Gigantism
Gigantism is a very rare condition due to growth hormone hypersecretion that causes children to grow abnormally fast and tall. Gigantism occurs when growth hormone (GH) hypersecretion occurs before the fusion of the long bone epiphysis and is characterized by tall stature 1. Gigantism should be suspected when the child’s height is 3 standard deviations above normal mean height or 2 standard deviations above the adjusted mean parental height 2. Children with gigantism will grow unusually tall, and many will experience delayed puberty. Acromegaly occurs when growth hormone hypersecretion occurs after the fusion of the epiphysis leading to large extremities and characteristic facies. Acromegaly is usually diagnosed in adults aged 30 to 50 but can affect people of any age. Despite these disparities, some degree of clinical overlap has been suggested by the observation that 10% of acromegalics have tall stature 3, indicating that the onset of growth hormone excess pre-dates epiphyseal fusion in many patients. Growth hormone excess during childhood and adolescence is extremely rare, with an estimated incidence of 8 per million person-years and the total number of reported cases thus far numbering only in the hundreds 4. The most common cause is a non-cancerous (benign) tumor of the pituitary gland, which may cause it to make too much growth hormone; an elevated insulin-like growth factor-1 (IGF-1) level established the diagnosis. Although rare, gigantism can be caused by other underlying conditions (which may cause a pituitary tumor) including Carney complex; McCune-Albright syndrome ; Multiple endocrine neoplasia type 1 (MEN-1) and type 4 (MEN-4); and Neurofibromatosis where an evaluation for neurofibromas with cafe au lait spots, optic gliomas, and skin lentigines should be done 4. In some cases gigantism is caused by mutations in the GPR101 gene. The first line of treatment is surgical excision of the pituitary tumor; however, this rarely results in a cure, and further medical treatment with somatostatin analogs or radiation is necessary 5. Children may still experience some symptoms, and need to have regular check-ups as they grow up.
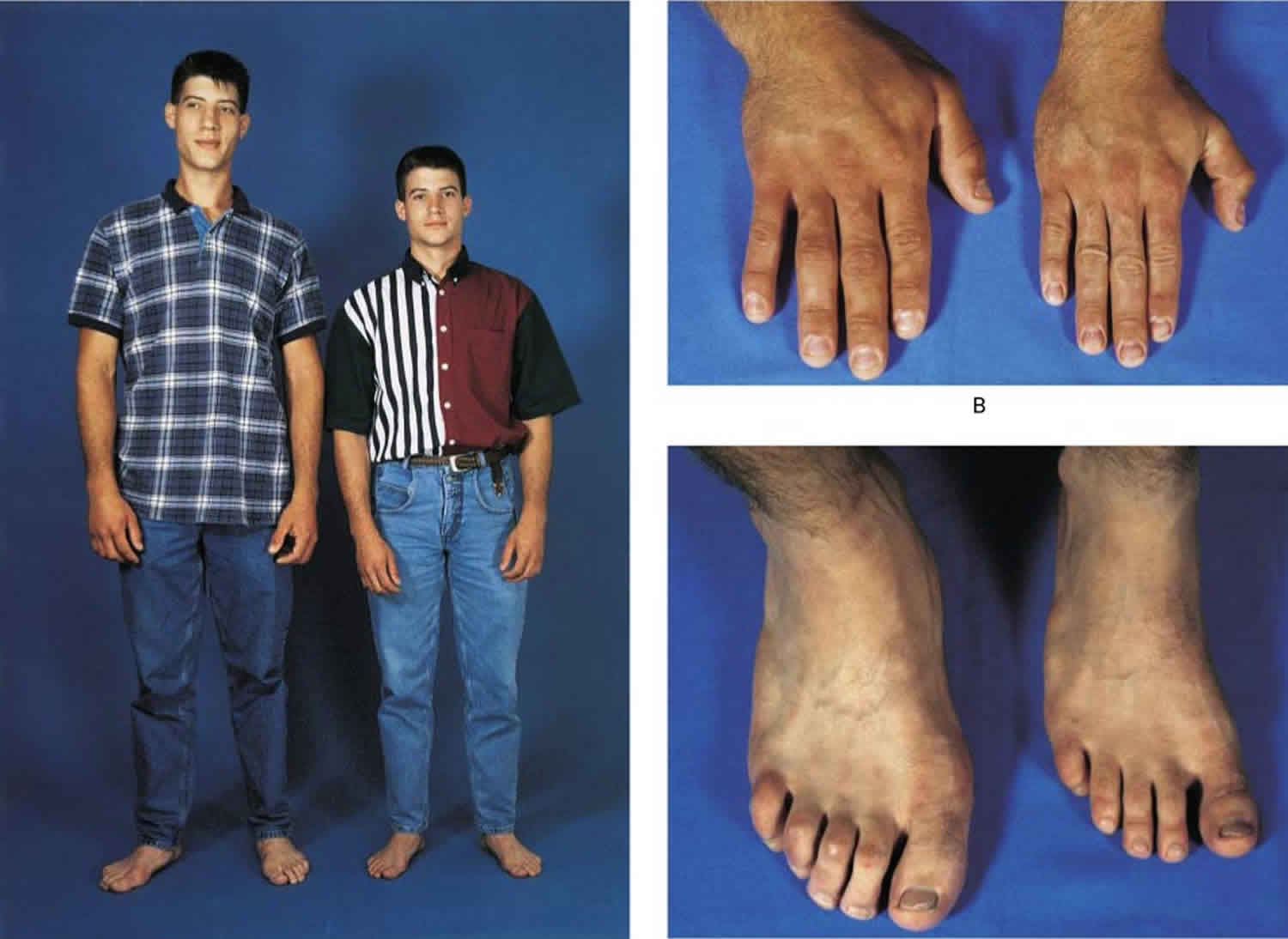
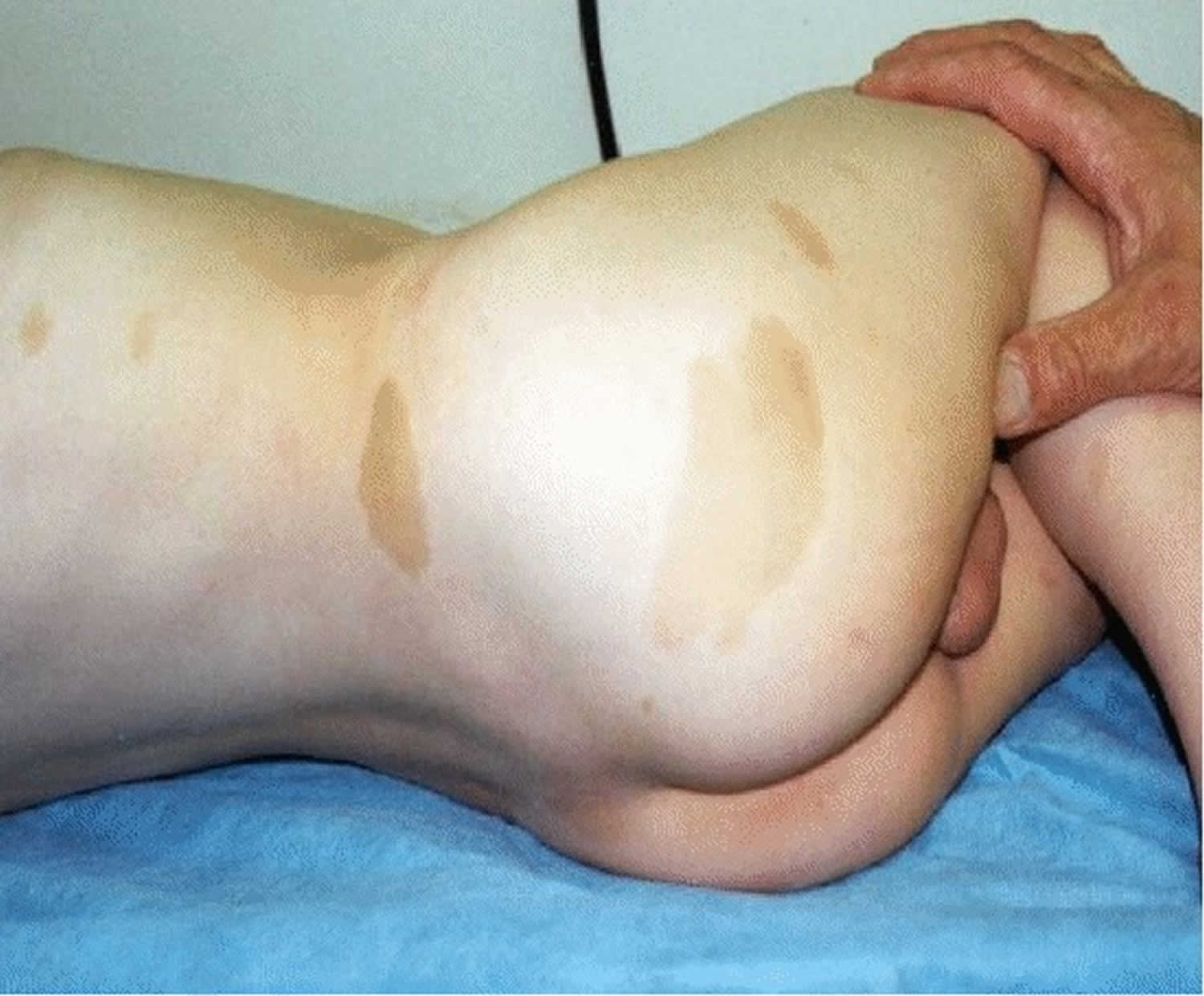
Figure 1. Pituitary gigantism
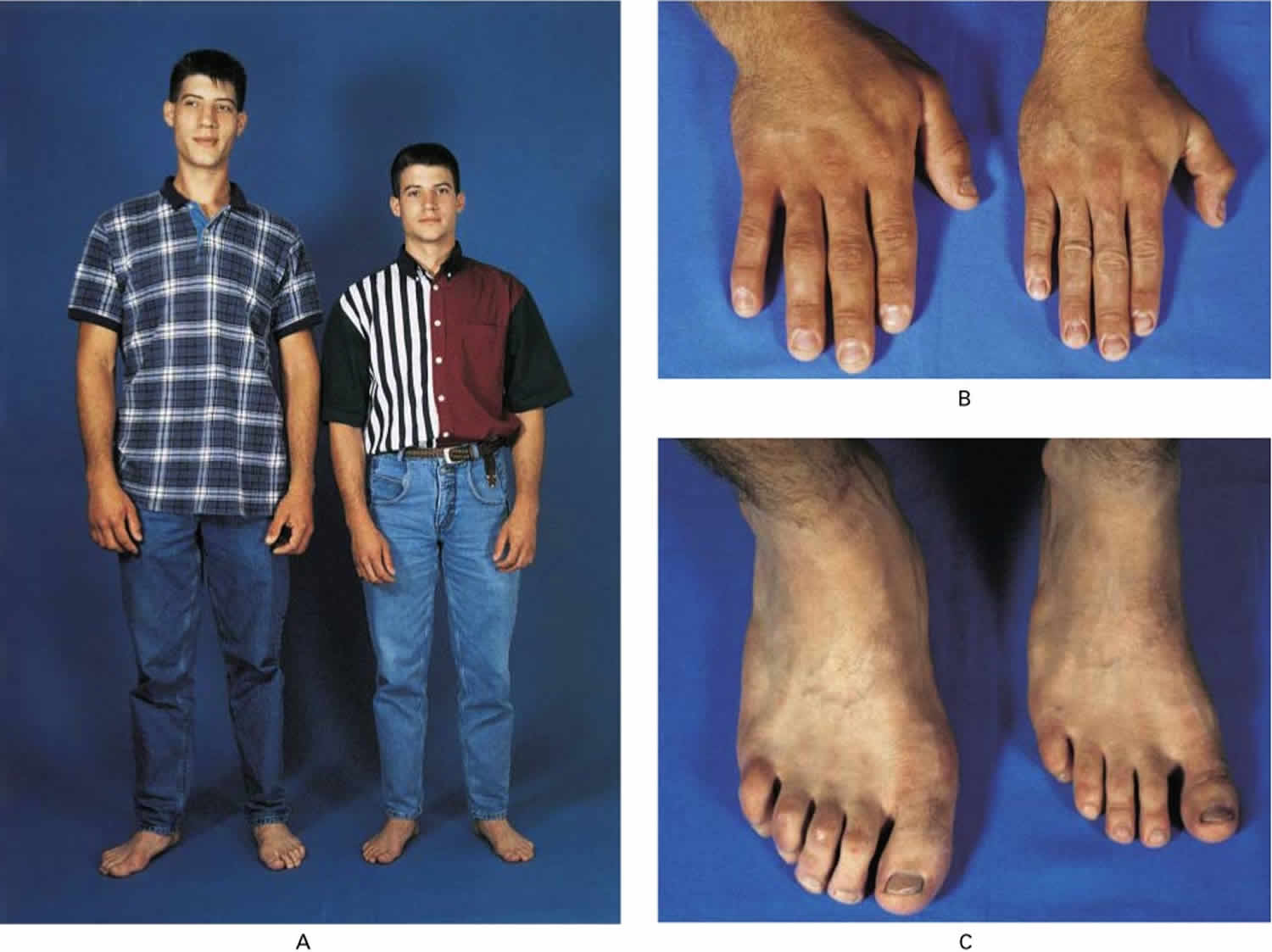
Footnote: A 22-year-old man with gigantism due to excess growth hormone is shown to the left of his identical twin. The increased height (Panel A) and enlarged hand (Panel B) and foot (Panel C) of the affected twin are apparent. Their height and features began to diverge at the age of approximately 13 years.
[Source 6 ]Hypothalamus and Pituitary Gland
The pituitary gland is a pea-shaped structure that measures 1–1.5 cm (0.5 in.) in diameter and lies in the hypophyseal fossa of the sella turcica of the sphenoid bone (see Figure 2). The pituitary gland attaches to the hypothalamus by a stalk, the infundibulum and has two anatomically and functionally separate portions:
- the anterior pituitary and
- the posterior pituitary.
The anterior pituitary (anterior lobe), also called the adenohypophysis, accounts for about 75% of the total weight of the gland and is composed of epithelial tissue. The anterior pituitary consists of two parts in an adult: The pars distalis is the larger portion, and the pars tuberalis forms a sheath around the infundibulum.
The posterior pituitary (posterior lobe), also called the neurohypophysis, is composed of neural tissue. It also consists of two parts: the pars nervosa, the larger bulbar portion, and the infundibulum.
A third region of the pituitary gland called the pars intermedia atrophies during human fetal development and ceases to exist as a separate lobe in adults. However, some of its cells migrate into adjacent parts of the anterior pituitary, where they persist.
Anterior Pituitary
The anterior pituitary secretes hormones that regulate a wide range of bodily activities, from growth to reproduction.
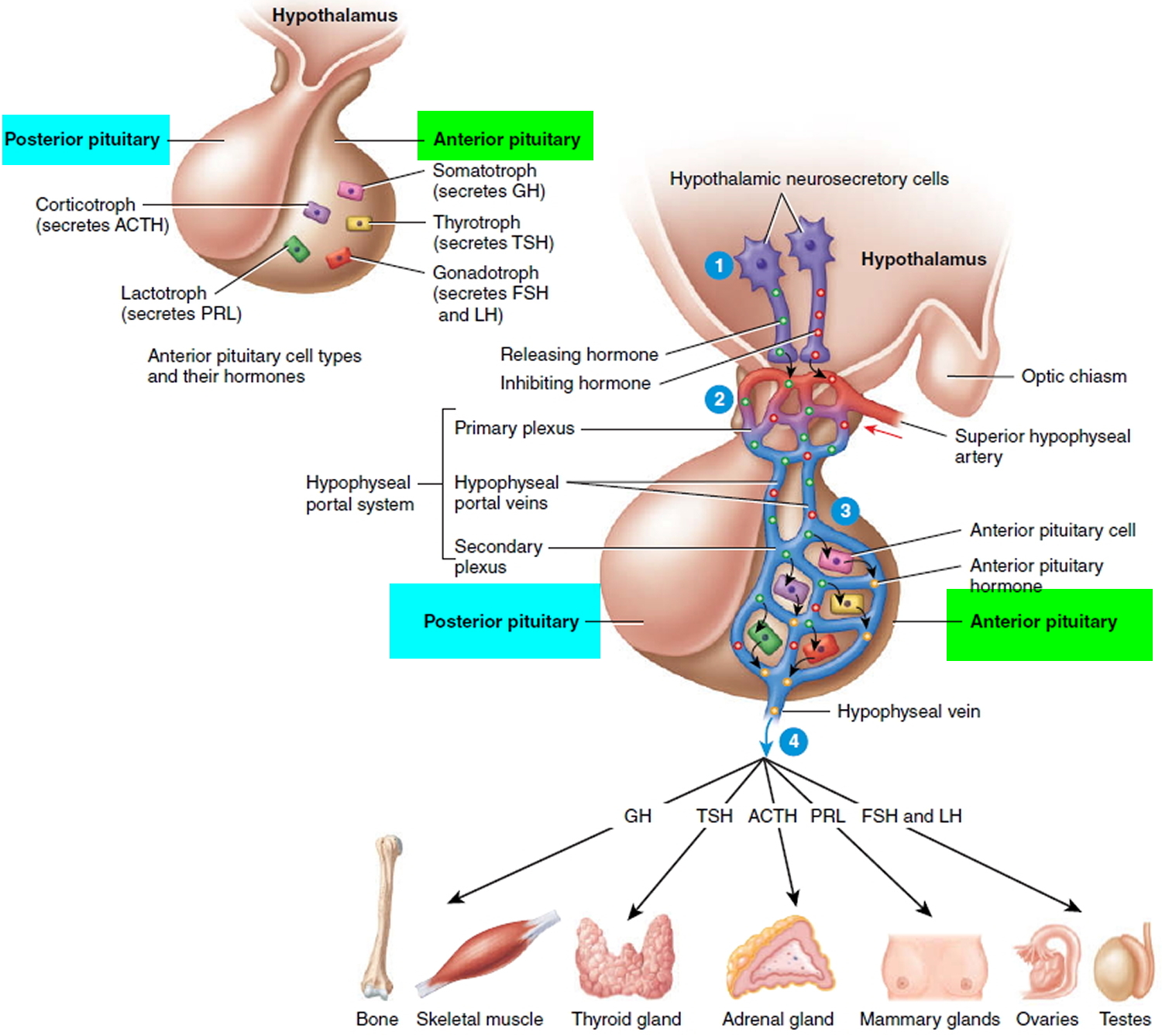
Types of Anterior Pituitary Cells and Their Hormones
Five types of anterior pituitary cells—somatotrophs, thyrotrophs, gonadotrophs, lactotrophs, and corticotrophs—secrete seven hormones:
- 1. Somatotrophs secrete growth hormone (GH), also known as human growth hormone (hGH) or somatotropin. Growth hormone stimulates general body growth and regulates aspects of metabolism.
- 2. Thyrotrophs secrete thyroid-stimulating hormone (TSH), also known as thyrotropin. TSH controls the secretions and other activities of the thyroid gland.
- 3. Gonadotrophs secrete two gonadotropins: follicle-stimulating hormone (FSH) and luteinizing hormone (LH). FSH and LH both act on the gonads (testes and ovaries). In men, they stimulate the testes to produce sperm and to secrete testosterone. In women, they stimulate the ovaries to mature oocytes (eggs) and to secrete estrogens and progesterone.
- 4. Lactotrophs secrete prolactin (PRL), which initiates milk production in the mammary glands.
- 5. Corticotrophs secrete adrenocorticotropic hormone (ACTH), also known as corticotropin, which stimulates the adrenal cortex to secrete glucocorticoids such as cortisol. Some corticotrophs, remnants of the pars intermedia, also secrete melanocyte stimulating hormone (MSH).
Hypothalamic Control of the Anterior Pituitary
Release of anterior pituitary hormones is regulated in part by the hypothalamus. The hypothalamus secretes five releasing hormones, which stimulate secretion of anterior pituitary hormones:
- 1. Growth hormone-releasing hormone (GHRH), also known as somatocrinin, stimulates secretion of growth hormone.
- 2. Thyrotropin-releasing hormone (TRH) stimulates secretion of thyroid-stimulating hormone.
- 3. Corticotropin-releasing hormone (CRH) stimulates secretion of adrenocorticotropic hormone.
- 4. Prolactin-releasing hormone (PRH) stimulates secretion of prolactin.
- 5. Gonadotropin-releasing hormone (GnRH) stimulates secretion of FSH and LH.
The hypothalamus also produces two inhibiting hormones, which suppress secretion of anterior pituitary hormones:
- 1. Growth hormone-inhibiting hormone (GHIH), also known as somatostatin, suppresses secretion of growth hormone.
- 2. Prolactin-inhibiting hormone (PIH), which is dopamine, suppresses secretion of prolactin.
Figure 2. Pituitary gland and hypothalamus
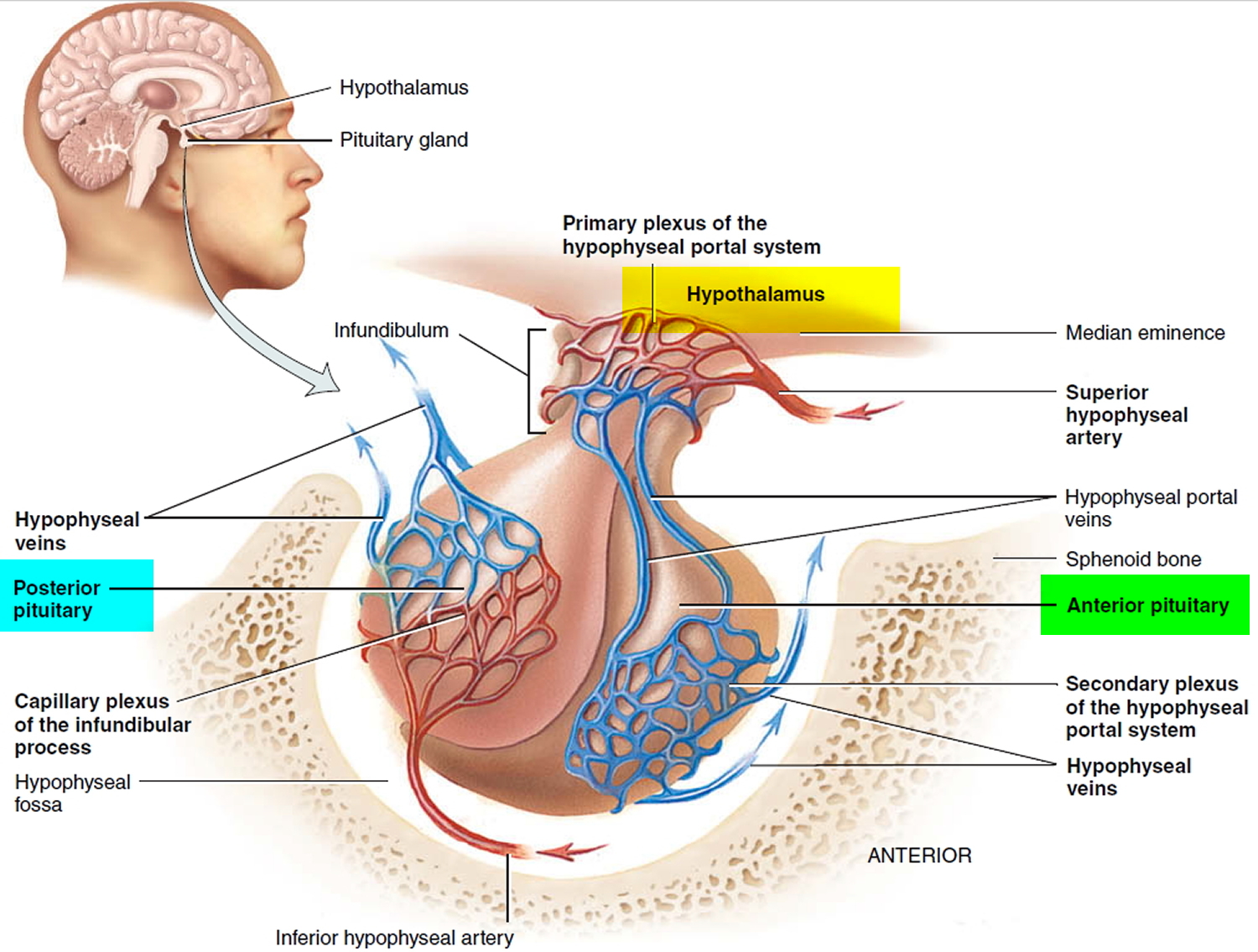
Figure 3. Hypothalamic Control of Pituitary Gland
Normal growth hormone physiology
- Growth hormone is secreted by the pituitary gland in a pulsatile fashion, meaning that its level goes up and down significantly throughout the day.
- For this reason, growth hormone measured at a random time of the day may be meaningless for diagnosis
- Growth hormone release normally falls dramatically after eating a large quantity of sugar, a phenomenon that is useful in making the diagnosis in acromegaly because tumor cells do not exhibit this response (see oral glucose tolerance test)
- Growth hormone has direct affects on the body, but like most pituitary hormones, acts on other specialized gland cells in the body to stimulate the release of another hormone
- Growth hormone acts on special cells in the liver, leading to the release into the blood stream a hormone called Insulin-like Growth Factor (IGF-1). IGF-1 is also called Somatomedin-C
- Because IGF-1 is released in a more steady fashion, its levels do not go up and down quickly and therefore it is a better hormone to measure to screen for acromegaly
Normal control of growth hormone production
A part of the brain called the hypothalamus controls the amount of growth hormone production by sending chemicals (“neuropeptides”) down the pituitary stalk to the pituitary gland.
- The main neuropeptide that stimulates growth hormone release is called growth hormone releasing hormone (GHRH).
- The main neuropeptide that inhibits growth hormone release is called somatostatin; drugs that mimic the effects of somatostatin (somatostatin analogs, or SSA’s) are used to treat acromegaly.
Gigantism causes
From the time someone is born, the way their body grows is controlled by hormones produced by the pituitary gland in the brain. The most important hormone for growth is called growth hormone, also known as human growth hormone (hGH or GH).
Most children with gigantism have too much growth hormone, which makes them grow too much, too fast.
In about 95% cases acromegaly and gigantism are secondary to a growth hormone secreting adenoma in the pituitary gland. Growth hormone-releasing hormone (GHRH) secretion from a hypothalamic adenoma or ectopic growth hormone-releasing hormone secretion from lung or pancreas neuroendocrine tumors can also cause acromegaly 1. Rarely, ectopic growth hormone secretion secondary to abdominal and hemopoietic malignancies can cause acromegaly. Genetic syndromes which have been associated with growth hormone hypersecretion are multiple endocrine neoplasia-1 (MEN-1), neurofibromatosis, Carney complex and McCune-Albright syndrome. Familial idiopathic pituitary adenomas (FIPA) which are due to aryl hydrocarbon protein interacting mutations can be associated with familial acromegaly. About 25% of cases with familial acromegaly present as teenagers with gigantism 7.
Table 1. Causes of growth hormone excess
| Sporadic | Syndromic/Familial | ||
| Disorder | Source of growth hormone (GH) | Disorder | Source or Genetic Mutation |
| Hypothalamic or pituitary growth hormone excess | Congenital GHRH excess (postulated) | Neurofibromatosis-1 | Tumor infiltration into somatostatinergic pathways (postulated) |
| Pituitary somatotroph or mammosomatotroph adenoma | McCune-Albright syndrome | Activating mutation of Gsα | |
| Pituitary hyperplasia | Multiple endocrineneoplasia Type-1 and Multiple endocrineneoplasia Type-4 | Defect in tumor suppression from mutations in menin and CDKN1 genes | |
| Hypothalamic gangliocytoma/neurocytoma | Carney complex | • Abnormality at 2p16 • Mutations inPRKAR1A at17q22-24 | |
| 3PA association | Succinate dehydrogenase defects | ||
| Ectopic growth hormone excess | • GHRH or growth hormone production by bronchial, carcinoid or pancreatic neoplasm Ectopic pituitary adenoma | • Familial somatotrophinomas • X-LAG syndrome | • Mutation in acylhydrocarbon receptor gene at 11q13.3 • Abnormality at 2p12-6 • Xq26.3 • duplications |
| GH = growth hormone; GHRH = growth hormone-releasing hormone; PRKARIA = protein kinase Aregulatory subunit 1 | |||
Sporadic growth hormone excess
Sporadic growth hormone excess may arise from central nervous system (CNS) pathology or rarely from ectopic growth hormone production 4. Traditionally, the term “primary growth hormone excess” has been used to differentiate an intrinsic pituitary source of growth hormone from other causes, including hypothalamic abnormalities. In actuality, it may be difficult to clearly distinguish the role of the pituitary from the hypothalamus, particularly in cases of early childhood growth hormone excess, as discussed below.
Hypothalamic-Pituitary Growth Hormone Excess
Unlike in acromegalic adults, in whom discreet pituitary adenomas are present in the overwhelming majority of patients 8, a number of different histopathologic mechanisms underlying childhood growth hormone hypersecretion have been suggested or observed. These relate to the concept that childhood growth hormone excess represents a distinct entity, with different characteristics in terms of pituitary morphology and function. Supporting this view have been reports of diffuse pituitary hyperplasia in the setting of early-onset gigantism, in which congenital growth hormone releasing-hormone (GHRH) excess has been proposed as the inciting cause 9. Additionally, the nearly ubiquitous finding of combined growth hormone and prolactin over-secretion in nearly all cases of early childhood gigantism, a feature not universally present in acromegaly, suggests that a separate pathologic process may be involved. This dual hormonal secretion has been attributed to the presence of mammosomatotrophs 10, which are rare in adulthood but predominate in fetal life. Even in cases of apparent pituitary microadenomas or macroadenomas arising during early childhood, this unique biochemical feature has been present 11. In contrast, prolactin levels are usually normal in cases of pituitary growth hormone-secreting adenomas originating during adolescence, which may be thought of as existing within the spectrum of adult growth hormone hypersecretion. Interestingly, a reversible transformation of pituitary somatotrophs into bihormonal mammosomatotrophs under the influence of ectopic overproduction of GHRH has been observed, lending additional support to the concept that hypothalamic GHRH excess may play a pivotal role in the genesis of early-onset gigantism 12. Although the etiology of sporadic gigantism is often unknown, a number of germline and somatic mutations in genes associated with syndromic and familial growth hormone hypersecretion have been reported in children and adolescents with pituitary gigantism 13.
An additional cause of sporadic growth hormone excess linked to CNS pathology is that which occurs in the setting of a hypothalamic gangliocytoma or neurocytoma. These rare tumors, comprised of large hypothalamic-like ganglion cells, have been demonstrated to produce GHRH 14 and to be found in close proximity to pituitary growth hormone-secreting adenomas 15. Normalization of serum growth hormone levels following resection of the hypothalamic tumor in some patients has further supported a central role for abnormal GHRH secretion in the development of gigantism or acromegaly in these cases 16.
Ectopic growth hormone excess
Ectopic growth hormone hypersecretion is a rare but important cause of acromegaly in adults, thought to represent less than 1% of all cases 17. In this condition, a paraneoplastic elaboration of GHRH or uncommonly growth hormone 18 occurs, with neuroendocrine tumors being the most common source 19. Specific lesions notorious for this capability include bronchial carcinoid and pancreatic neoplasms 20. Extra pituitary growth hormone excess has also been reported in the setting of an ectopic pituitary adenoma located within the sphenoid sinus or clivus 21, and in association with an empty sella 22. An ectopic source of GHRH or growth hormone leading to gigantism in a child has never been described 4.
Syndromic and familial forms of growth hormone excess
A second major category of childhood growth hormone hypersecretion is that which occurs in the setting of a well-recognized syndrome. In these cases, gigantism may be the sole presenting feature of the syndrome or it may be detected during on-going clinical follow-up for other endocrine or non- endocrine problems. Alternatively, biochemical evidence of sub-clinical growth hormone excess may be revealed through routine surveillance in a child known to be at risk for the development of gigantism. As is the case in sporadic growth hormone hypersecretion, a variety of different morphologic abnormalities involving the pituitary gland may be found. Paracrine pituitary GHRH secretion has also been implicated by the discovery of GHRH expression from clusters of cells in the hyperplastic pituitaries of two boys from a family with hereditary early-onset gigantism 23. Syndromes that are associated with the development of childhood growth hormone excess are reviewed below. Table 2 outlines the characteristics of the growth hormone excess and other clinical features in these disorders.
Table 2. Clinical characteristics in syndromes associated with growth hormone excess
| Disorder | Mode of Inheritance | Clinical Features | Frequency of Gigantism | Age of Onset of gigantism | Pituitary Morphology | Screening recommendations |
| Neurofibromatosis -1 | Autosomal Dominant or Sporadic | • Optic gliomas • Café au lait skin pigmentation | Extremely rare | 6 months on | Optic pathway tumor with normal to small pituitary | Not routine |
| McCune- Albright Syndrome | Sporadic | • Precocious Puberty • Café au lait skin pigmentation • Fibrous bone dysplasia • Multiple endocrinop athies | 15-20% | Early childhood on | Pituitary adenomas or diffuse pituitary hyperplasia or no visible abnormality | Annually |
| Multiple Endocrine Neoplasia Type 1 | Autosomal Dominant or Sporadic | Pituitary, pancreatic and parathyroid adenomas | 10-60% | 10% by age 40 but has occurred as early as age 5 | Pituitary adenoma | Annually beginning at age 5 |
| Multiple Endocrine Neoplasia Type 4 | Autosomal Dominant or Sporadic | Pituitary, pancreatic and parathyroid adenomas | Unknown | Unknown | Pituitary adenoma | Not established |
| Carney Complex | Autosomal Dominant or Sporadic | • Multiple endocrine tumors • Skin lentigines • Cardiac myxomas • Neural sheath tumors | 10% | Usually 3rd and 4th decade | Pituitary adenoma or pituitary hyperplasia | Annually beginning post pubertally |
| 3PA Association | Autosomal Dominant or Sporadic | Pheochromocytom paraganglioma, pituitary adenoma | Unknown | Usually 3rd& 4th decade | Pituitary adenoma with intracytoplasmic vacuoles | As clinically indicated in unaffected family members |
| Isolated Familial Somatotropinom as | Autosomal Dominant or Sporadic | Isolated growth hormone- secreting pituitary adenomas | 100% | Before 3rd decade and as early as age 5 | Pituitary adenoma | As clinically indicated in unaffected family members |
| X-linked Acrogigantism | Sporadic or X- linked | Isolated growth hormone excess | 100% | Early childhood with onset in late infancy or onset during adolescen ce | Pituitary adenoma or pituitary hyperplasi a or both | As clinically indicated in unaffected family members |
Neurofibromatosis-1
Beginning in the 1970’s, several reports of gigantism occurring in young children with neurofibromatosis-1 have appeared in the medical literature 24. In these cases, excessive somatic growth has been noted as early as 6 months of life 25. Neuroimaging in these patients typically reveals an optic glioma 26, usually with infiltration into the medial temporal lobe. However, growth hormone excess has frequently been reported to be a transient phenomenon in children with neurofibromatosis-1, raising questions as to the necessity of treatment 27. A number of investigations aimed at identifying the precise etiology of the gigantism in these children have been conducted. In all cases in which tumor tissue has been available, immunostaining for growth hormone, GHRH and somatostatin has been uniformly negative 28.
This, in conjunction with the known temporal lobe location of somatostatin-producing neurons, led to the hypothesis that growth hormone excess in these patients was the result of a hypothalamic regulatory defect. Specifically, tumor infiltration of somatostatinergic pathways would presumably result in reduced somatostatin tone leading to overproduction of GHRH-mediated pituitary growth hormone. Despite this plausible explanation, arginine-induced growth hormone stimulation in a patient with gigantism in the setting of neurofibromatosis-1 was normal, contrary to the expected lack of response to arginine, which is believed to act through somatostatin inhibition 29. Thus, the precise pathogenesis of gigantism in neurofibromatosis-1 remains unclear. Little information is available regarding the overall incidence of growth hormone hypersecretion in patients with neurofibromatosis-1 and optic gliomas, although studies have suggested that it may occur in over 10% of affected patients, some of whom have concurrent central precocious puberty 30. Interestingly, growth hormone excess has also been reported in children with sporadic optic pathway tumors without associated neurofibromatosis-1 31. Figure 2 demonstrates the café-au-lait pigmentation and linear growth acceleration observed in a young boy with neurofibromatosis-1 and gigantism.
Figure 4. Cafe-au-lait pigmentation in a young boy with neurofibromatosis-1 and gigantism
Footnote: Growth acceleration and characteristic “coast of California” café au lait macules in a child with neurofibromatosis and gigantism.
[Source 4 ]McCune-Albright syndrome
McCune-Albright syndrome is a complex and heterogenous disorder in which growth hormone excess may arise in conjunction with additional endocrinopathies and other abnormalities. In the classic form, McCune-Albright syndrome results in the triad of precocious puberty, café-au-lait skin pigmentation, and fibrous dysplasia of bone. It has long been recognized, however, that individuals with McCune-Albright syndrome have a propensity to develop a number of endocrine problems, including gigantism or acromegaly from excessive growth hormone secretion 32.
Elucidation of the molecular genetic defect in McCune-Albright syndrome in the early 1990’s 33 illuminated the underlying mechanism through which abnormal hormone secretion occurs in this condition. Activating mutations of Gsα, the stimulatory subunit of the heterotrimeric G-protein complex involved in intracellular signaling, have now been shown to form the basis for nearly all of the clinical manifestations of McCune-Albright syndrome 34. These mutations, which typically involve substitution of arginine at the 201 position with cysteine or histidine, result in unregulated signal transduction leading to increased intracellular cAMP accumulation and downstream gene transcription. The precise timing of the mutation during embryologic life, which occurs in a post-zygotic cell line, will ultimately determine the extent of abnormal cells and severity of the resultant clinical phenotype. The incidence of growth hormone excess in classic McCune-Albright syndrome has generally been reported to be 15-21% 35 and may be more common in males 35. However, enhanced recognition of the frequency of atypical or forme fruste variants of McCune-Albright syndrome have the potential to result in an increase of this estimated frequency. Indeed, a number of historical reports of extreme gigantism where fibrous bone dysplasia was also present strongly suggest a diagnosis of McCune-Albright syndrome in these individuals, a hypothesis confirmed by molecular genetic analysis in at least one case 36. Subclinical growth hormone excess has also been reported in McCune-Albright syndrome, in which the only clinical manifestation may be the presence of normal stature (rather than short stature) in the context of a history of untreated precocious puberty. Additional phenotypic features in this subgroup of patients with McCune-Albright syndrome include a higher incidence of vision and hearing deficits, TRH responsiveness, and hyperprolactinemia 37. Growth hormone excess in McCune-Albright syndrome is typically accompanied by skull base fibrous dysplasia and is notorious for being associated with increased craniofacial morbidity and macrocephaly 38.
However, early diagnosis and treatment has been found to decrease the risk of optic neuropathy in these patients 39. A variety of pituitary morphologic abnormalities have been noted in McCune-Albright syndrome patients with growth hormone hypersecretion 40, ranging from discrete pituitary adenomas 41 to diffuse pituitary hyperplasia 42, to no discernible radiographic abnormality 43. Of note is the fact that the identical Gsα mutation found in McCune-Albright syndrome has also been implicated in the pathogenesis of sporadic growth hormone-secreting pituitary adenomas, where it results in formation of the gsp oncogene. Up to 40% of somatotroph adenomas in adults have been demonstrated to contain either an Arg201 activating mutation, or a related point substitution of glutamine at position 227 44. Interestingly, these sporadic tumors as well as those from patients with McCune-Albright syndrome and acromegaly display the Gsα mutation exclusively from the maternal allele, providing evidence that the GNAS1 gene is subject to imprinting 45. Figure 5 demonstrates an area of classic café au lait skin pigmentation in a patient with McCune-Albright syndrome.
First-line treatment of acromegaly/gigantism related to McCune–Albright syndrome with pituitary hyperplasia is somatostatin analogues. In resistant cases, pegvisomant alone or in combination with octreotide or lanreotide is recommended. Pasireotide has also been used in the treatment of GH excess in McCune–Albright syndrome 46. If concomitant hyperprolactinemia occurs, a dopamine agonist should be added as well. In patients not responding to pharmacological therapy, pituitary surgery should be considered. Neurosurgery is challenging due to concomitant skull base fibrous dysplasia, as high vascularity of these bony lesions gives a high risk of haemorrhage. If operated, total hypophysectomy is suggested, as in most of the cases, the whole gland is involved. Radiotherapy of the pituitary gland should be carefully considered for severe disease if previously therapy options have failed. Radiation of associated bone lesions may lead to malignant transformation of sarcoma 47.
Figure 5. Cafe-au-lait pigmentation in a patient with McCune-Albright syndrome
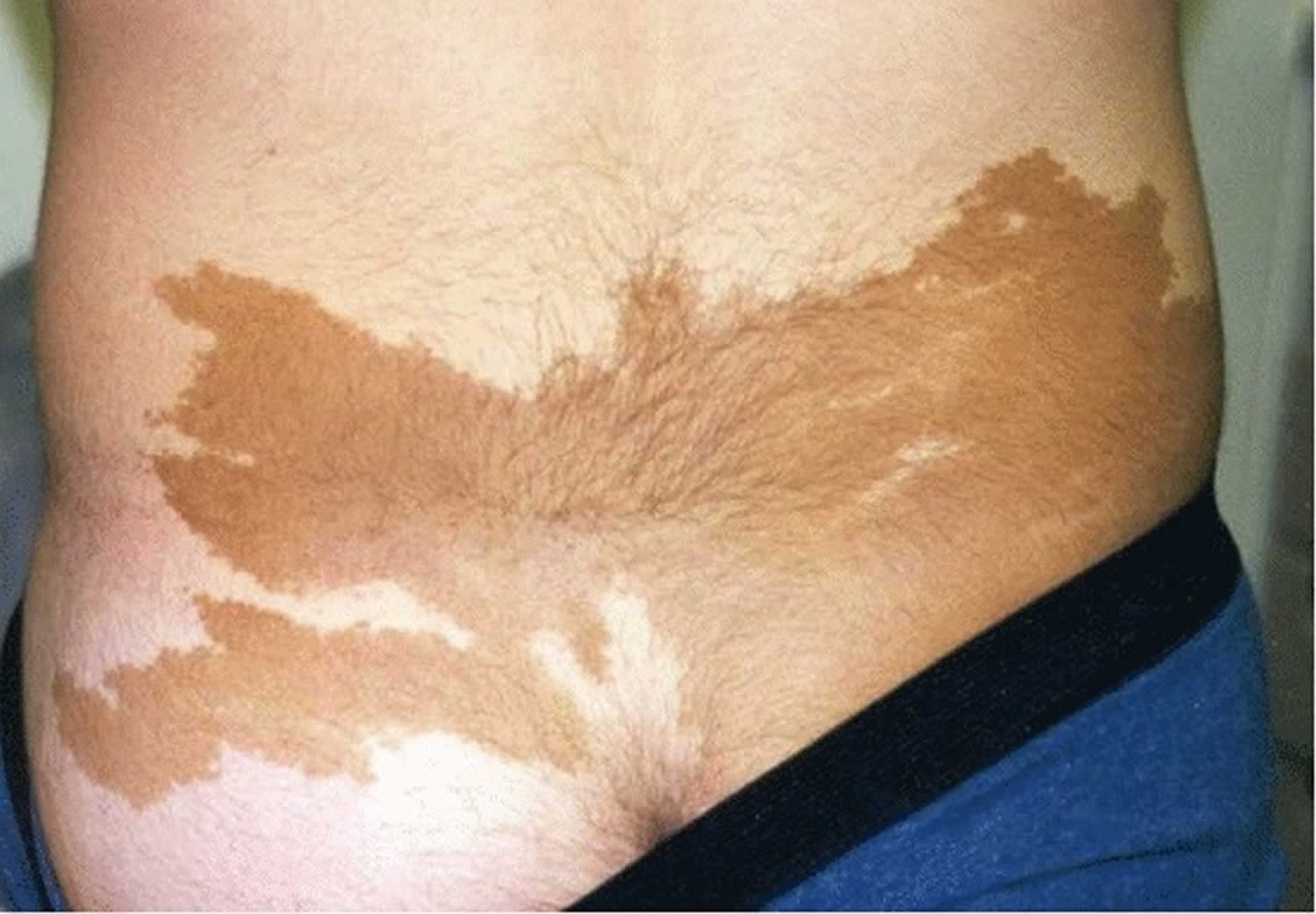
Footnote: Café au lait pigmentation in the typical “coast of Maine” configuration in an individual with McCune-Albright syndrome.
[Source 4 ]Multiple Endocrine Neoplasia-Type 1 (MEN-1)
Multiple Endocrine Neoplasia-Type 1 (MEN-1) is one of a number of familial cancer syndromes characterized by autosomal dominant inheritance and multi-endocrine gland involvement. Although significant clinical heterogeneity exists in terms of specific tumor combinations, the most frequent manifestations of MEN-1 are parathyroid, pancreatic, and pituitary adenomas 48. The gene for MEN-1, which had previously been mapped to chromosomal locus 11q13, has been cloned and demonstrated to encode for a 610 amino acid nuclear protein designated menin 49. Many different molecular genetic abnormalities within the menin gene have been identified in kindreds with MEN-1, including nonsense, missense, deletion, insertion and donor-splice mutations 50. Unfortunately, genotype/phenotype correlations have not been observed. In all cases of MEN-1, the development of neoplasia is thought to arise from a defect in normal tumor suppression via a 2-hit hypothesis. The first hit represents inheritance of a germline MEN-1 mutation, leading to a heterozygous loss of the menin gene in every cell 51. As menin is believed to function as a tumor suppressor protein, the second hit involves a somatic MEN-1 mutation in one cell, with subsequent abnormal cellular transformation and clonal expansion. Indeed, somatic biallelic MEN-1 mutations have been demonstrated to be present in at least 15% of sporadic pituitary adenomas, including somatotroph tumors 52. Anterior pituitary adenomas in individuals with known MEN-1 have a reported prevalence of 10-60% and are thought to represent the first clinical manifestation of the disease in up to 25% of sporadic cases 53. Of these, the majority are prolactinomas, with growth hormone-secreting adenomas developing in approximately 10% of individuals with MEN-1 by age 40. The youngest reported case of gigantism in MEN-1 occurred in a 5-year-old boy, who presented with growth acceleration and a growth hormone-secreting mammosomatotroph pituitary adenoma in the context of a known family history of MEN-1 54. Molecular genetic analysis confirmed the germline and tumor tissue MEN-1 mutations but failed to reveal an etiology for the accelerated presentation in this case. Nonetheless, current recommendations include screening for anterior pituitary hormone excess beginning at age 5 in all individuals with MEN-1, as well as ascertaining MEN-1 carrier status by germline mutation testing in a number of clinical situations 55. Interestingly, growth hormone excess due to ectopic elaboration of GHRH from a pancreatic neuroendocrine tumor has also been reported in several individuals with MEN-1 56.
Multiple Endocrine Neoplasia-Type 4 (MEN-4)
Multiple Endocrine Neoplasia-Type 4 (MEN-4) is a recently recognized entity that is caused by germline mutations in the CDKN1B gene which encodes the putative tumor suppressor p27Kip1 57. Affected patients are typically heterozygous for mutations in CDKN1B and exhibit a phenotype similar to that seen in MEN-1. Because the number of individuals who have been diagnosed with MEN-4 is low, screening protocols for patients and their family members have not yet been established 58.
Carney complex
Initially described in 1985, Carney complex is a rare autosomal dominant disorder in which the cardinal features include multiple endocrine tumors, skin lentigines (spotty pigmentation), cardiac myxomas and neural sheath tumors 59. Carney complex shares characteristics with several other syndromes, including MEN-1 (multiple endocrine tumors), McCune-Albright syndrome (endocrine hyperfunction and skin pigmentation) and Peutz-Jeghers (mucosal lentiginoses and gonadal tumors). It has now been demonstrated, however, to have a unique clinical and molecular genetic identity. Two distinct genetic abnormalities have been implicated in the pathogenesis of Carney complex. The first consists of a locus on 2p16 60, although a specific candidate gene within this region has not been identified. Additionally, mutations in the gene encoding for the protein kinase A regulatory subunit (1α) (PRKAR1A) at 17q22-24 have been demonstrated in 35-44% of both familial and sporadic cases of Carney complex 61. This protein, which is intricately involved in endocrine cell signaling pathways, is thought to function as a tumor suppressor gene. Supporting this theory has been the observation that tumors from patients with Carney complex (in which diminished levels of PRKAR1A are present) exhibit a 2-fold increase in cAMP responsiveness compared with control tumors 62. The identical mutation has also been found in some sporadic endocrine tumors. As with MEN-1, a germline mutation is thought to be the inciting event for eventual development of the disease. The clinical presentation of Carney complex is extremely heterogeneous, as is the age at diagnosis. The development of growth hormone excess is rare, occurring usually during the 3rd and 4th decades of life and typically found in only 10% of patients at the time of presentation 63. Thus, annual screening for growth hormone hypersecretion is recommended only in post pubertal patients. As in cases of gigantism/acromegaly in the setting of McCune-Albright syndrome, diffuse pituitary hyperplasia 64 and concomitant hyperprolactinemia 65 are frequently seen in individuals with Carney complex and growth hormone excess.
Diagnosis of Carney complex in a patient may be established clinically if two or more major criteria are present (characteristic skin lesions, cutaneous and heart myxomas, primary pigmented nodular adrenal disease, acromegaly, large-cell calcifying Sertoli cell tumour or characteristic calcification of testis, thyroid carcinoma or multiple hypoechoic nodules, breast ductal adenoma psammomatous melanotic schwannomas, blue nevus, osteochondromyxoma) 66. Another way to confirm Carney Comple diagnosis is the occurrence of one major criterion and an affected first-degree relative or a known inactivating PRKAR1A mutation. Genetic testing may be offered for patients with two major diagnostic criteria or for relatives of patients with Carney complex. Molecular techniques include Sanger sequencing. In negative cases, copy number variant analysis by comparative genomic hybridisation or deletion testing should be performed.
To date, there is no specific treatment approach for acromegaly in Carney Comple cases, and guideline on the management is the same as in sporadic cases. In a great majority of patients, surgery alone or combined with somatostatin analogs has been used. If multiple pituitary tumours are present, partial or complete hypophysectomy should be performed. Some authors suggest that due to overactivation of cAMP signalling in Carney Comple patients, the use of somatostatin analogs theoretically would be beneficial. However, resistance to somatostatin analogs treatment has been observed 67.
3PA Association
The constellation of paraganglioma, pheochromocytoma and pituitary adenoma is termed 3PA Association and has been shown to be due to germline mutations in a variety of subunits of succinate dehydrogenase 68. Growth hormone excess typically occurs in the 3rd and 4th decades of life 69. To date, no pediatric patients with gigantism in the setting of the 3PA phenotype have been reported.
Familial somatotropinomas
It has long been recognized that isolated pituitary gigantism or acromegaly may occur in a familial pattern. This phenomenon, termed “Familial Isolated Pituitary Adenomas” (FIPA), is defined as the development of growth hormone hypersecretion in two or more members of a family that does not exhibit features of MEN-1 or Carney complex. At least 46 different affected kindreds have been reported 70. Unlike in MEN-1 and CC, growth hormone excess tends to arise fairly early in life, with 70% of those with the disorder diagnosed before the 3rd decade. Early childhood gigantism in this setting has also occurred, involving sisters with abnormal linear growth since age 5 71 and a more virulent course than is seen in sporadic somatotropinomas has been suggested by a case series 72. Once assumed to represent a variant of MEN-1, mutations within the menin gene as the cause for Isolated Familial Somatotropinomas were conclusively excluded 73. However, the precise molecular genetic basis for the development of pituitary growth hormone-secreting adenomas in the majority of affected families has eluded detection. Initial investigation revealed loss of heterozygosity and linkage to a 9.7 Mb region of 11q13, suggesting the presence of an additional putative tumor suppressor gene in this region, distinct from that involved in MEN-1. Subsequent studies identified inactivating mutations in the gene encoding aryl hydrocarbon receptor interacting protein (AIP) at 11q13.3 in 15%-25% of families with Familial Isolated Pituitary Adenomas 74 making it the most common genetic defect found in these kindreds. Although the mechanism by which these mutations cause pituitary adenomas is unknown, the resulting phenotype is characterized by early-onset and aggressive disease. In an amazing case of medical sleuthing, a germline AIP mutation identified in DNA from the preserved teeth of an 18th century Irish giant was found to be an exact match for the mutation harbored by four contemporary Irish families with Familial Isolated Pituitary Adenomas, indicating a common ancestor dating back more than 50 generations ago 75. Interestingly, a second potential locus for Familial Isolated Pituitary Adenomas has mapped to 2p12-16, very close to the region implicated in a number of kindreds with Carney complex 76. Additional molecular genetic analysis performed in these patients has included a search for germline mutations within the GHRH receptor gene, Gsα and Gi2α genes, all of which were normal. Similar to observations in MEN-1, patients with Familial Isolated Pituitary Adenomas have discreet pituitary adenomas, the majority of which are comprised solely of somatotrophs 70. However, prolactinomas, gonadotropinomas and silent pituitary adenomas may all be seen in different members of the same kindred 77. Macroadenomas with invasion into the cavernous sinus are common in the setting of Familial Isolated Pituitary Adenomas, and treatment is notoriously difficult 77.
X-Linked Acrogigantism
An additional cause of familial gigantism and acromegaly has been linked to microduplication of Xq26.3 and termed X-linked acrogigantism (X-LAG). This genomic duplication was initially identified in 14 patients with gigantism and is associated with both sporadic and familial cases 78. Of the four genes contained in the duplicated region, the growth hormone excess appears to result from an abnormality of GPR101, a gene that encodes for an orphan G-protein coupled receptor 79. The GPR101 gene is markedly over-expressed in pituitary tissue from affected patients. Functional studies suggest a proliferative role for mutant GPR101, although the precise mechanism for how this aberration contributes to growth hormone hypersecretion is not yet clear. The condition can result from either germline or somatic duplications in GPR101 and has a female predominance (2/3 of the cases) 80. Mosaicism for GPR101 duplication resulting in X-LAG has also been reported in sporadic cases involving boys 81. Patients harboring the Xq26.3 microduplication exhibit a distinct phenotype characterized by strikingly early gigantism with a median age of onset of 12 months. In addition to hypersecretion of growth hormone, elevated circulating GHRH and prolactin have also been noted 82. Both pituitary adenomas and pituitary hyperplasia have been seen among cases testing positive for X-LAG. This discovery highlights new biological processes that will undoubtedly lead to novel insights regarding the central regulation of human growth.
The treatment of XLAG patients remains challenging and often requires a multimodal approach 83. Neurosurgery is the first line treatment among patients with pituitary tumors but often, further control of the disease requires additional medical therapy or radiotherapy. In cases of pituitary hyperplasia, total hypophysectomy could be an effective surgical treatment with the obvious disadvantage of complete hypopituitarism 84. In patients not controlled by surgery, pegvisomant alone or combined with somatostatin analogues or dopamine agonists is an effective treatment and successfully controls linear growth 85.
Overgrowth syndromes
The overgrowth syndromes comprise a diverse group of conditions with unique clinical, behavioral and molecular genetic features.
Sotos syndrome
Sotos syndrome, also known as cerebral gigantism, was first described in 1964 86. Since then, several hundred cases have been reported. Cardinal features of the disorder include early onset overgrowth, a characteristic facial configuration and stereotypical behavioral profile. The overgrowth in Sotos syndrome is of prenatal onset, with length being the most significantly affected parameter. After birth, acceleration of all growth parameters ensues, with occipital frontal head circumference measuring above the 97th percentile in nearly all affected infants by 12 months of age 87. Although the growth velocity slows by age 3 or 4, height invariably remains above the normal range throughout childhood, typically in association with somewhat lower weight percentiles. Children with Sotos syndrome are often described as being clumsy, with a tendency toward aggressive behavior 88. A minority have seizures, as well as structural abnormalities of the brain such as enlarged ventricles and absence of the corpus callosum.
In contrast, adult stature in Sotos syndrome is usually within the normal range for the general population 89, which has been attributed to the combination of an advanced bone age and a relatively early onset of puberty. Classic facial features include macrocephaly with dolichocephaly, hypertelorism, high-arched palate, prominent forehead and a pointed chin 90. Additional oral findings may include premature tooth eruption and supernumerary teeth 88. Developmental delay is ubiquitous, particularly in the area of speech and language acquisition 91.
Sotos syndrome is typically sporadic, although autosomal dominant transmission has been reported 88. Isolated cases of identical twin pairs who are concordant as well as discordant for the condition have also been described 88. Historically, the diagnosis was based entirely on clinical criteria. However, it is now known that Sotos syndrome is caused by a variety of molecular genetic alterations resulting in haploinsufficiency of the nuclear receptor-binding SET domain-containing protein 1 (NSD1) gene at 5q35 in ~90% of cases 92. The NSD1 gene encodes for a nuclear protein believed to function as a basic transcription factor and transcriptional regulator. Heterozygous mutations in the NFIX gene (Nuclear Factor I, X) have also been identified in some children with Sotos syndrome 93. While genotype-phenotype correlations have been suggested 94, this needs to be confirmed by additional studies of affected patients.
Beckwith-Wiedemann syndrome
Two physicians independently reported the first recognized cases of Beckwith-Wiedemann syndrome in the 1960’s 95, 96. Since that time, tremendous progress has been made in unraveling several aspects of this complex disorder. Beckwith-Wiedemann syndrome is typified by the combination of prenatal and postnatal overgrowth, congenital malformations and a predisposition to embryonal tumors. Characteristic features noted in the neonatal period include macroglossia, abdominal wall defects such as umbilical hernia, ear creases, visceromegaly and hyperinsulinemic hypoglycemia 97. A variety of additional abnormalities are found in a subset of patients, including hemihypertrophy or isolated facial asymmetry 98. While intelligence may be normal, mild to moderate developmental delay may also be present.
Although usually sporadic, several families manifesting heterogeneous inheritance patterns have been reported in whom there are several generations of affected individuals 99. The reported incidence of malignancy in children with Beckwith-Wiedemann syndrome varies between 4-21%, with the majority consisting of Wilms tumor 100. Therefore, frequent screening via abdominal ultrasonography during infancy and early childhood is essential 101, especially in patients with hemihypertrophy, which is known to be associated with an increased risk of cancer 102. Insights into the pathophysiology of the abnormal growth in this condition emerged with the discovery of abnormalities in imprinting of a number of growth regulatory genes within three regions of chromosome 11p15, including IGF-2, H19 and CDKN1C 103. The molecular genetic defects resulting in Beckwith-Wiedemann syndrome are extremely heterogeneous, and include maternal hypomethylation of 11p15, paternal uniparental disomy of this region, and unbalanced translocations leading to trisomy of the 11p15 locus 104. Enhanced understanding of the relationship between tumor risk and the molecular subtype in Beckwith-Wiedemann syndrome will result in improvements in targeted screening 105. Interestingly, an association has been noted between assisted reproduction and risk of imprinting disorders such as Beckwith-Wiedemann syndrome 106, although the risk appears to be small 107.
Simpson-Golabi-Behmel syndrome
Simpson-Golabi-Behmel syndrome is a complex X-linked overgrowth disorder sharing many features with Beckwith-Wiedemann syndrome. It is characterized by prenatal and postnatal overgrowth, coarse facial features and congenital anomalies. Some of the most commonly reported abnormalities include skeletal/hand defects, supernumerary nipples, macroglossia and visceromegaly. However, a wide spectrum in severity has been noted, ranging from mild features in carrier females to a lethal form of the disorder in affected males 108. Similarly, cognitive abilities vary from within the normal range to severe developmental delays. Approximately 36% of patients have a cardiac abnormality, the most common of which is a cardiovascular malformation 109. As is the case in Beckwith-Wiedemann syndrome, an increased incidence of embryonal tumors during early life is present. Delineation of the molecular genetic cause of Simpson-Golabi-Behmel syndrome has provided significant insight as to the reason for the striking similarities between this disorder and Beckwith-Wiedemann syndrome. Inactivating mutations of the glypican-3 (GPC3) gene at Xq26 have been demonstrated in 28-70% of individuals with Simpson-Golabi-Behmel syndrome 110. GPC3 is a member of a multigene family known to have critical roles in growth and development through the modulation of cellular responses to growth factors, including IGF-2 111. Exactly how abnormal levels of GPC3 promote tumorigenesis is poorly understood, but it may be through a disruption of the normal GPC3/IGF-2 complex, which is believed to be involved in IGF-2 modulation 112. An alternative proposal is that the physical manifestations of Simpson-Golabi-Behmel syndrome are due to abnormal interaction between GPC3 and CD26, a protein with important roles in the regulation of cell growth and immunologic response 113. Application of GPC3 mutational analysis in patients with unspecified overgrowth conditions has resulted in an extension of the Simpson-Golabi-Behmel syndrome phenotype 114 and the establishment of an international registry will be invaluable in providing information regarding the natural history and pathophysiology of this interesting condition. The oldest case of Simpson-Golabi-Behmel syndrome on record was discovered in an anatomical museum in the form of a macrosomic newborn who had died neonatally from unknown causes and was traced through following the family tree of a newly identified GPC3 mutation in an affected patient 115.
Weaver syndrome
Weaver syndrome is a rare condition that was first reported in 1974 116. Major features include prenatal or postnatal overgrowth, characteristic facies and advanced skeletal maturation. The typical appearance includes tall stature, macrocephaly, hypertelorism, large ears and micrognathia. A subset of patients have been reported to have cervical spine abnormalities 117 and the occasional development of neoplasia has also been noted in this population. The majority of individuals with Weaver syndrome have developmental delay, which is typically mild. Initially believed to be sporadic, multiple instances of familial occurrence have pointed strongly toward an autosomal dominant form of transmission 118. In 2011, mutations in the histone methytransferase, EZH2, were shown to cause Weaver syndrome 119. Heterozygous mutations in embryonic ectoderm development (EED) have also been identified in patients with Weaver syndrome 120. Significant phenotypic overlap between Weaver syndrome and Sotos syndrome often makes it difficult to differentiate between these overgrowth conditions 119. However, the availability of molecular genetic testing will aid in the diagnostic process.
Gigantism pathophysiology
Growth hormone is a 191 amino acid long protein with two disulfide bonds. It is secreted by the somatotroph cells in the anterior pituitary. It circulates for the most part as a 22kD protein and the remaining as a 20kD protein. It is secreted in a pulsatile manner, 4 to 11 pulses in a day. Due to this pulsatile nature of secretion measurement of random growth hormone levels are not useful. Growth hormone-releasing hormone (GHRH) stimulates the release of growth hormone from the pituitary. GHRH containing neurons are mainly seen in the arcuate nucleus and ventromedial nucleus. Somatostatin which is also secreted from the hypothalamus exerts an inhibitory action on the secretion of growth hormone. GHRH and somatostatin also regulate each other’s secretion in a paracrine manner. growth hormone stimulates the synthesis of IGF-1 from the liver. IGF-1 is a 70 amino acid protein which is similar to insulin. Besides, post-receptor signaling mechanisms involving tyrosine kinase and insulin receptor substrate-1 (IRS-1) are also similar for IGF-1 and insulin. IGF-1 circulates bound to IGF-1 binding proteins. IGF-1 exerts a negative feedback mechanism through GHRH and somatostatin.
Several other hormones can also modulate the secretion of growth hormone. Thyroid hormone facilitates the expression of the growth hormone gene. Hypothyroidism has been known to be associated with low growth hormone and IGF-1 which are reversible with thyroid hormone replacement therapy. Gonadal hormones can also upregulate the secretion of growth hormone as evident during the onset of puberty. Hypoglycemia decreases the secretion of somatostatin from the hypothalamus and increases the release of growth hormone. Mutations in the somatotrophs is a prerequisite for the abnormal response to GHRH. Point mutations in Arg 201 and Gly227 have been reported which are the sites for adenosine diphosphate (ADP) ribosylation and the binding domain of guanine triphosphate (GTP) respectively result in adenyl cyclase activation similar to GHRH. The tumorigenesis which leads to a monoclonal cell expansion is multifactorial, and the presence of a mutation alone is not enough 121.
Gigantism signs and symptoms
Gigantism can appear in a child of any age, from baby to teenager. Gigantism is very rare and should be suspected when the child’s height is 3 standard deviations above normal mean height or 2 standard deviations above the adjusted mean parental height.
The main symptom associated with gigantism is large body stature with accelerated growth or increased height compared to peers, which means the child will be unusually tall for their age. Muscles and organs may be enlarged as well.
Other gigantism symptoms include:
- large head
- prominent forehead
- protruding jaw
- coarse-looking facial features, such as a broad nose
- very large hands and feet, with thick fingers and toes
- excessive sweating
- a very large appetite
- general weakness
Some children also get headaches, nausea, problems with vision and delayed puberty.
Since gigantism is associated with various syndromes like neurofibromatosis, Carney complex and McCune Albright syndrome evaluation for neurofibromas, cafe au lait spots, optic gliomas, and skin lentigines should be done 122.
Serious conditions related to long-standing, untreated gigantism/acromegaly include:
- High blood pressure. Hypertension is seen in about 40% of patients with acromegaly and is usually mild. Anti-hypertensive treatment is similar to non-acromegalic patients. Good control of blood pressure important irrespective of the modality used for acromegalic treatment.
- Diabetes mellitus (adult-onset or Type 2).
- Heart disease, including heart failure due to heart enlargement. Cardiomyopathy is seen in most patients with acromegaly. Echocardiogram and electrocardiogram (ECG) should be done at baseline and repeated every year. Treatment of acromegaly improves the cardiomyopathy; however, this depends on the age, duration of disease and hypertension. In addition to echocardiogram and ECG, patients with gigantism will need Doppler of the peripheral arteries and veins.
- Obstructive Sleep Apnea (OSA). The prevalence of sleep apnea is 70% in patients with acromegaly. Prognathism, enlarged tongue and soft tissue accumulation in the upper airways all predispose to having OSA. Clinical assessment (Epworth score) and if needed polysomnography should be done and baseline and repeated every year. Surgical correction of prognathism may help, and referral to the maxillofacial surgeon should be considered.
- Arthropathy. Around 75% of patients with acromegaly are affected with arthropathy. Both small and large joints are affected. Bony expansion and soft tissue swelling can lead to nerve entrapment. Early diagnosis and aggressive treatment of acromegaly are essential to prevent arthropathy as these changes are irreversible.
- Colon polyps. Colon length is increased in acromegaly, and so are the mucosal folds. There is an increased prevalence of colonic polyps with acromegaly; however, the risk of colon cancer may or may not be increased. Patients should get a colonoscopy at baseline and every 5 years.
Giants are at increased risk of developing other tumors or lesions of the body:
- Thyroid cancer occurs at a higher frequency; talk to your doctor about screening
- You should also undergo regular colonoscopies to screen for colon polyps
Gigantism diagnosis
A doctor who sees a child who is growing unusually fast will need to ask some questions and do a physical examination, which might include checking height, weight, body proportions, senses, and stage of puberty.
Tests to diagnose gigantism include:
- Blood tests – to measure the level of hormones, and sometimes other substances
- Measurement of IGF-1 level is the initial test for the diagnosis of acromegaly as it is a stable molecule with a half-life of 15 hours. It should be measured in cases where there is clinical suspicion of acromegaly and pituitary masses – normal IGF-1 rules out acromegaly. False positive IGF-1 levels can be seen in pregnancy and adolescence, and false negative levels may be seen with estrogen therapy. Furthermore, hepatic failure, renal failure, hypothyroidism, malnutrition, sepsis, and poorly controlled diabetes mellitus can also influence IGF-1 levels. All cases with elevated IGF-1 levels need to have an oral glucose tolerance test (OGTT) with GH measurement to confirm the diagnosis of acromegaly or gigantism. A GH level of 1 mcg/lt or less 2 hours after a 75 gms of oral glucose tolerance test rules out acromegaly. Plasma glucose needs to be measured before and after the administration of glucose to make sure hyperglycemia has been achieved.
- Prolactin levels need to be assessed especially in the presence of galactorrhea or symptoms of hypogonadism. Anterior pituitary hormonal assessment needs to be done based on the clinical picture. Rarely, in the presence of a normal pituitary and biochemically confirmed acromegaly GHRH levels and imaging of the chest and abdomen need to be done to evaluate ectopic growth hormone or GHRH secretion 5.
- Oral glucose tolerance test (OGTT) – to see how growth hormone levels change when blood sugar level is increased
- An MRI or CT scan – to look at the pituitary gland. Pituitary Magnetic resonance imaging (MRI) is the preferred imaging modality for the diagnosis of acromegaly. The size, extent of the tumor, optic chiasmal compression, and cavernous sinus invasion can all be assessed on the MRI scan. Visual field testing is to be done in all cases where the tumor is in contact with the optic chiasma on the MRI scan.
- X-rays of the skull and jaw – to check bone thickness
It’s important to diagnose and treat gigantism as early as possible. If untreated, it can lead to problems such as diabetes, high blood pressure and arthritis.
Gigantism treatment
The aims of treatment in gigantism are:
- Removal of the pituitary tumor to relive mass effects
- Normalization of IGF-1 levels
- Monitoring of gigantism and associated comorbidities 121
There are three main modalities available for the treatment of gigantism each with its advantages and disadvantages, surgery, medical/drug therapy, and radiation therapy. The decision to use these modalities is made on a case by case basis.
Treatments for gigantism include:
- surgery – to remove or reduce the pituitary tumor
- radiotherapy – to reduce tumor growth and growth hormone levels
- drug therapy – to control growth hormones levels and symptoms, and shrink the tumor.
Hypopituitarism can occur as a result of surgery of radiation. Annual assessment of pituitary hormones and replace hormones as needed 123.
Surgery
Surgical excision of the tumor is the preferred initial treatment unless the patient is deemed unfit for surgery. Surgery is also the preferred modality in recurrence as long as the tumor remains accessible. The transsphenoidal approach involves accessing the tumor by getting to the sphenoid sinus either through a nasal or sublabial approach and removing the sellar floor. Even tumors with suprasellar extension can be removed with this approach. Endoscopic transsphenoidal resection has better tumor clearance, decreased morbidity, and complications as compared to microscopic approach. Post-surgical complications include diabetes insipidus and anterior pituitary deficiencies 124.
IGF-1 and growth hormone levels need to be measured 12 weeks after surgery. The goal is normalization of IGF-1 and an undetectable growth hormone level.
Surgical techniques
- Endoscopic Transsphenoidal Surgery: A minimally invasive surgery using a long, thin, flexible tube with a light and video camera at one end, called an endoscope, through a small cut inside your nose or behind your upper lip to remove the pituitary adenoma. Removing the tumor should instantly lower your levels of growth hormone and relieve pressure on the surrounding tissue. Often, facial features start to return to normal and swelling improves within a few days. The proposed benefit of using an endoscope is the enhanced ability to visualize the tumor in a panoramic fashion and not miss any residual tumor when resecting 125.
- Transsphenoidal Microscopic Surgery: The tumor is directly visualized using a microscope, and this is the traditional method of transsphenoidal surgery. A meta-analysis compared endoscopic and traditional microscopic transsphenoidal surgeries. It found they have comparable endocrine remission and complication rates. As expected, microadenomas had higher remission rates regardless of the surgical technique used, and the endoscopic technique confers the theoretical benefit of better visualization and, thus, better outcomes for macroadenomas. But, further studies are required to directly compare these surgical modalities 126.
- Craniotomy: This approach is reserved for those adenomas with suprasellar extension. Other reasons for choosing this approach include unfavorable para-sellar sinuses and internal or external carotid artery aneurysms 125.
When the tumor that is creating too much growth hormone is not located in the pituitary gland, other types of surgery are used to remove the tumor. Removing these nonpituitary tumors also lowers GH levels and improves acromegaly symptoms.
Figure 6. Endoscopic transnasal transsphenoidal surgery
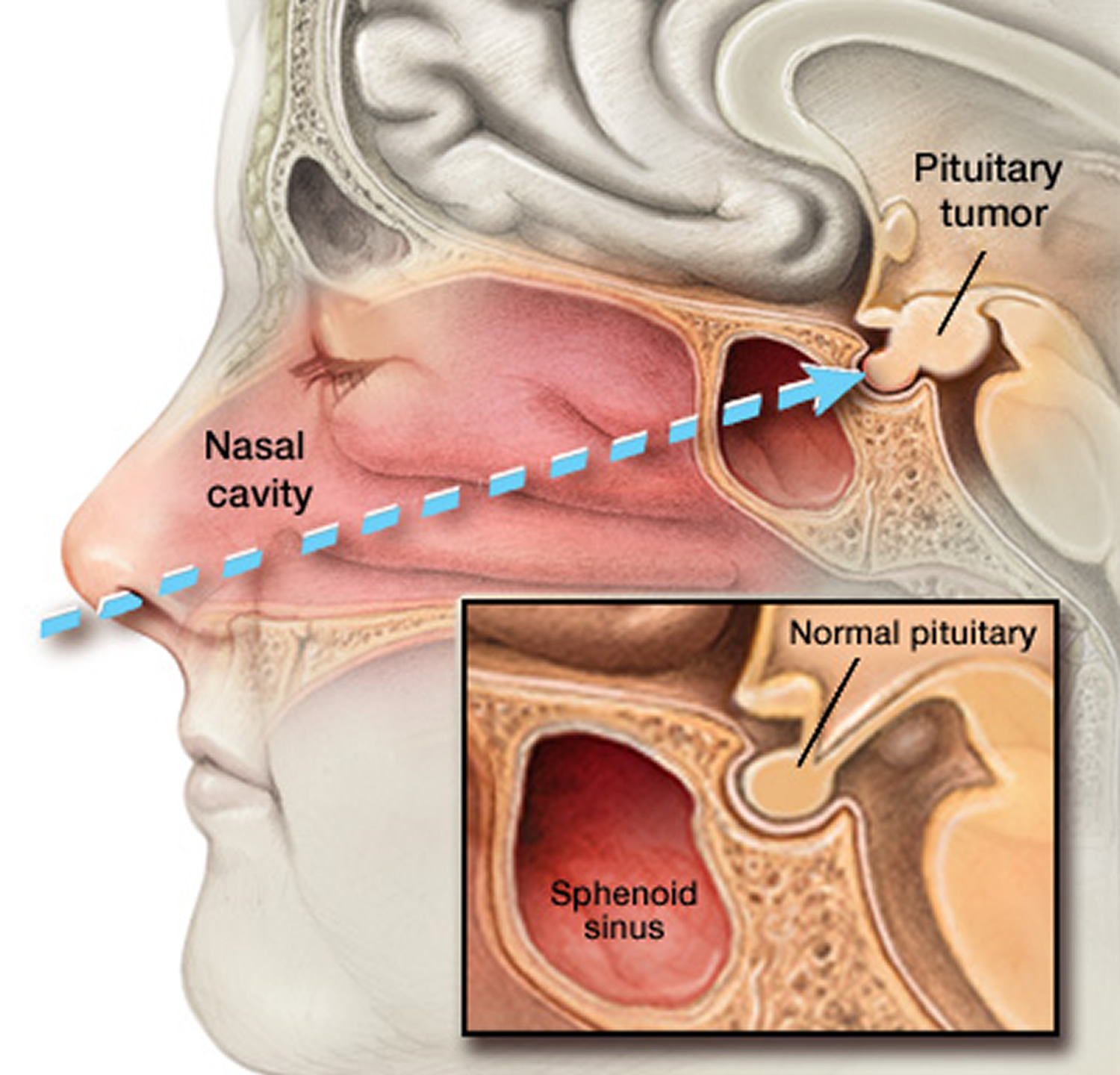
Note: In transnasal transsphenoidal endoscopic surgery, a surgical instrument is placed through the nostril and alongside the nasal septum to access a pituitary tumor.
With surgery, there is risk of:
- causing damage to healthy parts of your pituitary gland
- leakage of the fluid (cerebrospinal fluid) that surrounds and protects your brain
- meningitis – although this is rare
Your surgeon will discuss these risks with you and answer any questions you have.
Surgery outcomes
The surgery is considered a success if blood levels of GH and IGF-1 return to normal after 12 weeks. The cure rate right after surgery is about 85 percent for small tumors and 40 to 50 percent for large tumors 127.
When successful, the surgery relieves pressure on nearby areas of the brain and causes GH levels to drop right away. Soft tissue swelling may get better within a few days but facial changes may take longer to improve.
Surgery is most successful in people with smaller pituitary tumors. Success largely depends on the skill and experience of the surgeon, as well as the location of the tumor. Even experienced surgeons may not be able to remove the tumor if it’s too close to parts of the brain where surgery would be risky. However, surgeons may be able to remove part of the tumor.
Quality of life after treatment
Numerous studies have looked at the impact of different treatment modalities in first- or second-line therapies. Pituitary surgery compared with medical treatment seems superior in improving quality of life in patients with acromegaly 128 as in other types of pituitary tumor 129. A French study 130 found that patients who underwent surgery had a better quality of life than if they had received medical treatment only (65 ± 18% versus 54 ± 14%) and investigators concluded that neurosurgery was associated with greater improvement in quality of life when compared with medical therapy alone. Overall, greater GH suppression could be attained with surgery than with somatostatin receptor ligand injections 131. Patients with remission after surgery could avoid medication side effects and consequences of suboptimal biochemical control.
Postsurgery treatments
In most cases, levels of GH and IGF-1 improve but don’t go back to normal. If levels of these hormones are still too high or begin to rise again, you may need further treatment. Most often, this will involve taking medicines. In some cases, your doctor may recommend a second surgery.
An oral glucose tolerance test (OGTT) can be done 1 week post-operatively. A GH value of <0.4ng/mL is used as defining disease control 132. Serology of GH and IGF-1 can be measured by 3 to 6 months post-operatively as it can take this long for IGF-1 levels to normalize. Remission is then defined when normal IGF-1 levels are seen, and GH after OGTT is measured at <1ng/mL (although some recommend the use of <0.4ng/mL). If remission is confirmed, serology should be repeated at least annually, as relapse has been known to occur in some patients even as long as 10 or more years later. Post-operative imaging should be done a minimum of 3 months after surgery as the fat and gel foam packing can take that long to be resorbed 125. If residual disease is noted, the patient may need further treatment with repeat surgery if possible and appropriate, medical management, or radiotherapy. Pathology specimens are helpful for further management and prognostication as, for example, when the tumor is densely granulated, this may predict response to octreotide 133. Also, staining should be done for Ki67 and p21 as these levels have also shown to have prognostic value 134. Also, if the adenoma stains positive for prolactin, this could predict response to dopamine agonists.
Medical treatment
Various medical therapies are available for the management of gigantism. They are used in the treatment of persistent disease after surgery. The medicines may be used alone or in combination with each other.
- Somatostatin analogues: The drugs octreotide (Sandostatin) and lanreotide (Somatuline Depot) are synthetic versions of the brain hormone somatostatin. They can interfere with the excessive secretion of GH by the pituitary gland and the tumor may decrease in size in some people, thus can produce rapid declines in GH levels. When starting octreotide treatment, you initially inject yourself with a short-acting preparation under your skin (subcutaneously) three times a day to determine if you have any side effects from the medication and if it’s effective. Then, if it’s tolerated and effective, you can take a long-acting form that requires an injection into the muscles of your buttocks (gluteal muscles) by a health care professional, administered once a month. Octreotide was the first somatostatin receptor ligand to be introduced, and it preferentially bound to human somatostatin receptor type 2. The dose was 100-200 mg by subcutaneous injection three times a day. Long-acting somatostatin receptor ligand has become available in the form of octreotide LAR and lanreotide. Both have similar efficacy in suppressing growth hormone levels and normalizing IGF-1 levels. Octreotide LAR is given at a dose of 10 to 30 mg subcutaneously every four weeks while lanreotide is given at a dose of 120 mg subcutaneously every four weeks. Lanreotide is administered as a subcutaneous injection once a month. Side effects of somatostatin receptor ligand include abdominal pain, nausea, flatulence, diarrhea, and hyperglycemia. The primary role of somatostatin receptor ligand is as an adjunct to radiation after surgery however in certain instances it may be used to decrease the size of the tumor or improve cardiovascular function preoperatively 135. Several studies have shown that these drugs are safe and effective for long-term treatment, but scientists are currently studying other options, such as pills 136.
- Dopamine agonists. The medications cabergoline and bromocriptine (Parlodel) are taken as pills. However, their efficacy is limited, with normalization of IGF-1 seen only in 34% of patients. In some people, these drugs can lower levels of GH and IGF-1. The tumor may decrease in size in some people taking a dopamine agonist or somatostatin analogues. Dopamine agonists are most likely to work in people who have mild GH excess and those who have both acromegaly and hyperprolactinemia (too much of the hormone prolactin). Some people may develop compulsive behaviors, such as gambling, while taking these medications. Side effects can include nausea, vomiting, stuffed nose, tiredness, headache, dizziness when standing (postural hypotension), nightmares, and mood changes 137.
- Growth hormone-receptor antagonist. The medication pegvisomant (Somavert), a growth hormone antagonist, acts to block the effect of GH on body tissues. Pegvisomant may be particularly helpful for people who haven’t had good success with other forms of treatment. You administer this medication yourself daily by subcutaneous injection. This medication can normalize IGF-1 levels and relieve symptoms in most people with acromegaly, but doesn’t lower GH levels or reduce the tumor size. Pegvisomant can have a beneficial effect on glucose homeostasis and can improve insulin sensitivity 138. Van der Lely et al. 139 followed 152 patients treated with pegvisomant for 18 months and found normalization of IGF-1 in 90% of patients. Pegvisomant is administered at a dose of 10 to 30 mg subcutaneously every day. Due to the high expense, its role is limited to patients who do not respond to somatostatin analogues or have diabetes and worsening hyperglycemia with somatostatin analogues 140. Pegvisomant side effects can include liver problems, diarrhea and nausea.
Three different types of medicine are used:
- A monthly injection of either octreotide, lanreotide or pasireotide: this slows down the release of growth hormone and can sometimes also shrink tumors.
- A daily pegvisomant injection: this blocks the effects of growth hormone and can significantly improve symptoms.
- Bromocriptine or cabergoline tablets: these can stop growth hormone being produced, but they only work in a small proportion of people.
Each of these medications has different advantages and disadvantages. Speak to your doctor about the options available to you, and the benefits and risks of each.
Combination medical therapy has also been described. Combining cabergoline with a somatostatin analog has been seen to be effective in a subset of patients, even in the absence of hyperprolactinemia 132. Another common combination is that of a somatostatin analog with Pegvisomant. A study demonstrated that by adding Pegvisomant to a somatostatin analog in patients seemingly resistant to the somatostatin analog, IGF-1 levels normalized in 95% of patients 141. Importantly, if this combination is used, one needs to monitor hepatic function as the liver derangement is more common with the combination of drugs than with Pegvisomant alone 141.
A combination of a low-dose somatostatin analog with weekly pegvisomant is not an on-label or approved regimen for acromegaly; however, a study has shown promise for this novel dosing regimen to be effective in biochemical control in addition to being cost-effective in those requiring combination therapy for control of their acromegaly 142.
Radiation therapy
Radiation is often used as an adjunct for the treatment of persistent disease after surgery; rarely it may be used as a first-line treatment in patients unfit for surgery. Two commonly used radiation therapy modalities are external irradiation and stereotactic single high dose irradiation.
- External irradiation: Using linear accelerators radiation is focussed on the pituitary fossa. CT or MRI are used for dosimetry to minimize radiation exposure to the surrounding tissues. The total radiation dose (around 4500 cGy) is divided into fractions (generally 25 fractions of 180 cGy) and given over 6 weeks, this fractionation helps minimize injury to surrounding tissues. About 60% of patients achieve normalization of IGF-1 with radiation in 10 years. Hypopituitarism is a known side effect of radiation and can occur years after cessation of radiation, therefore annual clinical and laboratory assessment for hypopituitarism should be done. With external irradiation, there is a resolution of even radiologically invisible disease.
- Stereotactic single high dose irradiation: Using a gamma knife or stereotactic multiple arc radiotherapy a high dose of radiation is delivered to a previously mapped area. Care should be taken to limit the exposure of radiation (< 8 cGy) to the optic chiasma. Since the radiation is delivered to a mapped area radiologically invisible disease will not be resolved. The rate of development of hypopituitarism is the same as that of external irradiation 143.
Treatment of tall stature
Depending on the absolute height and the degree of growth potential remaining, one of the goals in the treatment of gigantism may be prevention of further linear growth. When this is the case, acceleration of epiphyseal fusion can be achieved with exogenous sex steroids 144. Short-term administration of both high dose testosterone and estrogen have been utilized for this purpose in children with gigantism, resulting in significant improvements in terms of adult height 145.
However, such an approach would require great caution given reports of subfertility in women who were treated with high dose estrogen during adolescence with the goal of attenuating growth in the setting of constitutional tall stature 146.
Living with gigantism
When the condition is successfully treated, children with gigantism can have a normal life expectancy and avoid most of the complications caused by it. However, they may still have symptoms such as muscle weakness and restricted movement, and some may also have psychological problems. Because of their size, it may also be hard to buy them items such as clothes and shoes. Regular medical follow-up is needed to monitor the condition over time.
- Bello MO, Garla VV. Gigantism And Acromegaly. [Updated 2019 Dec 20]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538261[↩][↩]
- Bello MO, Garla VV. Gigantism And Acromegaly. [Updated 2021 Dec 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK538261[↩]
- Sotos JF. Overgrowth. Hormonal Causes. Clin Pediatr (Phila) 1996; 35(11):579-590.[↩]
- Eugster E. Gigantism. [Updated 2018 Apr 17]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279155[↩][↩][↩][↩][↩][↩][↩][↩]
- Katznelson L, Laws ER, Melmed S, Molitch ME, Murad MH, Utz A, Wass JA., Endocrine Society. Acromegaly: an endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2014 Nov;99(11):3933-51.[↩][↩]
- Pituitary Gigantism. N Engl J Med 1999; 340:524 DOI: 10.1056/NEJM199902183400705[↩]
- Adelman DT, Liebert KJ, Nachtigall LB, Lamerson M, Bakker B. Acromegaly: the disease, its impact on patients, and managing the burden of long-term treatment. Int J Gen Med. 2013;6:31-8.[↩]
- Melmed S. Acromegaly. N Engl J Med 1990; 322(14):966-977.[↩]
- Zimmerman D, Young WF, Jr., Ebersold MJ et al. Congenital gigantism due to growth hormone-releasing hormone excess and pituitary hyperplasia with adenomatous transformation. J Clin Endocrinol Metab 1993; 76(1):216-222.[↩]
- Dubuis JM, Deal CL, Drews RT et al. Mammosomatotroph adenoma causing gigantism in an 8-year old boy: a possible pathogenetic mechanism. Clin Endocrinol (Oxf) 1995; 42(5):539-549.[↩]
- Gelber SJ, Heffez DS, Donohoue PA. Pituitary gigantism caused by growth hormone excess from infancy. J Pediatr 1992; 120(6):931-934.[↩]
- Vidal S, Horvath E, Kovacs K, Lloyd RV, Smyth HS. Reversible transdifferentiation: interconversion of somatotrophs and lactotrophs in pituitary hyperplasia. Mod Pathol 2001; 14(1):20-28.[↩]
- Hannah-Shmouni F, Trivellin G, Stratakis CA. Genetics of gigantism and acromegaly. Growth Horm IGF Res 2016; 30-31:37-41.[↩]
- Isidro ML, Iglesias DP, Matias-Guiu X, Cordido F. Acromegaly due to a growth hormone-releasing hormone-secreting intracranial gangliocytoma. J Endocrinol Invest 2005; 28(2):162-165.[↩]
- Asada H, Otani M, Furuhata S, Inoue H, Toya S, Ogawa Y. Mixed pituitary adenoma and gangliocytoma associated with acromegaly–case report. Neurol Med Chir (Tokyo) 1990; 30(8):628-632.[↩]
- Asa SL, Scheithauer BW, Bilbao JM et al. A case for hypothalamic acromegaly: a clinicopathological study of six patients with hypothalamic gangliocytomas producing growth hormone-releasing factor. J Clin Endocrinol Metab 1984; 58(5):796-803.[↩]
- Ghazi AA, Amirbaigloo A, Dezfooli AA et al. Ectopic acromegaly due to growth hormone releasing hormone. Endocrine 2013; 43(2):293-302.[↩]
- Beuschlein F, Strasburger CJ, Siegerstetter V et al. Acromegaly caused by secretion of growth hormone by a non-Hodgkin’s lymphoma. N Engl J Med 2000; 342(25):1871-1876.[↩]
- Garby L, Caron P, Claustrat F et al. Clinical characteristics and outcome of acromegaly induced by ectopic secretion of growth hormone-releasing hormone (GHRH): a French nationwide series of 21 cases. J Clin Endocrinol Metab 2012; 97(6):2093-2104.[↩]
- Biswal S, Srinivasan B, Dutta P et al. Acromegaly caused by ectopic growth hormone: a rare manifestation of a bronchial carcinoid. Ann Thorac Surg 2008; 85(1):330-332.[↩]
- Appel JG, Bergsneider M, Vinters H, Salamon N, Wang MB, Heaney AP. Acromegaly due to an ectopic pituitary adenoma in the clivus: case report and review of literature. Pituitary 2012; 15 Suppl 1:S53-S56.[↩]
- Osella G, Orlandi F, Caraci P et al. Acromegaly due to ectopic secretion of GHRH by bronchial carcinoid in a patient with empty sella. J Endocrinol Invest 2003; 26(2):163-169.[↩]
- Glasker S, Vortmeyer AO, Lafferty AR et al. Hereditary pituitary hyperplasia with infantile gigantism. J Clin Endocrinol Metab 2011; 96(12):E2078-E2087.[↩]
- Costin G, Fefferman RA, Kogut MD. Hypothalamic gigantism. J Pediatr 1973; 83(3):419-425.[↩]
- Drimmie FM, MacLennan AC, Nicoll JA, Simpson E, McNeill E, Donaldson MD. Gigantism due to growth hormone excess in a boy with optic glioma. Clin Endocrinol (Oxf) 2000; 53(4):535-538.[↩]
- Duchowny MS, Katz R, Bejar RL. Hypothalamic mass and gigantism in neurofibromatosis: treatment with bromocriptine. Ann Neurol 1984; 15(3):302-304.[↩]
- Sani I, Albanese A. Endocrine Long-Term Follow-Up of Children with Neurofibromatosis Type 1 and Optic Pathway Glioma. Horm Res Paediatr 2017; 87(3):179-188.[↩]
- Fuqua JS, Berkovitz GD. Growth hormone excess in a child with neurofibromatosis type 1 and optic pathway tumor: a patient report. Clin Pediatr (Phila) 1998; 37(12):749-752.[↩]
- Waguespack SG, Eugster EA, Pescovitz OH. Growth hormone (GH) excess in a child with neurofibromatosis type 1 (NF1) an optic pathway glioma. Pediatric Research 49[6 Suppl 2 of 2], 82A. 2001.[↩]
- Cambiaso P, Galassi S, Palmiero M et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am J Med Genet A 2017; 173(9):2353-2358.[↩]
- Josefson JL, Listernick R, Charrow J, Habiby RL. Growth Hormone Excess in Children with Optic Pathway Tumors Is a Transient Phenomenon. Horm Res Paediatr 2016; 86(1):35-38.[↩]
- Salenave S, Boyce AM, Collins MT, Chanson P. Acromegaly and McCune-Albright syndrome. J Clin Endocrinol Metab 2014; 99(6):1955-1969.[↩]
- Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Engl J Med 1991; 325(24):1688-1695.[↩]
- Lumbroso S, Paris F, Sultan C. McCune-Albright syndrome: molecular genetics. J Pediatr Endocrinol Metab 2002; 15 Suppl 3:875-882.[↩]
- Yao Y, Liu Y, Wang L et al. Clinical characteristics and management of growth hormone excess in patients with McCune-Albright syndrome. Eur J Endocrinol 2017; 176(3):295-303.[↩][↩]
- Tinschert S, Gerl H, Gewies A, Jung HP, Nurnberg P. McCune-Albright syndrome: clinical and molecular evidence of mosaicism in an unusual giant patient. Am J Med Genet 1999; 83(2):100-108.[↩]
- Akintoye SO, Chebli C, Booher S et al. Characterization of gsp-mediated growth hormone excess in the context of McCune-Albright syndrome. J Clin Endocrinol Metab 2002; 87(11):5104-5112.[↩]
- Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis 2012; 7 Suppl 1:S4.[↩]
- Boyce AM, Glover M, Kelly MH et al. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab 2013; 98(1):E126-E134.[↩]
- Vortmeyer AO, Glasker S, Mehta GU et al. Somatic GNAS mutation causes widespread and diffuse pituitary disease in acromegalic patients with McCune-Albright syndrome. J Clin Endocrinol Metab 2012; 97(7):2404-2413.[↩]
- Zumkeller W, Jassoy A, Lebek S, Nagel M. Clinical, endocrinological and radiography features in a child with McCune-Albright syndrome and pituitary adenoma. J Pediatr Endocrinol Metab 2001; 14(5):553-559.[↩]
- Moran A, Asa SL, Kovacs K et al. Gigantism due to pituitary mammosomatotroph hyperplasia. N Engl J Med 1990; 323(5):322-327.[↩]
- Cuttler L, Jackson JA, Saeed uz-Zafar M, Levitsky LL, Mellinger RC, Frohman LA. Hypersecretion of growth hormone and prolactin in McCune-Albright syndrome. J Clin Endocrinol Metab 1989; 68(6):1148-1154.[↩]
- Shimon I, Melmed S. Genetic basis of endocrine disease: pituitary tumor pathogenesis. J Clin Endocrinol Metab 1997; 82(6):1675-1681.[↩]
- Mantovani G, Bondioni S, Lania AG et al. Parental origin of Gsalpha mutations in the McCune-Albright syndrome and in isolated endocrine tumors. J Clin Endocrinol Metab 2004; 89(6):3007-3009.[↩]
- Wong SC, Zacharin M. Long-term health outcomes of adults with McCune-Albright syndrome. Clin Endocrinol (Oxf). 2017 Nov;87(5):627-634. doi: 10.1111/cen.13419[↩]
- Salenave, S., Boyce, A. M., Collins, M. T., & Chanson, P. (2014). Acromegaly and McCune-Albright syndrome. The Journal of clinical endocrinology and metabolism, 99(6), 1955–1969. https://doi.org/10.1210/jc.2013-3826[↩]
- Skogseid B, Rastad J, Oberg K. Multiple endocrine neoplasia type 1. Clinical features and screening. Endocrinol Metab Clin North Am 1994; 23(1):1-18.[↩]
- Guru SC, Goldsmith PK, Burns AL et al. Menin, the product of the MEN1 gene, is a nuclear protein. Proc Natl Acad Sci U S A 1998; 95(4):1630-1634.[↩]
- Bassett JH, Forbes SA, Pannett AA et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet 1998; 62(2):232-244.[↩]
- Mutch MG, Dilley WG, Sanjurjo F et al. Germline mutations in the multiple endocrine neoplasia type 1 gene: evidence for frequent splicing defects. Hum Mutat 1999; 13(3):175-185.[↩]
- Boggild MD, Jenkinson S, Pistorello M et al. Molecular genetic studies of sporadic pituitary tumors. J Clin Endocrinol Metab 1994; 78(2):387-392.[↩]
- Carty SE, Helm AK, Amico JA et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery 1998; 124(6):1106-1113.[↩]
- Stratakis CA, Schussheim DH, Freedman SM et al. Pituitary macroadenoma in a 5-year-old: an early expression of multiple endocrine neoplasia type 1. J Clin Endocrinol Metab 2000; 85(12):4776-4780.[↩]
- Brandi ML, Gagel RF, Angeli A et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86(12):5658-5671.[↩]
- Sala E, Ferrante E, Verrua E et al. Growth hormone-releasing hormone-producing pancreatic neuroendocrine tumor in a multiple endocrine neoplasia type 1 family with an uncommon phenotype. Eur J Gastroenterol Hepatol 2013; 25(7):858-862.[↩]
- Lee M, Pellegata NS. Multiple endocrine neoplasia type 4. Front Horm Res 2013; 41:63-78.[↩]
- Lodish MB, Trivellin G, Stratakis CA. Pituitary gigantism: update on molecular biology and management. Curr Opin Endocrinol Diabetes Obes 2016; 23(1):72-80.[↩]
- Carney JA, Gordon H, Carpenter PC, Shenoy BV, Go VL. The complex of myxomas, spotty pigmentation, and endocrine overactivity. Medicine (Baltimore) 1985; 64(4):270-283.[↩]
- Stratakis CA, Carney JA, Lin JP et al. Carney complex, a familial multiple neoplasia and lentiginosis syndrome. Analysis of 11 kindreds and linkage to the short arm of chromosome 2. J Clin Invest 1996; 97(3):699-705.[↩]
- Kirschner LS, Carney JA, Pack SD et al. Mutations of the gene encoding the protein kinase A type I-alpha regulatory subunit in patients with the Carney complex. Nat Genet 2000; 26(1):89-92.[↩]
- Sandrini F, Stratakis C. Clinical and molecular genetics of Carney complex. Mol Genet Metab 2003; 78(2):83-92.[↩]
- Stratakis CA, Kirschner LS, Carney JA. Clinical and molecular features of the Carney complex: diagnostic criteria and recommendations for patient evaluation. J Clin Endocrinol Metab 2001; 86(9):4041-4046.[↩]
- Pack SD, Kirschner LS, Pak E, Zhuang Z, Carney JA, Stratakis CA. Genetic and histologic studies of somatomammotropic pituitary tumors in patients with the “complex of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Clin Endocrinol Metab 2000; 85(10):3860-3865.[↩]
- Raff SB, Carney JA, Krugman D, Doppman JL, Stratakis CA. Prolactin secretion abnormalities in patients with the “syndrome of spotty skin pigmentation, myxomas, endocrine overactivity and schwannomas” (Carney complex). J Pediatr Endocrinol Metab 2000; 13(4):373-379.[↩]
- Stratakis C.A., Kirschner L.S., Carney J.A. Clinical and molecular features of the Carney complex: Diagnostic criteria and recommendations for patient evaluation. J. Clin. Endocrinol. Metab. 2001;86:4041–4046. doi: 10.1210/jcem.86.9.7903[↩]
- Cuny T., Mac T.T., Romanet P., Dufour H., Morange I., Albarel F., Lagarde A., Castinetti F., Graillon T., North M.O., et al. Acromegaly in Carney complex. Pituitary. 2019;22:456–466. doi: 10.1007/s11102-019-00974-8[↩]
- O’Toole SM, Denes J, Robledo M, Stratakis CA, Korbonits M. 15 YEARS OF PARAGANGLIOMA: The association of pituitary adenomas and phaeochromocytomas or paragangliomas. Endocr Relat Cancer 2015; 22(4):T105-T122.[↩]
- Denes J, Swords F, Rattenberry E et al. Heterogeneous genetic background of the association of pheochromocytoma/paraganglioma and pituitary adenoma: results from a large patient cohort. J Clin Endocrinol Metab 2015; 100(3):E531-E541.[↩]
- Soares BS, Frohman LA. Isolated familial somatotropinoma. Pituitary 2004; 7(2):95-101.[↩][↩]
- Matsuno A, Teramoto A, Yamada S et al. Gigantism in sibling unrelated to multiple endocrine neoplasia: case report. Neurosurgery 1994; 35(5):952-955.[↩]
- Nozieres C, Berlier P, Dupuis C et al. Sporadic and genetic forms of paediatric somatotropinoma: a retrospective analysis of seven cases and a review of the literature. Orphanet J Rare Dis 2011; 6:67.[↩]
- Jorge BH, Agarwal SK, Lando VS et al. Study of the multiple endocrine neoplasia type 1, growth hormone-releasing hormone receptor, Gs alpha, and Gi2 alpha genes in isolated familial acromegaly. J Clin Endocrinol Metab 2001; 86(2):542-544.[↩]
- Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (FIPA) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (AIP) gene. Endocr Rev 2013; 34(2):239-277.[↩]
- Chahal HS, Stals K, Unterlander M et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med 2011; 364(1):43-50.[↩]
- Gadelha MR, Une KN, Rohde K, Vaisman M, Kineman RD, Frohman LA. Isolated familial somatotropinomas: establishment of linkage to chromosome 11q13.1-11q13.3 and evidence for a potential second locus at chromosome 2p16-12. J Clin Endocrinol Metab 2000; 85(2):707-714.[↩]
- Cansu GB, Taskiran B, Trivellin G, Faucz FR, Stratakis CA. A novel truncating AIP mutation, p.W279*, in a familial isolated pituitary adenoma (FIPA) kindred. Hormones (Athens ) 2016; 15(3):441-444.[↩][↩]
- Iacovazzo D, Korbonits M. Gigantism: X-linked acrogigantism and GPR101 mutations. Growth Horm IGF Res 2016; 30-31:64-69.[↩]
- Trivellin, G., Daly, A. F., Faucz, F. R., Yuan, B., Rostomyan, L., Larco, D. O., Schernthaner-Reiter, M. H., Szarek, E., Leal, L. F., Caberg, J. H., Castermans, E., Villa, C., Dimopoulos, A., Chittiboina, P., Xekouki, P., Shah, N., Metzger, D., Lysy, P. A., Ferrante, E., Strebkova, N., … Stratakis, C. A. (2014). Gigantism and acromegaly due to Xq26 microduplications and GPR101 mutation. The New England journal of medicine, 371(25), 2363–2374. https://doi.org/10.1056/NEJMoa1408028[↩]
- Iacovazzo D, Caswell R, Bunce B et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: a clinico-pathological and genetic study. Acta Neuropathol Commun 2016; 4(1):56.[↩]
- Daly AF, Yuan B, Fina F et al. Somatic mosaicism underlies X-linked acrogigantism syndrome in sporadic male subjects. Endocr Relat Cancer 2016; 23(4):221-233.[↩]
- Daly AF, Lysy PA, Desfilles C et al. GHRH excess and blockade in X-LAG syndrome. Endocr Relat Cancer 2016; 23(3):161-170.[↩]
- Iacovazzo D., Caswell R., Bunce B., Jose S., Yuan B., Hernández-Ramírez L.C., Kapur S., Caimari F., Evanson J., Ferrau F., et al. Germline or somatic GPR101 duplication leads to X-linked acrogigantism: A clinico-pathological and genetic study. Acta Neuropathol Commun. 2016;4:56. doi: 10.1186/s40478-016-0328-1[↩]
- Moran A., Larson R., Kovacs K., Horvath E., Singer W., Sagman U., Reubi J.C., Wilson C.B., Pescovitz O.H. Gigantism due to pituitary mammosomatotroph hyperplasia. N. Engl. J. Med. 1990;323:322–327. doi: 10.1056/NEJM199008023230507[↩]
- Rodd C., Millette M., Iacovazzo D., Stiles C.E., Barry S., Evanson J., Albrecht S., Caswell R., Bunce B., Josen S., et al. Somatic GPR101 duplication causing X-linked acrogigantism (XLAG)-Diagnosis and management. J. Clin. Endocrinol. Metab. 2016;101:1927–1930. doi: 10.1210/jc.2015-4366[↩]
- Sotos JF, DODGE PR, MUIRHEAD D, CRAWFORD JD, TALBOT NB. CEREBRAL GIGANTISM IN CHILDHOOD. A SYNDROME OF EXCESSIVELY RAPID GROWTH AND ACROMEGALIC FEATURES AND A NONPROGRESSIVE NEUROLOGIC DISORDER. N Engl J Med 1964; 271:109-116.[↩]
- Cole TR, Hughes HE. Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31(1):20-32.[↩]
- Raitz R, Laragnoit A. Supernumerary teeth and dental management in Sotos syndrome. J Dent Child (Chic ) 2009; 76(3):246-250.[↩][↩][↩][↩]
- Agwu JC, Shaw NJ, Kirk J, Chapman S, Ravine D, Cole TR. Growth in Sotos syndrome. Arch Dis Child 1999; 80(4):339-342.[↩]
- Sotos JF. Overgrowth. Section VI. Genetic syndromes and other disorders associated with overgrowth. Clin Pediatr (Phila) 1997; 36(3):157-170.[↩]
- Lane C, Milne E, Freeth M. Cognition and Behaviour in Sotos Syndrome: A Systematic Review. PLoS One 2016; 11(2):e0149189.[↩]
- Saugier-Veber P, Bonnet C, Afenjar A et al. Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum Mutat 2007; 28(11):1098-1107.[↩]
- Sotos JF. Sotos syndrome 1 and 2. Pediatr Endocrinol Rev 2014; 12(1):2-16.[↩]
- Cecconi M, Forzano F, Milani D et al. Mutation analysis of the NSD1 gene in a group of 59 patients with congenital overgrowth. Am J Med Genet A 2005; 134(3):247-253.[↩]
- Beckwith J. Macroglossia, omphalocete, adrenal cytomegaly:fifantism and hyperplastic visceromegaly. Birth Defects 1969; 5:188-196.[↩]
- WIEDEMANN HR. [FAMILIAL MALFORMATION COMPLEX WITH UMBILICAL HERNIA AND MACROGLOSSIA–A “NEW SYNDROME”?]. J Genet Hum 1964; 13:223-232.[↩]
- Pettenati MJ, Haines JL, Higgins RR, Wappner RS, Palmer CG, Weaver DD. Wiedemann-Beckwith syndrome: presentation of clinical and cytogenetic data on 22 new cases and review of the literature. Hum Genet 1986; 74(2):143-154.[↩]
- Sathienkijkanchai A, Prucka SK, Grant JH, Robin NH. Isolated facial hemihyperplasia: manifestation of Beckwith-Wiedemann syndrome. J Craniofac Surg 2008; 19(1):279-283.[↩]
- Wangler MF, An P, Feinberg AP, Province M, Debaun MR. Inheritance pattern of Beckwith-Wiedemann syndrome is heterogeneous in 291 families with an affected proband. Am J Med Genet A 2005; 137(1):16-21.[↩]
- Rump P, Zeegers MP, van Essen AJ. Tumor risk in Beckwith-Wiedemann syndrome: A review and meta-analysis. Am J Med Genet A 2005; 136(1):95-104.[↩]
- Weksberg R, Shuman C, Beckwith JB. Beckwith-Wiedemann syndrome. Eur J Hum Genet 2010; 18(1):8-14.[↩]
- Debaun MR, Tucker MA. Risk of cancer during the first four years of life in children from The Beckwith-Wiedemann Syndrome Registry. J Pediatr 1998; 132(3 Pt 1):398-400.[↩]
- Cerrato F, Sparago A, Verde G et al. Different mechanisms cause imprinting defects at the IGF2/H19 locus in Beckwith-Wiedemann syndrome and Wilms’ tumour. Hum Mol Genet 2008; 17(10):1427-1435.[↩]
- Cohen MM, Jr. Beckwith-Wiedemann syndrome: historical, clinicopathological, and etiopathogenetic perspectives. Pediatr Dev Pathol 2005; 8(3):287-304.[↩]
- Brioude F, Lacoste A, Netchine I et al. Beckwith-Wiedemann syndrome: growth pattern and tumor risk according to molecular mechanism, and guidelines for tumor surveillance. Horm Res Paediatr 2013; 80(6):457-465.[↩]
- Uyar A, Seli E. The impact of assisted reproductive technologies on genomic imprinting and imprinting disorders. Curr Opin Obstet Gynecol 2014; 26(3):210-221.[↩]
- Bowdin S, Allen C, Kirby G et al. A survey of assisted reproductive technology births and imprinting disorders. Hum Reprod 2007; 22(12):3237-3240.[↩]
- Zimmermann N, Stanek J. Perinatal Case of Fatal Simpson-Golabi-Behmel Syndrome with Hyperplasia of Seminiferous Tubules. Am J Case Rep 2017; 18:649-655.[↩]
- Lin AE, Neri G, Hughes-Benzie R, Weksberg R. Cardiac anomalies in the Simpson-Golabi-Behmel syndrome. Am J Med Genet 1999; 83(5):378-381.[↩]
- Cottereau E, Mortemousque I, Moizard MP et al. Phenotypic spectrum of Simpson-Golabi-Behmel syndrome in a series of 42 cases with a mutation in GPC3 and review of the literature. Am J Med Genet C Semin Med Genet 2013; 163C(2):92-105.[↩]
- Debaun MR, Ess J, Saunders S. Simpson Golabi Behmel syndrome: progress toward understanding the molecular basis for overgrowth, malformation, and cancer predisposition. Mol Genet Metab 2001; 72(4):279-286.[↩]
- Mariani S, Iughetti L, Bertorelli R et al. Genotype/phenotype correlations of males affected by Simpson-Golabi-Behmel syndrome with GPC3 gene mutations: patient report and review of the literature. J Pediatr Endocrinol Metab 2003; 16(2):225-232.[↩]
- Davoodi J, Kelly J, Gendron NH, MacKenzie AE. The Simpson-Golabi-Behmel syndrome causative glypican-3, binds to and inhibits the dipeptidyl peptidase activity of CD26. Proteomics 2007; 7(13):2300-2310.[↩]
- Li M, Shuman C, Fei YL et al. GPC3 mutation analysis in a spectrum of patients with overgrowth expands the phenotype of Simpson-Golabi-Behmel syndrome. Am J Med Genet 2001; 102(2):161-168.[↩]
- Kehrer C, Hoischen A, Menkhaus R et al. Whole exome sequencing and array-based molecular karyotyping as aids to prenatal diagnosis in fetuses with suspected Simpson-Golabi-Behmel syndrome. Prenat Diagn 2016; 36(10):961-965.[↩]
- Weaver DD, Graham CB, Thomas IT, Smith DW. A new overgrowth syndrome with accelerated skeletal maturation, unusual facies, and camptodactyly. J Pediatr 1974; 84(4):547-552.[↩]
- Kelly TE, Alford BA, Abel M. Cervical spine anomalies and tumors in Weaver syndrome. Am J Med Genet 2000; 95(5):492-495.[↩]
- Proud VK, Braddock SR, Cook L, Weaver DD. Weaver syndrome: autosomal dominant inheritance of the disorder. Am J Med Genet 1998; 79(4):305-310.[↩]
- Cooney E, Bi W, Schlesinger AE, Vinson S, Potocki L. Novel EED mutation in patient with Weaver syndrome. Am J Med Genet A 2017; 173(2):541-545.[↩][↩]
- Imagawa E, Higashimoto K, Sakai Y et al. Mutations in genes encoding polycomb repressive complex 2 subunits cause Weaver syndrome. Hum Mutat 2017; 38(6):637-648.[↩]
- Carroll PV, Jenkins PJ. Acromegaly. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. MDText.com, Inc.; South Dartmouth (MA): Feb 1, 2016.[↩][↩]
- Eugster E. Gigantism. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. MDText.com, Inc.; South Dartmouth (MA): Apr 17, 2018.[↩]
- Melmed S, Casanueva FF, Klibanski A, Bronstein MD, Chanson P, Lamberts SW, Strasburger CJ, Wass JA, Giustina A. A consensus on the diagnosis and treatment of acromegaly complications. Pituitary. 2013 Sep;16(3):294-302.[↩]
- Melmed S, Casanueva F, Cavagnini F, Chanson P, Frohman LA, Gaillard R, Ghigo E, Ho K, Jaquet P, Kleinberg D, Lamberts S, Laws E, Lombardi G, Sheppard MC, Thorner M, Vance ML, Wass JA, Giustina A. Consensus statement: medical management of acromegaly. Eur. J. Endocrinol. 2005 Dec;153(6):737-40.[↩]
- Marquez, Y., Tuchman, A., & Zada, G. (2012). Surgery and radiosurgery for acromegaly: a review of indications, operative techniques, outcomes, and complications. International journal of endocrinology, 2012, 386401. https://doi.org/10.1155/2012/386401[↩][↩][↩]
- Chen, C. J., Ironside, N., Pomeraniec, I. J., Chivukula, S., Buell, T. J., Ding, D., Taylor, D. G., Dallapiazza, R. F., Lee, C. C., & Bergsneider, M. (2017). Microsurgical versus endoscopic transsphenoidal resection for acromegaly: a systematic review of outcomes and complications. Acta neurochirurgica, 159(11), 2193–2207. https://doi.org/10.1007/s00701-017-3318-6[↩]
- Katznelson L, Laws ER Jr, Melmed S, et al. Acromegaly: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism. 2014;99(11):3933–3951.[↩]
- Geraedts, V. J., Andela, C. D., Stalla, G. K., Pereira, A. M., van Furth, W. R., Sievers, C., & Biermasz, N. R. (2017). Predictors of Quality of Life in Acromegaly: No Consensus on Biochemical Parameters. Frontiers in endocrinology, 8, 40. https://doi.org/10.3389/fendo.2017.00040[↩]
- Waddle, M. R., Oudenhoven, M. D., Farin, C. V., Deal, A. M., Hoffman, R., Yang, H., Peterson, J., Armstrong, T. S., Ewend, M. G., & Wu, J. (2019). Impacts of Surgery on Symptom Burden and Quality of Life in Pituitary Tumor Patients in the Subacute Post-operative Period. Frontiers in oncology, 9, 299. https://doi.org/10.3389/fonc.2019.00299[↩]
- Matta MP, Couture E, Cazals L, Vezzosi D, Bennet A, Caron P. Impaired quality of life of patients with acromegaly: control of GH/IGF-I excess improves psychological subscale appearance. Eur J Endocrinol. 2008 Mar;158(3):305-10. doi: 10.1530/EJE-07-0697[↩]
- Rubeck KZ, Madsen M, Andreasen CM, Fisker S, Frystyk J, Jørgensen JO. Conventional and novel biomarkers of treatment outcome in patients with acromegaly: discordant results after somatostatin analog treatment compared with surgery. Eur J Endocrinol. 2010 Nov;163(5):717-26. doi: 10.1530/EJE-10-0640[↩]
- Adigun OO, Nguyen M, Fox TJ, et al. Acromegaly. [Updated 2021 Dec 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK431086[↩][↩]
- Bhayana S, Booth GL, Asa SL, Kovacs K, Ezzat S. The implication of somatotroph adenoma phenotype to somatostatin analog responsiveness in acromegaly. J Clin Endocrinol Metab. 2005 Nov;90(11):6290-5. doi: 10.1210/jc.2005-0998[↩]
- Alimohamadi, M., Ownagh, V., Mahouzi, L., Ostovar, A., Abbassioun, K., & Amirjmshidi, A. (2014). The impact of immunohistochemical markers of Ki-67 and p53 on the long-term outcome of growth hormone-secreting pituitary adenomas: A cohort study. Asian journal of neurosurgery, 9(3), 130–136. https://doi.org/10.4103/1793-5482.142732[↩]
- Caron P, Beckers A, Cullen DR, Goth MI, Gutt B, Laurberg P, Pico AM, Valimaki M, Zgliczynski W. Efficacy of the new long-acting formulation of lanreotide (lanreotide Autogel) in the management of acromegaly. J. Clin. Endocrinol. Metab. 2002 Jan;87(1):99-104.[↩]
- Paragliola RM, Salvatori R. Novel somatostatin receptor ligands therapies for acromegaly. Frontiers in Endocrinology. 2018;9:78.[↩]
- Abs R, Verhelst J, Maiter D, Van Acker K, Nobels F, Coolens JL, Mahler C, Beckers A. Cabergoline in the treatment of acromegaly: a study in 64 patients. J. Clin. Endocrinol. Metab. 1998 Feb;83(2):374-8.[↩]
- Giustina, A., Arnaldi, G., Bogazzi, F., Cannavò, S., Colao, A., De Marinis, L., De Menis, E., Degli Uberti, E., Giorgino, F., Grottoli, S., Lania, A. G., Maffei, P., Pivonello, R., & Ghigo, E. (2017). Pegvisomant in acromegaly: an update. Journal of endocrinological investigation, 40(6), 577–589. https://doi.org/10.1007/s40618-017-0614-1[↩]
- van der Lely AJ, Hutson RK, Trainer PJ et al. Long-term treatment of acromegaly with pegvisomant, a growth hormone receptor antagonist. Lancet 2001; 358(9295):1754-1759.[↩]
- Trainer PJ, Drake WM, Katznelson L, Freda PU, Herman-Bonert V, van der Lely AJ, Dimaraki EV, Stewart PM, Friend KE, Vance ML, Besser GM, Scarlett JA, Thorner MO, Parkinson C, Klibanski A, Powell JS, Barkan AL, Sheppard MC, Malsonado M, Rose DR, Clemmons DR, Johannsson G, Bengtsson BA, Stavrou S, Kleinberg DL, Cook DM, Phillips LS, Bidlingmaier M, Strasburger CJ, Hackett S, Zib K, Bennett WF, Davis RJ. Treatment of acromegaly with the growth hormone-receptor antagonist pegvisomant. N. Engl. J. Med. 2000 Apr 20;342(16):1171-7.[↩]
- Feenstra J, de Herder WW, ten Have SM, van den Beld AW, Feelders RA, Janssen JA, van der Lely AJ. Combined therapy with somatostatin analogues and weekly pegvisomant in active acromegaly. Lancet. 2005 May 7-13;365(9471):1644-6. doi: 10.1016/S0140-6736(05)63011-5. Erratum in: Lancet. 2005 May;365(9471):1620.[↩][↩]
- Bonert V, Mirocha J, Carmichael J, Yuen KCJ, Araki T, Melmed S. Cost-Effectiveness and Efficacy of a Novel Combination Regimen in Acromegaly: A Prospective, Randomized Trial. J Clin Endocrinol Metab. 2020 Sep 1;105(9):dgaa444. doi: 10.1210/clinem/dgaa444[↩]
- Jenkins PJ, Bates P, Carson MN, Stewart PM, Wass JA. Conventional pituitary irradiation is effective in lowering serum growth hormone and insulin-like growth factor-I in patients with acromegaly. J. Clin. Endocrinol. Metab. 2006 Apr;91(4):1239-45.[↩]
- Drop SL, De Waal WJ, De Muinck Keizer-Schrama SM. Sex steroid treatment of constitutionally tall stature. Endocr Rev 1998; 19(5):540-558.[↩]
- Minagawa M, Yasuda T, Someya T, Kohno Y, Saeki N, Hashimoto Y. Effects of octreotide infusion, surgery and estrogen on suppression of height increase and 20K growth hormone ratio in a girl with gigantism due to a growth hormone-secreting macroadenoma. Horm Res 2000; 53(3):157-160.[↩]
- Albuquerque EV, Scalco RC, Jorge AA. MANAGEMENT OF ENDOCRINE DISEASE: Diagnostic and therapeutic approach of tall stature. Eur J Endocrinol 2017; 176(6):R339-R353.[↩]
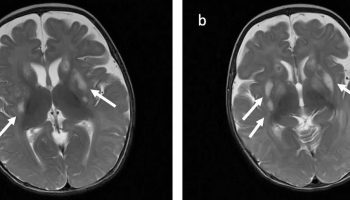
{kind=link}