lesions in two individuals who did not have systemic involvement.
There have been reports of benefit from experimental treatments using eculizumab (monoclonal antibody binding C5 complement) and treprostinil (prostacyclin analogue) 3. The initial enthusiasm for eculizumab as monotherapy may have faded. Burgin et al 4 note that eculizumab (900 mg/d) failed as treatment for advanced systemic atrophic papulosis with GI involvement, with fatality as the outcome. Shapiro et al 5 note that only two patients initially given eculizumab as monotherapy are still alive; they instead use a combination of subcutaneous treprostinil and eculizumab. Eculizumab, if given early enough, may help abate malignant atrophic papulosis, but as monotherapy it is not instrumental in controlling it. With four patients treated, there have had no deaths. The first two are now six-year survivors. Each of them had been critically ill in ICU settings prior to therapy. This information is important because malignant atrophic papulosis without treatment or hope of treatment is lethal and patients should be directed towards those experienced with treatment as eculizumab alone may not work.
Other reports 6 have supported the use of eculizumab for systemic Degos disease but informal communication to the author noted that despite treatment with eculizumab, some patients with systemic Degos disease have died. One report noted 2 patients with Degos disease in whom eculizumab failed 7. One had overlapping lupus and the other had pulmonary hypertension. The pulmonary hypertension patient was treated with treprostinil and cutaneous Degos disease lesions resolved but disabling digital pain manifested. In the patient with Degos disease and lupus, treprostinil temporally resulted in clearing of hematuria, CNS symptoms and MRI finding improvements.
A presentation by Magro et al 8 reported that eculizumab, which has been approved for paroxysmal nocturnal hemoglobinuria, improved systemic Degos disease in 3 patients. This small series was uncontrolled, but the results suggest promise for eculizumab as a treatment. The effectiveness of eculizumab also may support the idea that Degos disease is not a collagen-vascular disease, but instead is an endothelial- or complement-mediated disease. Eculizumab is expensive, which may hinder its acceptance as widely used therapy.
An article published in 2013 9 found that exploratory treatment with eculizumab did not help the development or progression of Degos disease systemic manifestations, with 5 of 7 patients’ disease initially improving but then regressing, with progression of disease in 1 immediately and the 4 others after a period of time.
A proposed new treatment for Degos disease is treprostinil, a medication for pulmonary hypertension. Shapiro et al 10 report a series of 3 patients treated with subcutaneous treprostinil (dose not reported). One patient had systemic lupus erythematosus (SLE), overlap scleroderma, extensive malignant atrophic papulosis lesions, and severe pulmonary hypertension without CNS or bowel involvement. She was placed on therapy with treprostinil for her pulmonary hypertension, but in the months subsequent to initiation of treatment, dramatic and complete resolution of cutaneous malignant atrophic papulosis–like lesions and disabling digital pain occurred. The second patient had pure malignant atrophic papulosis but progressive organ (CNS, bladder, neurological, and gross hematuria) dysfunction despite eculizumab treatment, but therapy with treprostinil was temporally associated with clearing of hematuria, resolution of CNS symptoms, and improvement in MRI findings.
Other treatments that have been tried without real effect include the following: topical corticosteroids, phenformin and ethylestrenol, iodohydroxyquinoline, aspirin and dipyridamole, phenylbutazone, arsenic, sulfonamides, dextran, corticosteroids, heparin, warfarin, niacin, streptomycin, corticotropin, azathioprine, methotrexate, cyclosporine, tacrolimus, mycophenolate mofetil, pentoxifylline, and clopidogrel.
Hohwy et al 11 treated a patient with fatal systemic malignant atrophic papulosis (MAP) with narrow-band ultraviolet (UVB) light and prednisolone and, later, with aspirin, pentoxifylline, and warfarin. Treatment was not effective. Interestingly, the patient has a mutation of factor V Leiden and lupus anticoagulant.
Care is primarily supportive. Admission to the hospital can be necessary in cases of gastrointestinal bleeding or other systemic complications.
One case reported in the medical literature demonstrated improvement of skin lesions and gastrointestinal symptoms upon the administration of nicotine patches. Some researchers 12 have advocated the use of intravenous immunoglobulin as a treatment for affected individuals. More research is necessary to determine the long-term safety and effective of such therapies in individuals with Degos disease.
In diagnosed cases of Degos disease, examination of the gastrointestinal tract on a regular basis may detect intestinal perforation before symptoms of acute complications (i.e., peritonitis) appear. Some associated complications such as gastrointestinal bleeding, intestinal perforation, bowel infarction, or intracranial bleeding proper surgical intervention is necessary.
Degos disease prognosis
Degos disease (malignant atrophic papulosis) is generally regarded as a serious vascular disease because it leads to the involvement of multiple organs and results in death within 2–3 years. The most common cause of death is sepsis due to peritonitis and perforation (occurring in approximately 60% of reported cases) developing within two to three years of internal organ involvement 13.
Patients with only skin lesions may have a good prognosis. Gastrointestinal involvement may occur in as many as 60% of patients, and death in such cases is likely. About 15% of patients with Degos disease have long-term survival, with disease often limited to the skin and with no manifestations of fatal bowel or central nervous system (CNS) involvement.
Systemic Degos disease is frequently fatal within 2-3 years from the onset of systemic involvement. The cause of death is usually intestinal perforation. However, the range of survival time from time of diagnosis varies from less than 1 year to more than 12 years. Other causes of death include bowel infarction, pleuropericardial pathology and neurologic infarction and hemorrhage 14.
Wilson et al 15 reviewed the 24 reported instances of malignant atrophic papulosis malignant/systemic type and benign/purely cutaneous type in pediatric patients. They found that 14 cases (58%) were fatal. Patients died an average of 3.6 years after the diagnosis of malignant atrophic papulosis.
In one patient, in whom skin and abdominal symptoms occurred at the same time, death from bowel hemorrhage followed in 6 months.
In 1996, Subbiah et al 16 described the neurologic features of a series of 15 patients with Degos disease at the Mayo Clinic. Each patient had the white papules that are the hallmark of Degos disease (biopsy proven). Long-term follow-up revealed 6 patients were dead. Nine patients with skin lesions only were nearly asymptomatic. Immunosuppressive and antiplatelet agents did not halt disease progression. Central nervous system (brain) infarcts and hemorrhages with intravascular thrombi and without evidence of vasculitis were notable findings at autopsy.
Notash et al 17 reported a 48-year-old Iranian man with lethal systemic Degos disease.
Two reports describe acute abdomen associated with Degos disease, one fatal 18.
When associated with gastrointestinal tract or central nervous system involvement, patients with Degos disease have a poor prognosis and a high mortality.
In patients with benign cutaneous Degos disease, immunosuppression (in 1 patient with cyclosporine) can cause Degos disease to evolve into systemic fatal disease.
- Degos disease. https://rarediseases.org/rare-diseases/degos-disease/[↩]
- Degos disease. https://emedicine.medscape.com/article/1087180-overview[↩]
- Malignant Atrophic Papulosis Is Challenging to Diagnose, Treat. https://www.the-rheumatologist.org/article/malignant-atrophic-papulosis-is-challenging-to-diagnose-treat[↩]
- Burgin S, Stone JH, Shenoy-Bhangle AS, McGuone D. Case records of the Massachusetts General Hospital. Case 18-2014. A 32-Year-old man with a rash, myalgia, and weakness. N Engl J Med. 2014 Jun 12. 370(24):2327-37.[↩]
- Shapiro L, Whelan P, Magro C. Case 18-2014: A man with a rash, myalgia, and weakness. N Engl J Med. 2014 Oct 2. 371(14):1361.[↩]
- Magro CM, Wang X, Garrett-Bakelman F, Laurence J, Shapiro LS, DeSancho MT. The effects of Eculizumab on the pathology of malignant atrophic papulosis. Orphanet J Rare Dis. 2013 Nov 26. 8:185.[↩]
- Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Köhlmeier-Degos disease) with treprostinil–early experience. Orphanet J Rare Dis. 2013 Apr 4. 8:52.[↩]
- Magro C, Shapiro L, Johnson B, Salmon J, Whelan P. Rheumatology Grand Rounds Hospital for Special Surgery (New York City) Degos Disease. Feb 10, 2010. In press.[↩]
- Theodoridis A, Makrantonaki E, Zouboulis CC. Malignant atrophic papulosis (Kohlmeier-Degos disease) – A review. Orphanet J Rare Dis. 2013 Jan 14. 8(1):10.[↩]
- Shapiro LS, Toledo-Garcia AE, Farrell JF. Effective treatment of malignant atrophic papulosis (Kohlmeier-Degos disease) with treprostinil — early experience. Orphanet J Rare Dis. 2013 Apr 4. 8(1):52. [↩]
- Hohwy T, Jensen MG, Tottrup A, Steiniche T, Fogh K. A fatal case of malignant atrophic papulosis (Degos’ disease) in a man with factor V Leinden mutation and lupus anticoagulant. Acta Derm Venereol. 2006. 86(3):245-7.[↩]
- Zhu KJ, Zhou Q, Lin AH, Lu ZM, Cheng H. The use of intravenous immunoglobulin in cutaneous and recurrent perforating intestinal Degos disease (malignant atrophic papulosis). Br J Dermatol. 2007 Jul. 157(1):206-7.[↩]
- Heymann W: Degos disease: Considerations for reclassification. J Am Acad Dermatol. 2009;61(3):505–506.[↩]
- Ali YN, Hamed M, Azita N. Lethal systemic degos disease with prominent cardio-pulmonary involvement. Indian J Dermatol. 2011 Sep-Oct. 56(5):564-7.[↩]
- Wilson J, Walling HW, Stone MS. Benign cutaneous Degos disease in a 16-year-old girl. Pediatr Dermatol. 2007 Jan-Feb. 24(1):18-24.[↩]
- Subbiah P, Wijdicks E, Muenter M, Carter J, Connolly S. Skin lesion with a fatal neurologic outcome (Degos’ disease). Neurology. 1996 Mar. 46(3):636-40.[↩]
- Notash AY, Mazoochy H, Mirshams M, Nikoo A. Lethal systemic Degos disease with prominent cardio-pulmonary involvement. Saudi Med J. 2008 Jan. 29(1):133-7.[↩]
- Kim DW, Kang SB, Lee KH, Choe GY, Park SY, Nicholay M. Degos’ disease (malignant atrophic papulosis) as a fatal cause of acute abdomen: report of a case. Surg Today. 2008. 38(9):866-70.[↩]


{kind=link}