What is dyskeratosis congenita
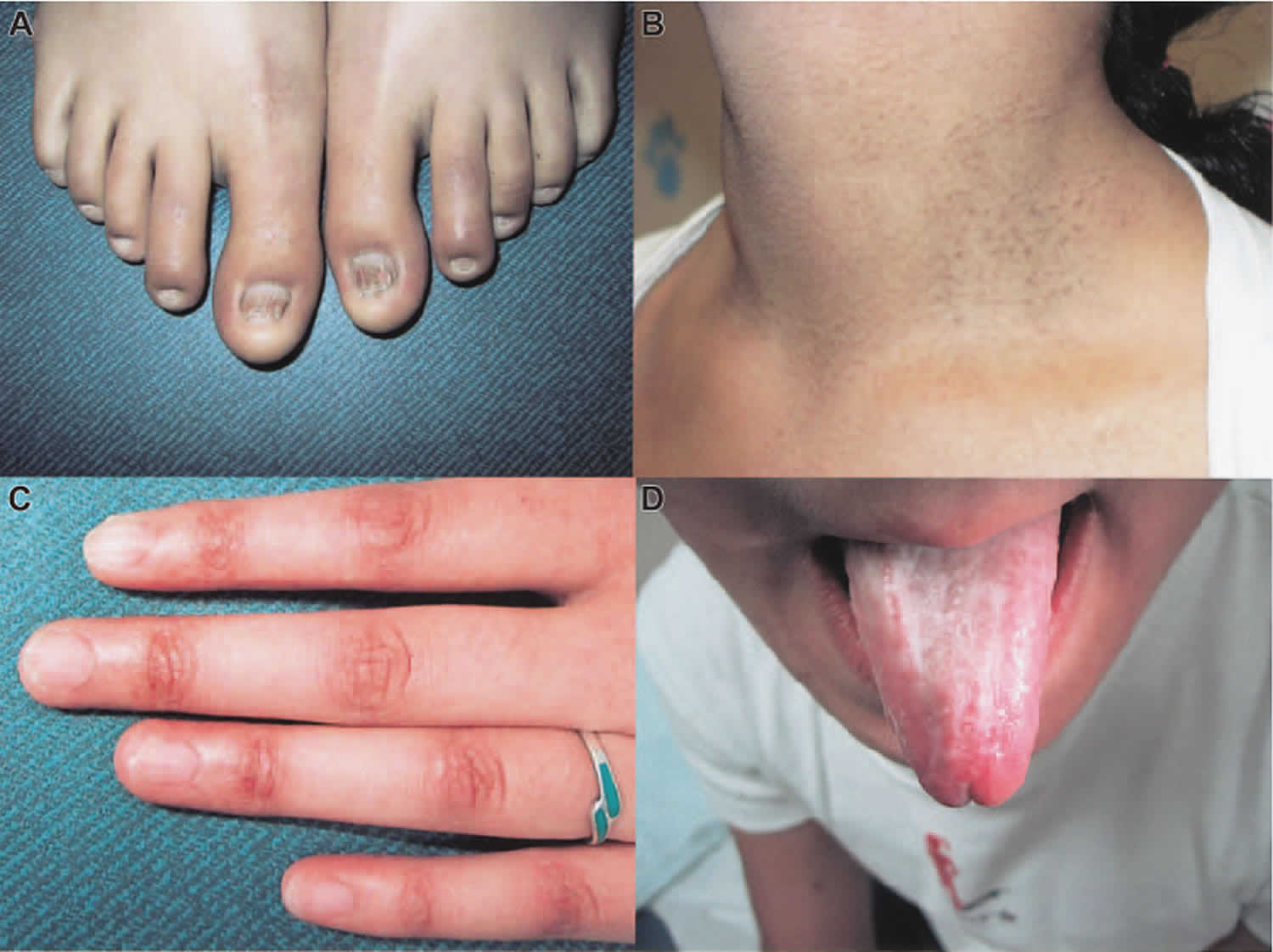
Dyskeratosis congenita is also known as Zinsser–Engman–Cole syndrome, is a group of genetic diseases that most commonly manifest with mucocutaneous signs, bone marrow failure and/or lung or liver fibrosis. There are three features that are characteristic of dyskeratosis congenita: fingernails and toenails that grow poorly or are abnormally shaped (nail dystrophy); changes in skin coloring (pigmentation), especially on the neck and chest, in a pattern often described as “lacy”; and white patches inside the mouth (oral leukoplakia).
There is considerable variability in the severity, age at onset and organ involvement, even within individual families. The various clinical features are now known to be due to ‘telomere shortening’ (unstable chromosomes), one of the causes of premature ageing. Since identifying the genetic changes, it has been discovered that a number of other conditions are due to changes in the same genes.
The telomere is a region of repetitive DNA that forms a cap over the end of a chromosome in normal cells. It protects the end of the chromosome and ensures its stability. The normal cell divides about 50 times in its lifetime. The telomere gets shorter with every cell division. Eventually telomere shortening results in damaged DNA that can’t divide any more; hence telomere shortening is associated with cellular ageing.
An enzyme called telomerase prevents telomere shortening. It is secreted by some stem cells and nearly all cancer cells, allowing them to keep on dividing.
Mutations have currently been identified in six genes coding for proteins involved in the maintenance of telomeres.
- Most mutations affect telomerase. Without effective telomerase, the telomere length gets shorter with each cell division and quickly becomes so short the cell dies much sooner than normal. This is most important in cells that divide a lot, such as in the bone marrow and skin.
- Other mutations affect the protein ‘shelterin’ which shelters the telomeres from damage by DNA repair mechanisms.
The exact prevalence of dyskeratosis congenita is unknown. It is estimated to occur in approximately 1 in 1 million people 1. To date eleven genes (DKC1, TERC, TERT, TINF2, ACD, CTC1, NHP2 (NOLA2), NOP10 (NOLA3), PARN, RTEL1, and WRAP53 gene) when mutated have been shown to be responsible for dyskeratosis congenita. Approximately 70% of those who meet the clinical diagnostic criteria for dyskeratosis congenita have a mutation in one of these genes 2. However, mutations in these genes only account for about one half of patients with classical clinical manifestations of dyskeratosis congenita, suggesting that there are additional genes that when mutated cause dyskeratosis congenita.
People with dyskeratosis congenita have an increased risk of developing several life-threatening conditions. They are especially vulnerable to disorders that impair bone marrow function. These disorders disrupt the ability of the bone marrow to produce new blood cells. Affected individuals may develop aplastic anemia, also known as bone marrow failure, which occurs when the bone marrow does not produce enough new blood cells. They are also at higher than average risk for myelodysplastic syndrome, a condition in which immature blood cells fail to develop normally; this condition may progress to a form of blood cancer called leukemia. People with dyskeratosis congenita are also at increased risk of developing leukemia even if they never develop myelodysplastic syndrome. In addition, they have a higher than average risk of developing other cancers, especially cancers of the head, neck, anus, or genitals.
People with dyskeratosis congenita may also develop pulmonary fibrosis, a condition that causes scar tissue (fibrosis) to build up in the lungs, decreasing the transport of oxygen into the bloodstream. Additional signs and symptoms that occur in some people with dyskeratosis congenita include eye abnormalities such as narrow tear ducts that may become blocked, preventing drainage of tears and leading to eyelid irritation; dental problems; hair loss or prematurely grey hair; low bone mineral density (osteoporosis); degeneration (avascular necrosis) of the hip and shoulder joints; or liver disease. Some affected males may have narrowing (stenosis) of the urethra, which is the tube that carries urine out of the body from the bladder. Urethral stenosis may lead to difficult or painful urination and urinary tract infections.
The severity of dyskeratosis congenita varies widely among affected individuals. The least severely affected individuals have only a few mild physical features of the disorder and normal bone marrow function. More severely affected individuals have many of the characteristic physical features and experience bone marrow failure, cancer, or pulmonary fibrosis by early adulthood.
While most people with dyskeratosis congenita have normal intelligence and development of motor skills such as standing and walking, developmental delay may occur in some severely affected individuals. In one severe form of the disorder called Hoyeraal Hreidaarsson syndrome, affected individuals have an unusually small and underdeveloped cerebellum, which is the part of the brain that coordinates movement. Another severe variant called Revesz syndrome involves abnormalities in the light-sensitive tissue at the back of the eye (retina) in addition to the other symptoms of dyskeratosis congenita.
The classical mucocutaneous triad of dyskeratosis congenita was originally defined by the following three mucocutaneous features:
- Nail dystrophy
- Longitudinal ridging
- Loss of nails
- Koilonychia (thin spoon-shaped nails)
- Abnormal skin pigmentation
- Reticulate (lace-like) hyperpigmentation of skin creases, described as ‘dirty neck’ (>90%)
- Oral leukoplakia
- Appears later in childhood
- White patches in the mouth
The first two features usually appear by the age of 10 years.
Other skin changes can include:
- Early hair greying or hair loss
- Sparse eyelashes
- Nail ridging
- Hyperhidrosis (excessive sweating)
- Squamous cell carcinomas (SCC), often arising in teens or twenties (50% by 40 years). Squamous cell carcinoma is often found in mucosal sites including oral cancer
Bone marrow failure develops by the age of 30 years in 80-90%.
In addition to skin cancer, many other forms of cancer occur commonly in this syndrome, usually developing in the twenties:
- Mouth – oral cancer especially squamous cell carcinoma of the tongue
- Airways – carcinomas of larynx or bronchus
- Digestive tract – carcinomas of oesophagus, colon, pancreas and anorectal
- Bone marrow – leukaemias, in particular, acute myeloid leukaemia is 200 times more common than in the general population.
- Lymphomas
The new classification of dyskeratosis congenita
With the identification of the genetic mutations of dyskeratosis congenita, other previously unconnected manifestations have now been linked. A proposed modification of the definition is:
- One or more features of the classic mucocutaneous triad
- Bone marrow failure, such as aplastic anemia
- Two or more of the other internal changes known to occur in dyskeratosis congenita:
- eyes – excessive tears
- nervous system – learning difficulties, developmental/mental retardation, ataxia (unsteadiness), underdeveloped cerebellum, microcephaly (small brain), deafness
- lungs – pulmonary fibrosis (scarring), abnormal blood vessels
- bones – osteoporosis (thin bones), aseptic necrosis (bone death), scoliosis (twisted spine), short stature
- teeth – extensive dental loss and decay, decreased root/crown ratios, mild taurodontism (enlarged tooth and pulp chamber)
- digestive system – liver fibrosis/failure, esophageal stricture (narrowing), peptic ulcer
- genitourinary system – hypogonadism (small testes), undescended testes, phimosis (tight foreskin), urethral stricture (narrowing)
The combinations of clinical features, the age of onset and severity vary with different mutations and within families.
A number of other conditions have now been shown to have the same genetic mutations as classic dyskeratosis congenita. These include:
- Hoyeraal–Hreidarsson syndrome – very short stature, bone marrow failure, immunodeficiency, underdeveloped cerebellum
- Idiopathic aplastic anemia (anemia due to bone marrow failure)
- Myelodysplasia (bone marrow failure)
- Idiopathic pulmonary fibrosis
- Paroxysmal nocturnal hemoglobinuria
- Revesz syndrome – bilateral exudative retinopathy (eye disease), bone marrow hypoplasia (failure), nail dystrophy, fine hair, cerebellar hypoplasia, growth retardation
Figure 1. Dyskeratosis congenita
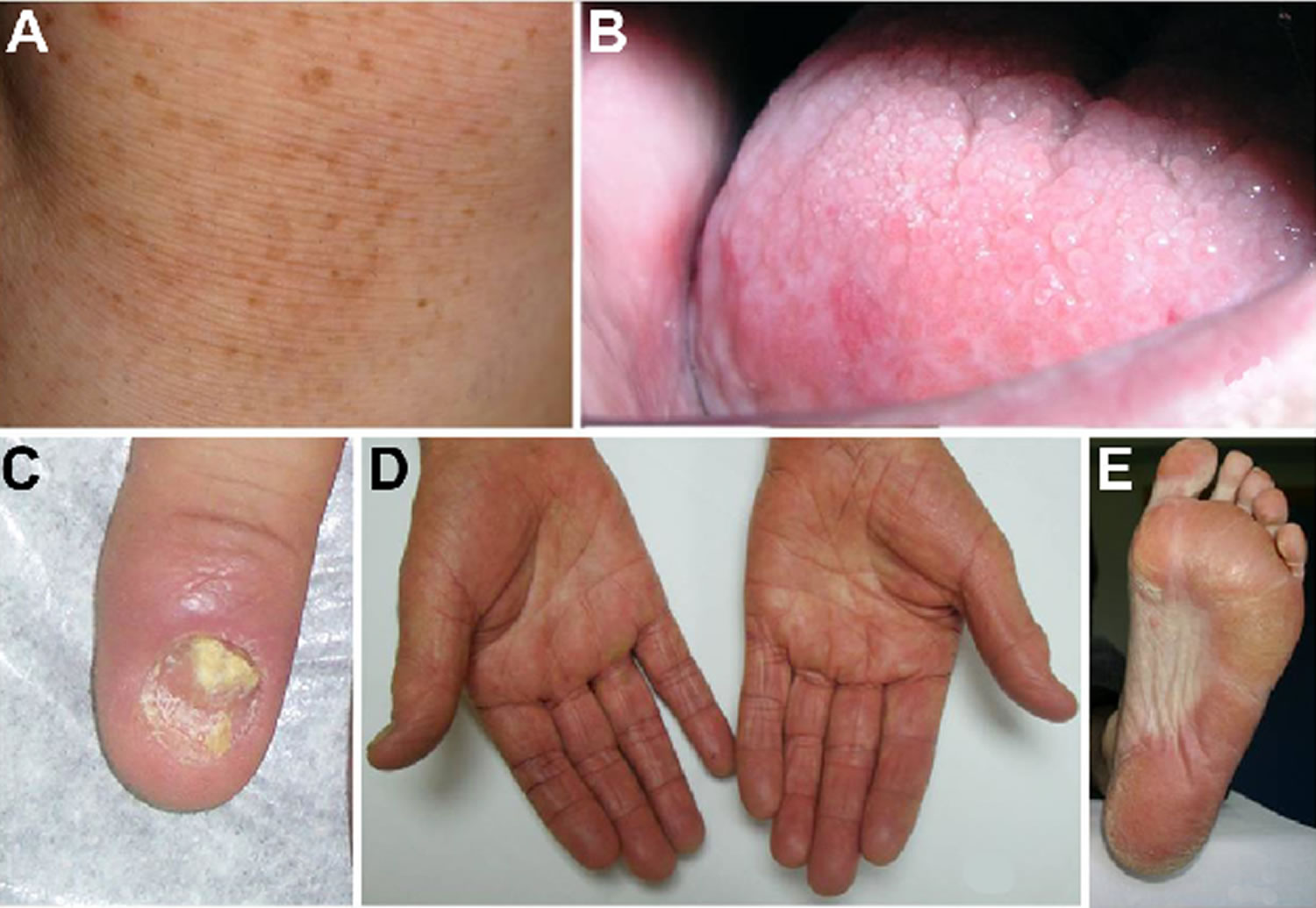
Footnote: Dyskeratosis congenita clinical findings. (A) Abnormal skin pigmentation. (B) Leukoplakia. (C) Nail dystrophy. (D and E) Hyperkeratosis and hyperpigmentation of the palms and soles.
[Source 3 ]Dyskeratosis congenita causes
In 70 percent of people with dyskeratosis congenita, the disorder is caused by mutations in the DKC1, TERC, TERT, TINF2, ACD, CTC1, NHP2 (NOLA2), NOP10 (NOLA3), PARN, RTEL1, and WRAP53 gene. These genes provide instructions for making proteins that help maintain structures known as telomeres, which are found at the ends of chromosomes. In a small number of individuals with dyskeratosis congenita, mutations in other genes involved with telomere maintenance have been identified. Other affected individuals have no mutations in any of the genes currently associated with dyskeratosis congenita. In these cases, the cause of the disorder is unknown, but other unidentified genes related to telomere maintenance are likely involved.
Telomeres help protect chromosomes from abnormally sticking together or breaking down (degrading). In most cells, telomeres become progressively shorter as the cell divides. After a certain number of cell divisions, the telomeres become so short that they trigger the cell to stop dividing or to self-destruct (undergo apoptosis).
Telomeres are maintained by two important protein complexes called telomerase and shelterin. Telomerase helps maintain normal telomere length by adding small repeated segments of DNA to the ends of chromosomes each time the cell divides. The main components of telomerase, called hTR and hTERT, are produced from the TERC and TERT genes, respectively. The hTR component is an RNA molecule, a chemical cousin of DNA. It provides a template for creating the repeated sequence of DNA that telomerase adds to the ends of chromosomes. The function of the hTERT component is to add the new DNA segment to chromosome ends. The DKC1 gene provides instructions for making another protein that is important in telomerase function. This protein, called dyskerin, attaches (binds) to hTR and helps stabilize the telomerase complex.
The shelterin complex helps protect telomeres from the cell’s DNA repair process. Without the protection of shelterin, the repair mechanism would sense the chromosome ends as abnormal breaks in the DNA sequence and either attempt to join the ends together or initiate apoptosis. The TINF2 gene provides instructions for making a protein that is part of the shelterin complex.
TERT, TERC, DKC1, or TINF2 gene mutations result in dysfunction of the telomerase or shelterin complexes, leading to impaired maintenance of telomeres and reduced telomere length. Cells that divide rapidly are especially vulnerable to the effects of shortened telomeres. As a result, people with dyskeratosis congenita may experience a variety of problems affecting quickly dividing cells in the body such as cells of the nail beds, hair follicles, skin, lining of the mouth (oral mucosa), and bone marrow.
Breakage and instability of chromosomes resulting from inadequate telomere maintenance may lead to genetic changes that allow cells to divide in an uncontrolled way, resulting in the development of cancer in people with dyskeratosis congenita.
The severity of dyskeratosis congenita depends on telomere length – the shorter the telomeres the more severe the disease, and this can often be linked to the type of inheritance. Generally, the X-linked form is the most severe, the autosomal dominant form the mildest and the autosomal recessive type can be anywhere in between the two extremes.
Dyskeratosis congenita inheritance pattern
Dyskeratosis congenita can have different inheritance patterns.
Autosomal dominant dyskeratosis congenita
- TERT and TERC mutations affect telomerase.
- TINF2 gene mutations affect shelterin.
Autosomal recessive dyskeratosis congenita
- NHP2, NOP10,TERT, TERC, TINF2 mutations.
X-linked dyskeratosis congenita
- DKC1 mutations affect dyskerin protein, a component of telomerase.
Hoyeraal–Hreidarsson syndrome / Revesz syndrome / Some cases of idiopathic aplastic anemia
- TINF2 mutations
When dyskeratosis congenita is caused by DKC1 gene mutations, it is inherited in an X-linked recessive pattern. The DKC1 gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a mutation would have to occur in both copies of the gene to cause the disorder. Because it is unlikely that females will have two altered copies of this gene, males are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
When dyskeratosis congenita is caused by mutations in other genes, it can be inherited in an autosomal dominant or autosomal recessive pattern. Autosomal dominant means one copy of the altered gene in each cell is sufficient to cause the disorder. Autosomal recessive means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Dyskeratosis congenita symptoms
The symptoms and onset of symptoms in patients with dyskeratosis congenita varies greatly depending on the gene mutated, the nature of the mutation, for how many generations the mutation has been inherited, and possibly other genetic and environmental factors. However, even among members of the same family the symptoms and onset may vary to some degree. In some families disease seems to become more severe and manifests earlier in life with subsequent generations. One of the characteristics is that, with the exception of the very severe forms (Hoyeraal-Hreidarsson and to some extent the Revesz syndrome), clinical symptoms are not present at birth, but develop during childhood, adolescence, and in some cases only late in life. In general, the earlier the disease becomes apparent, the more likely it is that the disease is severe and rapidly progressing. Likewise, the later in life clinical symptoms appear, the milder the form of disease and the slower the progression of disease. The exception to this is the risk of cancer and leukemia, which increases with age and is more common in patients with moderate to mild forms of disease. Patients with the classic form of dyskeratosis congenita are those who present with the originally described skin, nail and mouth abnormalities. In these patients the skin and nail abnormalities usually appear before 10 years of age and bone marrow failure by 20 years of age. In approximately 80-90 percent of patients with classic dyskeratosis congenita bone marrow failure occurs by age 30. In some cases, bone marrow failure appears before skin, nail or mucous membrane symptoms. Patients with the mild form of dyskeratosis congenita may have no apparent symptoms (asymptomatic) into their 30s or 40s and often present only with one of the clinical features associated with dyskeratosis congenita such as bone marrow failure, pulmonary fibrosis, liver fibrosis, or osteoporosis. Skin and nail changes, might be so mild that they are overlooked, or not noticed.
The X-linked form and some of the sporadic forms often present as classic dyskeratosis congenita, whereas individuals with the autosomal dominant form of dyskeratosis congenita often tend to have fewer abnormalities and later onset of symptoms. The skin and mucous membranes abnormalities are usually milder in the autosomal dominant form. The autosomal recessive form can vary dramatically with some individuals experiencing bone marrow failure early during childhood, while others have no blood abnormalities into their 40s. Although these findings are typical of many cases, individual cases may turn out differently.
Skin, nail, and mouth changes
The skin abnormalities associated with dyskeratosis congenita include abnormal dark discoloration of the skin with a distribution pattern that resembles a net (reticulate hyperpigmentation). Affected areas may appear as grayish, flat spots (macules) on a degenerated (atrophic) or light colored patch of skin. The face, neck and shoulders are most often affected.
Nail abnormalities usually affect the fingernails before the toenails and are characterized by fissures, underdevelopment (hypoplasia) and eventually degeneration and distortion of the affected nails. Some individuals may ultimately lose the affected nails.
The development of white, thickened patches on mucous membranes of the mouth (oral leukoplakia) usually develops slowly appearing anywhere during the second, third or fourth decade. Although the mouth is predominantly affected, the mucous membranes of the anus and the urethra may become involved in rare cases.
Bone marrow failure
Most individuals with dyskeratosis congenita eventually develop bone marrow failure marked by deficiency of all three of the main types of blood cells (i.e., red cells, white cells, and platelets) a condition called pancytopenia. The bone marrow produces specialized cells (hematopoietic stem cells) that grow and eventually develop into red blood cells (erythrocytes), white blood cells (leukocytes), and platelets. The cells are released into the bloodstream to travel throughout the body performing their specific functions. Red blood cells deliver oxygen to the body, white blood cells help in fighting off infections, and platelets allow the body to form clots to stop bleeding. The degree of bone marrow failure can greatly vary from very mild with only one type of blood cell affected to very severe with low counts in all blood cell lineages. Bone marrow examination shows a reduced number of blood cell producing progenitor cells (hyopcelular or empty bone marrow). Sometimes it is not only the blood counts that are abnormal but the blood cells themselves can show abnormalities, such as chromosomal (karyotypic) differences. These findings are usually described as myelodysplasia or myelodysplastic syndrome (MDS). Patients with myelodysplastic syndrome are at a higher risk of developing leukemia particularly if they are associated with certain karyotypic abnormities such as only one chromosome 7 (monosomy 7) over a long period of time. In rare cases myelodysplastic syndrome or leukemia may be the first manifestation of disease.
Pancytopenia (low blood cell count of all blood cell lineages) may result in a variety of symptoms. Bruising, small red spots on the skin (petechiae), paleness of the skin (pallor) and frequent infections may be the first signs of bone marrow failure. The specific symptoms and progression of the disorder vary from case to case. Some individuals may have mild symptoms that remain stable for many years; others may have serious symptoms that can progress to life-threatening complications. Bone marrow failure may develop during childhood or not become severe until well into adulthood.
Individuals with anemia may experience tiredness, increased need for sleep, weakness, lightheadedness, dizziness, irritability, headaches, pale skin color, difficulty breathing (dyspnea), and cardiac symptoms. Individuals with low white blood cell counts (leukopenia) have an increased risk of contracting bacterial and fungal infections. Individuals with low platelet counts (thrombocytopenia) are more susceptible to excessive bruising following minimal injury and to spontaneous bleeding from the mucous membranes, especially those of the gums and nose.
Some patients with the same genetic abnormality responsible for dyskeratosis congenita may present with bone marrow failure only. The severity of bone marrow failure in these patients can vary greatly, from affecting only one or two blood values in peripheral blood, to the full picture with low blood counts in all blood cell lineages, a condition termed aplastic anemia. Features of the skin or other symptoms associated with dyskeratosis congenita may not be present or be so mild that they are not appreciated. These patients are often initially misdiagnosed as having idiopathic aplastic anemia (see also below). Whether patients with the genetic abnormality for dyskeratosis congenita but only showing bone marrow failure should be classified as having dyskeratosis congenita is controversial, alternative classifications used are atypical dyskeratosis congenita, or aplastic anemia with short telomeres. It is important that in these individuals the treatment plan, response to treatment, disease surveillance, and prognosis differs from patients with idiopathic aplastic anemia. In addition because of the inherited nature of the disease, members of the family of the affected individual may also be at risk.
Leukemia and cancer
Individuals with dyskeratosis congenita also have a predisposition to develop leukemia and cancer (malignancy) especially squamous cell carcinoma of the head and neck, and especially at the site of leukoplakia. If cancer occurs, it usually does not develop until the age of about 30. Thus, leukemia and cancer are more common in individuals who have a moderate or milder form of dyskeratosis congenita. Individuals who underwent a stem cell or bone marrow transplant for the treatment of their bone marrow failure are also at risk of developing cancer later in life. In rare cases leukemia or cancer may be the first manifestation of disease.
Lung disease
The development of lung disease (pulmonary fibrosis) is often found in patients with dyskeratosis congenita. It usually develops later than the skin abnormalities and bone marrow failure, however in some patients with mild disease pulmonary fibrosis may be the first or only obvious manifestation. In these patients disease usually manifest at the age of 50 to 60 years of age. The cause of pulmonary fibrosis in patients with dyskeratosis congenita is not fully understood. Breathing difficulties and a decreased lung function may be signs of lung disease. Smoking seems to accelerate the progression of pulmonary disease.
Other symptoms
A variety of additional symptoms have been reported in individuals with dyskeratosis congenita. These symptoms occur with much less frequency than the abovementioned symptoms. These less common symptoms include excessively watery eyes due to obstruction of the tear ducts (epiphora), excessive sweating (hyperhidrosis) of the palms of the hands and the soles of the feet, cavities and tooth loss, narrowing of the esophagus (esophageal stricture), urinary tract anomalies, especially hypospadism, early graying and premature hair loss, lung disease and short stature, liver disease, underdeveloped testes (hypogonadism), failure of the testes to descend into the scrotum, and skeletal abnormalities.
Some affected children may experience delays in attaining developmental milestones and learning disabilities. Additional symptoms have been reported that occur in less than 10 percent of cases including deafness, or abnormalities of the eye retina.
Hoyeraal-Hreidarsson syndrome
Once considered a separate disorder, Hoyeraal-Hreidarsson syndrome is now identified as a severe variant of dyskeratosis congenita. Symptoms usually occur during the first year of life and include severe growth retardation that occurs before birth (intrauterine growth retardation), bone marrow failure, immune system deficiencies, underdevelopment of the cerebellum (cerebellar hypoplasia), clumsiness caused by the inability to coordinate voluntary movements (ataxia), and microcephaly, a condition that indicates that head circumference is smaller than would be expected for an infant’s age and sex. Abnormalities of the gut varying from malabsorbtion to severe inflammation with ulcers may be present in these children. Bone marrow failure and immune system deficiency may result in life-threatening complications. Because of the complexity and because multiple organs are severely impaired the prognosis of children diagnosed with Hoyeraal-Hreidarsson syndrome is usually poor. These children often die before they develop the characteristic nail and skin abnormalities.
Revesz syndrome
Revesz syndrome is another severe form of dyskeratosis congenita that may present similar to the Hoyeraal-Hreidarsson syndrome but in addition is associated with abnormalities of the eye (bilateral exudative retinopathy, Coats retinopathy).
Dyskeratosis congenita diagnosis
The diagnosis of dyskeratosis congenita is based on the definition above.
Gene mutations have so far only been identified in approximately 50% of cases. Molecular genetic tests to determine mutations in the DKC1, TERC, TERT, TINF2 NHP2, or NOP10 gene can confirm a diagnosis of dyskeratosis congenita. However, clinical genetic testing is expensive and for some genes only available on a research basis. Furthermore, genetic testing usually does not test for large gene deletions, thus patients with disease due to a large gene deletion are usually missed. Difficult may also be the proof that the identified sequence variant is in fact responsible for disease might be difficult. Not all the mutations described in the literature are in fact responsible for disease (rare polymorphisms) or cause disease in all individuals (variable penetrance). In about 50% of patients no mutation is identified, despite the presence of classic clinical manifestations.
It may be possible to distinguish dyskeratosis congenita by flow-fluorescence in situ hybridization (FISH) analysis due to the very short telomeres compared to age-matched controls.
It is important to consider the diagnosis in cases of bone marrow failure or lung fibrosis where no other cause has been identified, oral leukoplakia in a young person with no history of tobacco use and in early-onset cancers.
Dyskeratosis congenita treatment
There is no cure at this time for dyskeratosis congenita. There is no consensus about how to treat patients with dyskeratosis congenita. The literature is biased toward treatment and treatment outcome of patients who present with the classical form of disease. Little is still known about the treatment and disease monitoring of individuals with atypical or silent disease.
The treatment of dyskeratosis congenita is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, adult internists and hematologist, dermatologists, dental specialists, medical geneticist, cancer specialists (oncologists) and other healthcare professionals may need to systematically and comprehensively plan an affected individual’s treatment.
General treatment recommendations for individuals with dyskeratosis congenita include avoiding smoking and alcohol to preserve the lungs and liver and the use of moisturizing creams to prevent damage to the skin. Good dental hygiene may help to prevent early tooth loss and delay the development of malignancy of the tongue. In certain patients marrow failure and immunodeficiency transiently respond to androgens and hematopoietic growth hormones.
Treatment is aimed at maintaining bone marrow function as this is the major cause of death:
- Oxymetholone – an anabolic steroid that helps bone marrow function in two-thirds of patients for several years. Androgen (e.g., oxymetholone), which is artificial male hormone, may increase red blood cell and, less often, platelet production in some individuals. Androgen therapy may be supplemented with corticosteroid (e.g., prednisone) therapy, which may delay growth acceleration potentially associated with androgen therapy and reduce bleeding associated with thrombocytopenia.
- Hematopoietic growth factors – erythropoietin, granulocyte macrophage colony-stimulating factor (GM-CSF) and granulocyte colony-stimulating factor (G-CSF). A class of drugs known as hematopoietic growth factors has been used to treat individuals with dyskeratosis congenita, specifically granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF). These growth factors may transiently increase the production of certain white blood cells (neutrophils). In rare cases, treatment with these drugs also increases red blood cell and platelet levels.
- Bone marrow transplant – this was invariably fatal in dyskeratosis congenita due to the rapid development of liver failure or lung fibrosis post-transplant. It has now been recognized that this was due to intolerance to the pretransplant chemotherapy and/or radiotherapy. Patients with dyskeratosis congenita have an increased sensitivity towards radiation and certain chemotherapy drugs. An alternative conditioning regiment is without irradiation or certain chemotherapy drugs such as busulfan or melphalan should be avoided. Pulmonary complications after hematopoietic stem cell transplantation are not uncommon and may be fatal. Bone marrow transplants have now been performed successfully in a small number of dyskeratosis congenita patients with less aggressive pretransplant therapy. If a compatible donor can be located, hematopoietic stem cell transplantation can potentially cure the blood abnormalities associated with dyskeratosis congenita. Hematopoietic stem cell transplantation should be considered in patients who present with mainly bone marrow failure. Hematopoietic stem cell transplant does not improve tissues affected by dyskeratosis cogenita.
In most cases, the benefits of androgens and growth factors are only temporary. The specific amount of time these treatments improve bone marrow function varies in each individual case.
The hypersensitivity of individuals with dyskeratosis congenita to radiation and chemotherapy encumbers the treatment of cancer in these individuals. Surgical resection of the cancer is probably the first line of treatment.
Although dyskeratosis congenita would seem to be an ideal condition for gene therapy, no progress has been made in this direction yet.
Affected individuals should be monitored for the development of lung disease and cancer.
Genetic counseling is important for the planning of future pregnancies. Ante-natal diagnosis has been achieved successfully.
Dyskeratosis congenita life expectancy
Dyskeratosis congenita is a multisystem disorder that carries a poor prognosis (mean survival of 30 years), with most deaths related to infections, bleeding, and malignancy. The degree of severity and age at death is quite variable. Some forms are milder with survival into the forties; others are fatal in infancy. The main causes of death are bone marrow failure in 75-80% (due to increased susceptibility to infection or hemorrhage), lung fibrosis in 10-15% or cancer in 10%.
In the dyskeratosis congenita registry, approximately 70% of affected individuals died of bone marrow failure or its complications, and these deaths occurred at a median age of 16 years. Therapeutic interventions are mostly palliative, but bone marrow transplant and stem cell transplant for aplastic anemia have been tried with variable success. Wide variation in clinical phenotype may occur in individuals, suggesting that other genetic or environmental factors may be contributory. The prognosis is worse for the X-linked and autosomal forms compared with the autosomal dominant form.
Hoyeraal-Hreidarsson syndrome is also associated with mutations in dyskeratosis congenita1. Mutations in this gene have been described in patients with Hoyeraal-Hreidarsson syndrome, which is characterized by intrauterine growth restriction, microcephaly, mental retardation, cerebellar malformation, and progressive bone marrow failure. Mucosal ulcerations have been found in a few patients, and some authorities hypothesize that Hoyeraal-Hreidarsson syndrome may be a severe variant of dyskeratosis congenita in which affected individuals die before the development of mucocutaneous findings. One study found that patients with Hoyeraal-Hreidarsson syndrome have significantly shorter telomeres than those with the milder form of disease.
In addition, studies have also found that not only are shortened telomeres associated with Hoyeraal-Hreidarsson syndrome, but more so are telomere dysfunction and telomere protection 4. The severe neurologic deficits in this severe form point to an important role of the dyskeratosis congenita1 gene in brain function.
The biology of telomere shortening is not only associated with dyskeratosis congenita, but it has comorbid associations with neuropsychiatric conditions. A cohort study with 6 pediatric and 8 adult subjects showed that 83% of the children and 88% of the adult had a comorbid neuropsychiatric condition. These conditions include schizophrenia, anxiety, intellectual disability, attention-deficit/hyperactivity disorder, adjustment disorder, mood disorders, or pervasive developmental disorders 5.
Mortality/morbidity
In an analysis of individuals with dyskeratosis congenita, approximately 70% of patients died either directly from bone marrow failure or from its complications at a median age of 16 years. Eleven percent died from sudden pulmonary complications; a further 11% died of pulmonary disease in the bone marrow transplantation (BMT) setting. Seven percent died from malignancy (eg, Hodgkin disease, pancreatic carcinoma). Fatal opportunistic infections such as Pneumocystis carinii pneumonia and cytomegalovirus infection have been reported.
- Dyskeratosis congenita. https://ghr.nlm.nih.gov/condition/dyskeratosis-congenita[↩]
- Savage SA. Dyskeratosis Congenita. 2009 Nov 12 [Updated 2016 May 26]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22301[↩]
- García, M.S., & Teruya-Feldstein, J. (2014). The diagnosis and treatment of dyskeratosis congenita: a review. Journal of blood medicine.[↩]
- Touzot F, Gaillard L, Vasquez N, et al. Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J Allergy Clin Immunol. 2012 Feb. 129(2):473-82, 482.e1-3[↩]
- Rackley S, Pao M, Seratti GF, et al. Neuropsychiatric conditions among patients with dyskeratosis congenita: a link with telomere biology?. Psychosomatics. 2012 May-Jun. 53(3):230-5.[↩]



{kind=link}