Hepatoerythropoietic Porphyria
Hepatoerythropoietic porphyria (HEP) also called UROD-related hepatoerythropoietic porphyria is an extremely rare inherited disorder of the heme-biosynthetic pathway known as cutaneous porphyrias caused by mutations on both copies of a person’s UROD gene that encodes uroporphyrinogen decarboxylase (UROD) enzyme that is crucial in the fifth step of the heme biosynthesis pathway, which means that hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16. In hepatoerythropoietic porphyria (HEP), the uroporphyrinogen decarboxylase (UROD) enzyme activity is usually less than 10% its normal levels 17. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in the body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. When porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with hepatoerythropoietic porphyria (HEP). The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
Most affected individuals with hepatoerythropoietic porphyria (HEP) have a profound deficiency of uroporphyrinogen decarboxylase (UROD) enzyme and onset of hepatoerythropoietic porphyria (HEP) is usually during infancy or early childhood. However, some individuals may have a mild form that can go undiagnosed until adulthood. The childhood form of hepatoerythropoietic porphyria (HEP) is often associated skin photosensitivity with painful, blistering skin lesions that develop on sun-exposed skin (photosensitivity). Affected areas of skin can scar often with mutilation and loss of facial features and fingers and become discolored 15. Bacteria may infect the damaged skin and contribute to mutilation and scarring. The signs and symptoms of childhood form of hepatoerythropoietic porphyria (HEP) resemble congenital erythropoietic porphyria (CEP), with symptoms of skin blistering on sun-exposed skin that usually begin in infancy. Abnormal, excessive hair (hypertrichosis), red-brown discoloration of teeth (erythrodontia), and reddish-colored urine. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D deficiency. Red blood cells have a shortened life-span with mild or severe hemolytic anemia. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with abnormal enlargement of the liver and/or spleen (hepatosplenomegaly). Mild cases of hepatoerythropoietic porphyria (HEP) may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), the most common form of porphyria in humans where the porphyria cutanea tarda (PCT) that may be acquired in 75% to 80% of cases (also known as porphyria cutanea tarda type 1 or sporadic porphyria cutanea tarda) or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance or porphyria cutanea tarda type 2 or familial porphyria cutanea tarda). Hepatoerythropoietic Porphyria (HEP) is the autosomal recessive form of familial porphyria cutanea tarda (F-PCT). Skin photosensitivity is generally much more severe in hepatoerythropoietic porphyria (HEP) than in porphyria cutanea tarda (PCT).
Hepatoerythropoietic porphyria (HEP) is an extremely rare disorder that affects males and females in equal numbers. Approximately less than 100 cases have been reported in the medical literature 3, 18, 19, 20. The exact incidence or prevalence of hepatoerythropoietic porphyria (HEP) in the general population is unknown 20. The frequency of hepatoerythropoietic porphyria (HEP) can only be inferred based on that of familial porphyria cutanea tarda (F-PCT), which occurs in one in 20,000 individuals 1. Over a ten-year period from 2007 to 2017, a referral center or porphyria-specific diagnostic laboratory provided molecular diagnostic testing on 4 unrelated individuals with hepatoerythropoietic porphyria (HEP), identifying one novel variant 21. Two founder variants have been identified in Norway 22.
A diagnosis of hepatoerythropoietic porphyria (HEP) is based upon identification of characteristic symptoms, a detailed medical history, a thorough clinical evaluation and a variety of specialized tests. Hepatoerythropoietic porphyria (HEP) may be considered in infants and children with chronic, blistering photosensitivity.
Diagnosis of hepatoerythropoietic porphyria (HEP) can be made by demonstrating significant elevations of specific porphyrins in your urine and stool, as well as identification of a specific fluorescence emission peak in plasma. DNA genetic testing to identify the specific mutations in an individual’s UROD genes is the most specific and sensitive test to confirm the diagnosis of hepatoerythropoietic porphyria (HEP).
The treatment of hepatoerythropoietic porphyria (HEP) is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with hepatoerythropoietic porphyria (HEP). Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance and/or protection from sunlight will benefit individuals with hepatoerythropoietic porphyria (HEP) and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies (removal of blood) to lower the amount of porphyrins in the liver, which are used to treat individuals with porphyria cutanea tarda (PCT), are generally ineffective in individuals with hepatoerythropoietic porphyria (HEP) since elevated iron levels are not a feature of the disorder. Another treatment for porphyria cutanea tarda (PCT), the antimalarial drug hydroxychloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin (rEPO) a synthetic version of the naturally occurring hormone erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with hepatoerythropoietic porphyria (HEP) whose anemia was not associated with increased red cell destruction.
Figure 1. Hepatoerythropoietic porphyria (HEP)
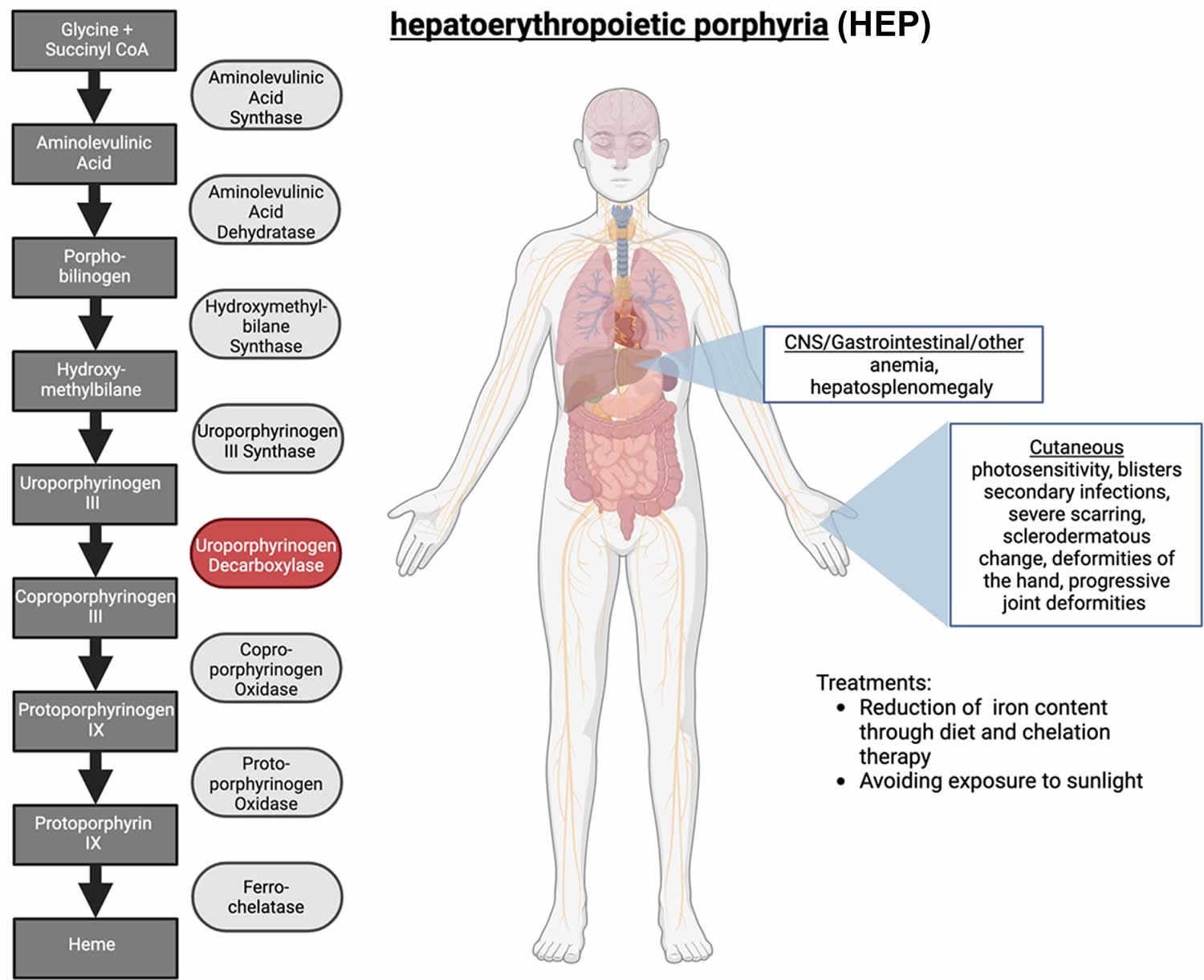
Figure 2. Hepatoerythropoietic porphyria (HEP)
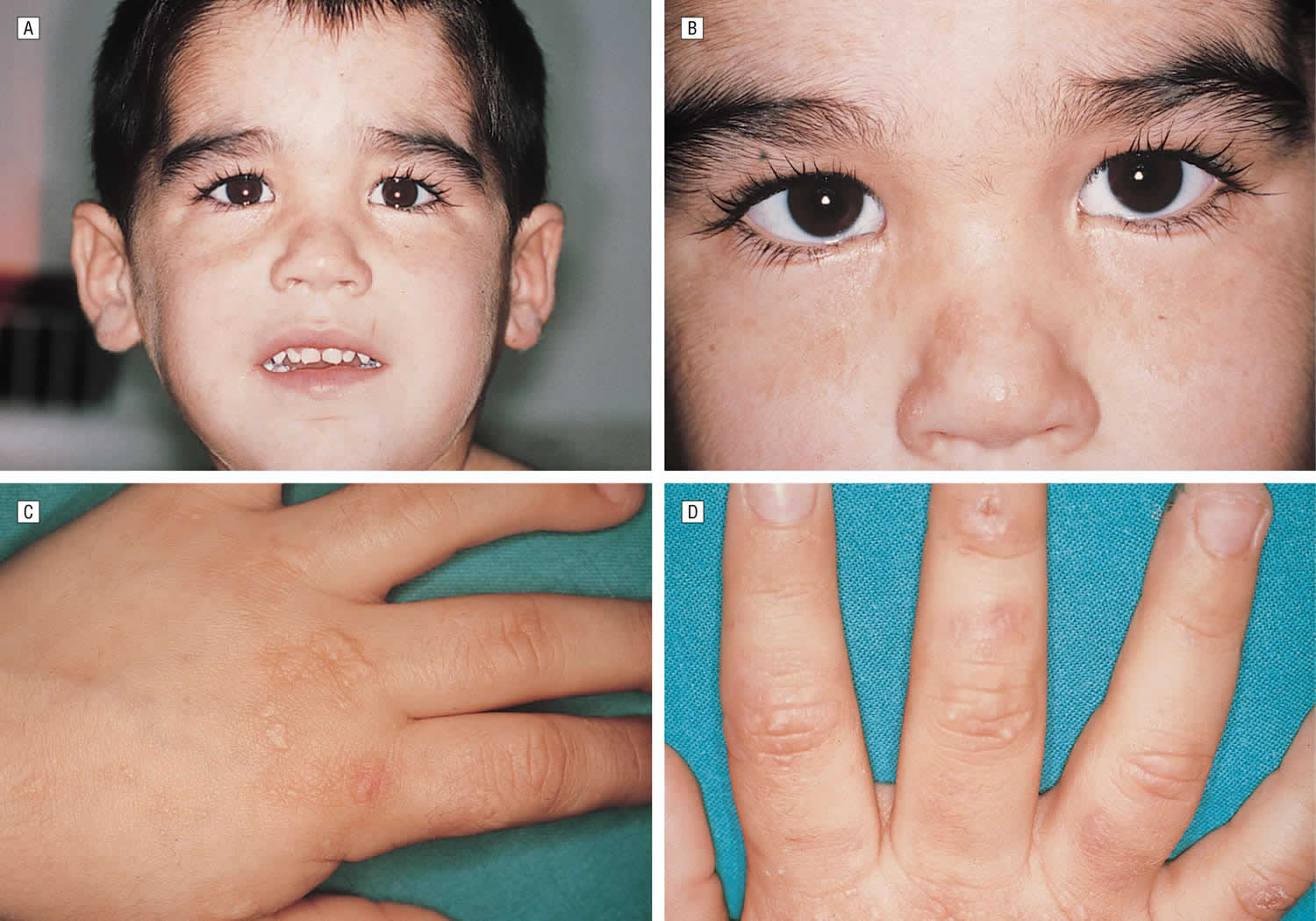
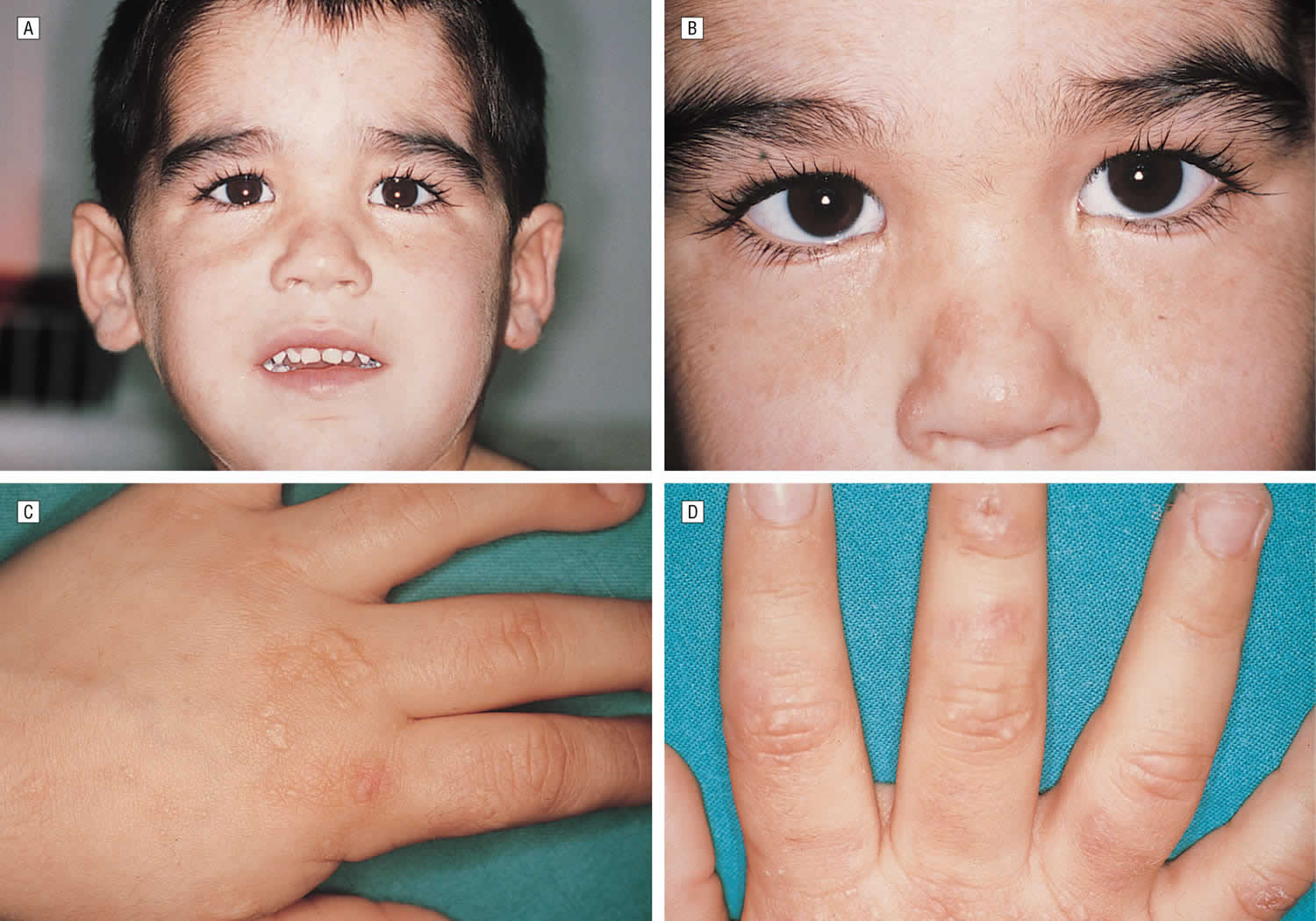
Footnotes: A 5-year-old boy, with no relevant personal or family history, had a syndrome of skin hyperfragility and photosensitivity since 2 years of age. His urine was dark. Skin lesions appeared as vesicles, blisters, and erosions on the face and the back of the hands. Lesions resolved with superficial scars and milia cysts. He presented with hypertrichosis on the face, limbs, and trunk. In the past 3 years, the patient has not presented with any active lesions, and only some superficial scars and mild hypertrichosis remained visible. (A) and (B), The patient, aged 5 years, with mild hypertrichosis of the forehead. (C) and (D) Superficial scars, after blisters and erosions, on the back of the hands. The young boy had a typical profile of porphyrin accumulation with an excess of urinary uroporphyrin and the presence of isocoproporphyrin in feces. Erythrocytic UROD catalytic activity was dramatically decreased. The diagnosis of hepatoerythropoietic porphyria (HEP) was therefore made and confirmed by UROD gene analysis. A point mutation in the third exon of the UROD gene was found at codon 46, a phenylalanine-to-leucine substitution (F46L). The boy was homozygous and the parents were heterozygous for the mutation.
[Source 9 ]Figure 3. Hepatoerythropoietic Porphyria dark urine

Figure 4. Porphyrin molecular structure
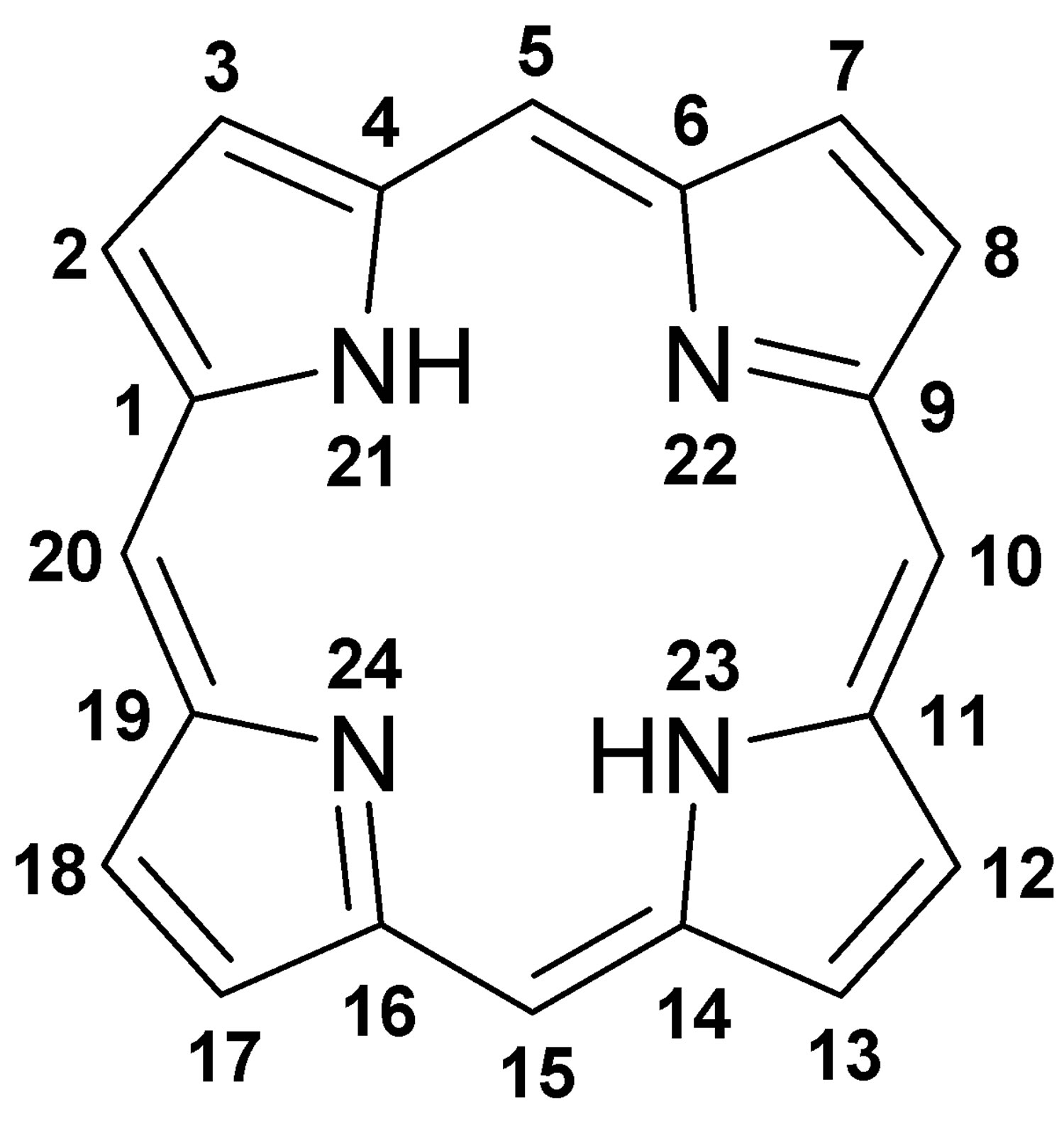
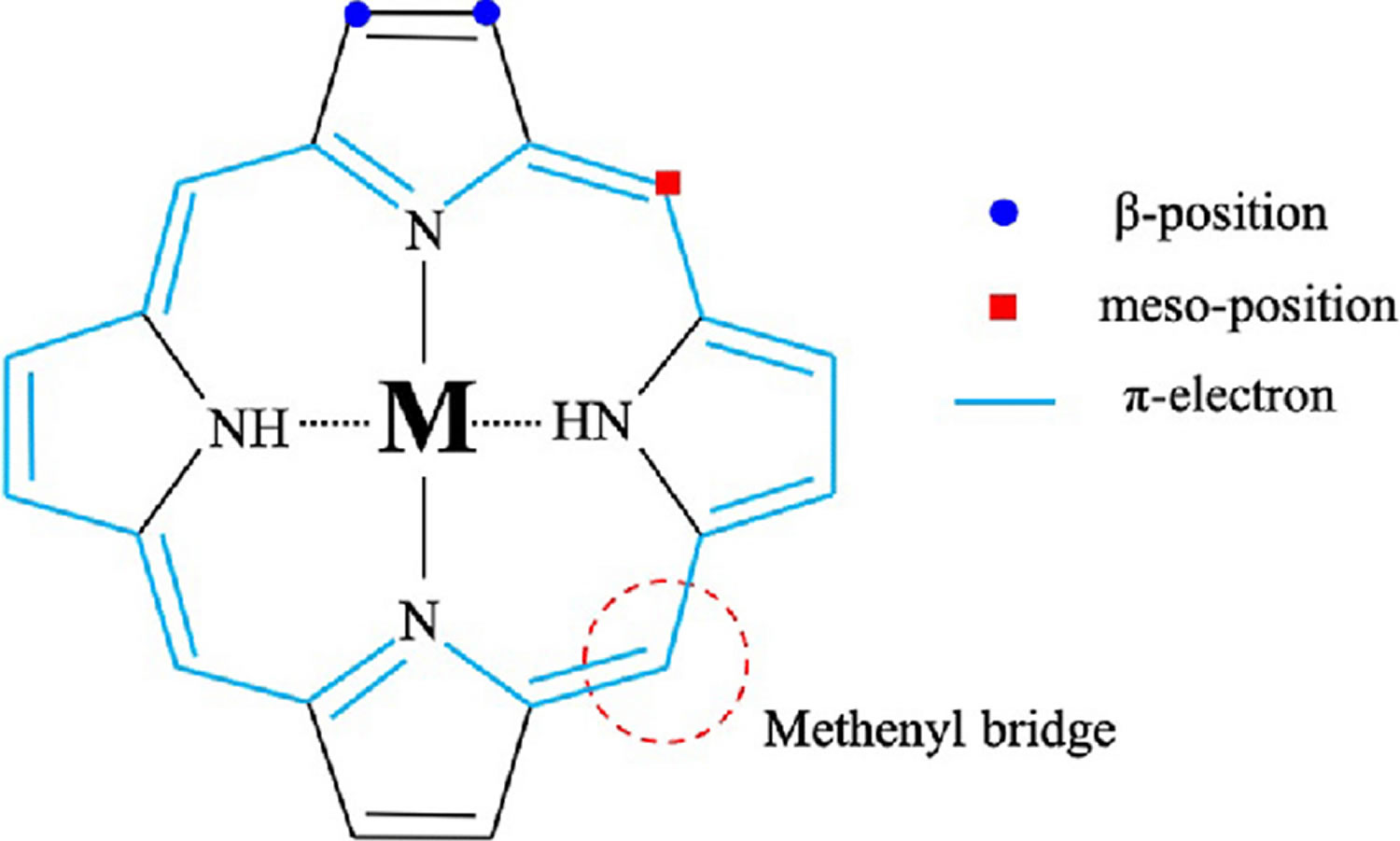
Footnote: Molecular structure of porphyrin (M represent metal ions, such as Mg, Cu, Fe, Zn, etc.).
[Source 23 ]Figure 5. Hemoglobin molecular structure
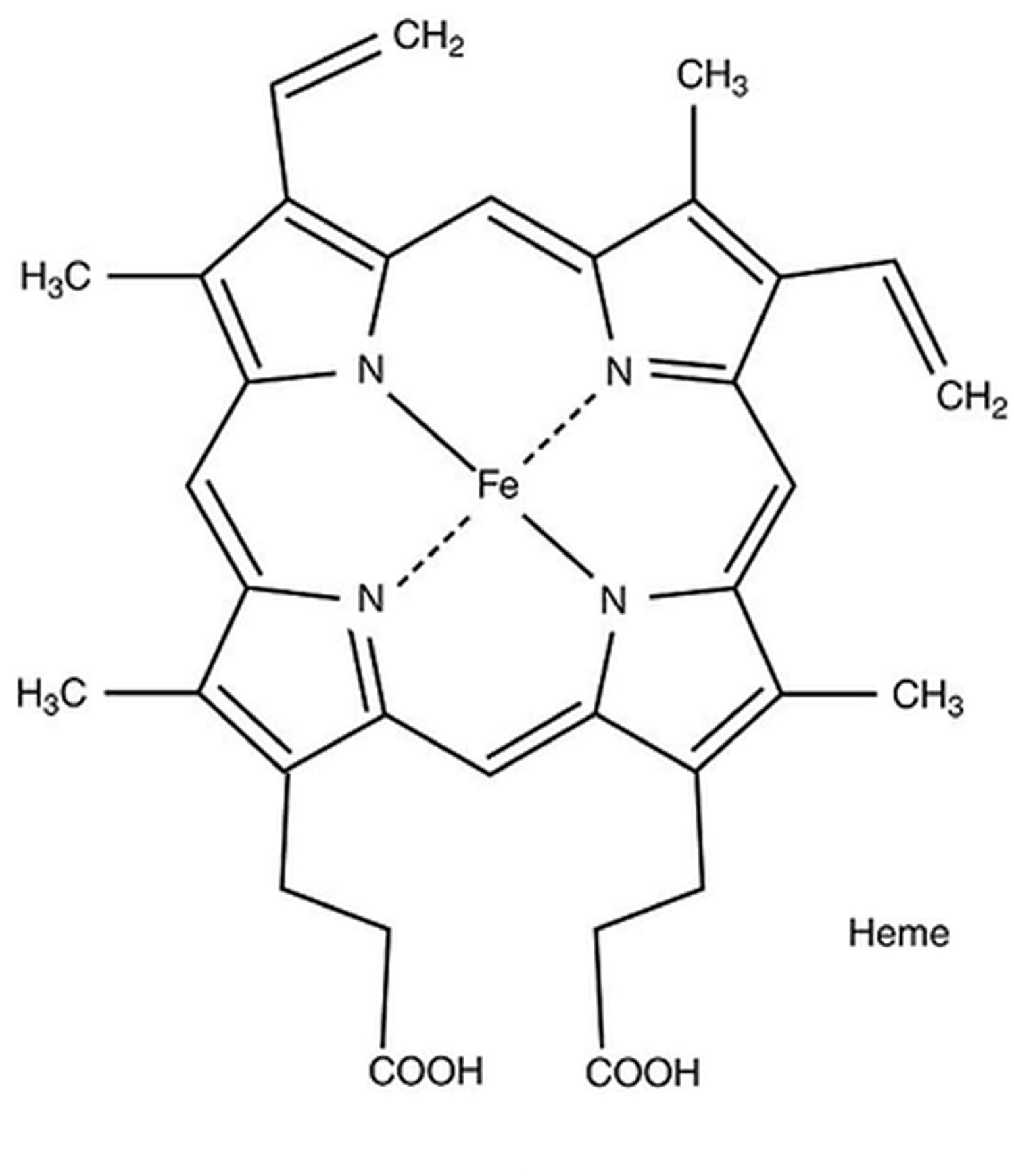
Figure 6. Heme biosynthesis pathway
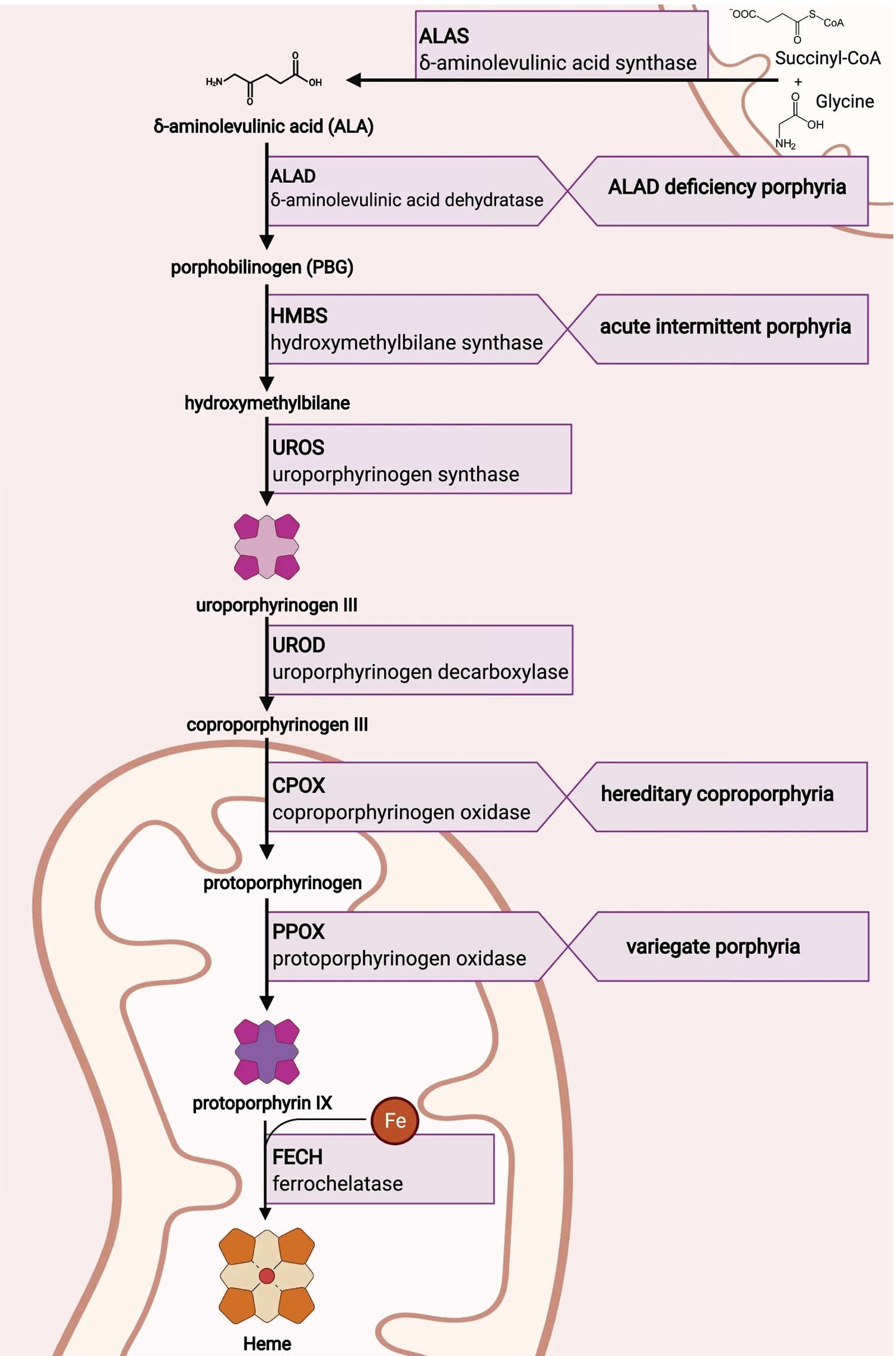
Figure 7. Heme synthesis pathway
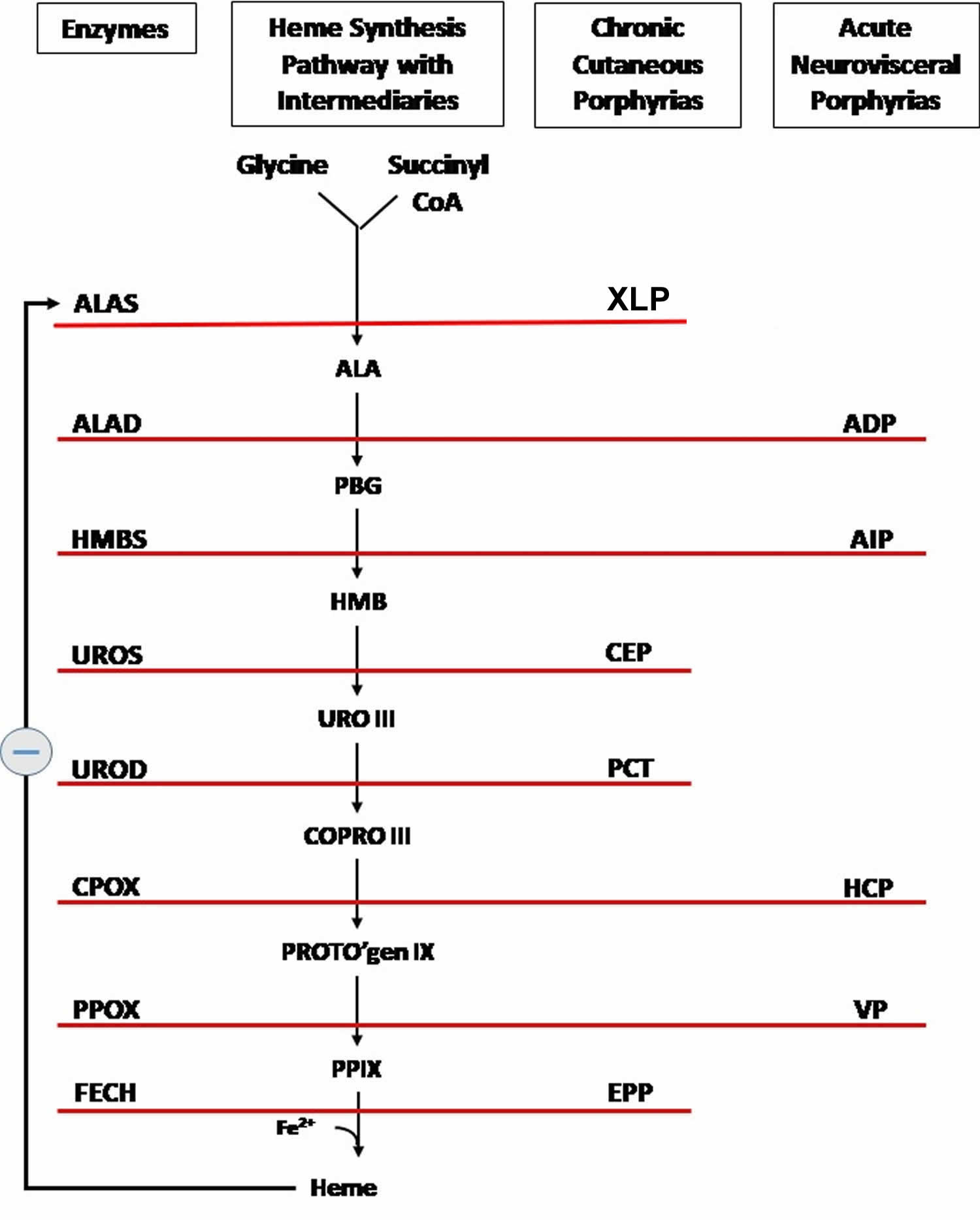
Footnotes: The heme biosynthetic pathway requires 8 enzymatic steps. Heme synthesis pathway showing the enzymes involved in the heme synthesis pathway and the associated porphyrias with the disruption of each specific enzyme. Gain-of-function variants in ALAS2 result in X-linked protoporphyria (XLP), and loss-of-functions variants in FECH result in erythropoietic protoporphyria (EPP). In both X-linked protoporphyria (XLP) and erythropoietic protoporphyria (EPP), metal-free protoporphyrin IX (PPIX) accumulates in erythroblasts, erythrocytes, the plasma, and the biliary system. Metal-free protoporphyrin IX (PPIX) is photosensitive, particularly to visible light in the blue range, and the light-mediated activation of metal-free protoporphyrin IX (PPIX) produces free radicals that damage the surrounding tissues.
Enzymes, encoded by genes, catalyze each of the steps. Gene mutations cause deficient enzyme production. Disruptions are indicated by red lines connecting enzymes with the resultant porphyrias. ALAS (ALAS2) = aminolevulinate synthase (aminolevulinate synthase 2); ALAD = delta-aminolevulinic acid dehydratase; PBGD = porphobilinogen dehydratase; HMBS = hydroxymethylbilane synthase; UROS = uroporphyrinogen-III synthase; UROD = uroporphyrinogen III decarboxylase; CPOX = coproporphyrinogen-III oxidase; PPOX = protoporphyrinogen oxidase; FECH = ferrochelatase.
Porphyrias resulting from disruption of enzyme production. XLP (X-linked protoporphyria); ADP (aminolevulinic acid dehydratase porphyria); AIP (acute intermittent porphyria); CEP (congenital erythropoietic porphyria); PCT (porphyria cutanea tarda); HCP (hereditary coproporphyria); VP (variegate porphyria); EPP (erythropoietic protoporphyria).
Abbreviations: ALA = aminolevulinic acid; PBG = porphobilinogen; HMB = hydroxymethylbilane; URO III = uroporphyrinogen III; COPRO III = coproporphyrinogen III; PROTO’gen IX protoporphyrinogen IX; PPIX = protoporphyrin IX; Fe2+ = iron.
[Source 25 ]Hepatoerythropoietic Porphyria Cause
Hepatoerythropoietic porphyria (HEP) is caused by mutations on both copies of a person’s UROD gene that provides instructions for making an enzyme known as uroporphyrinogen decarboxylase (UROD) that is crucial in the fifth step of the heme biosynthesis pathway, which means that hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait 1, 5. Heme (haem) is vital for all of the body’s organs, although it is most abundant in the blood, bone marrow, and liver. Heme is an essential component of iron-containing proteins called hemoproteins, including hemoglobin (the protein that carries oxygen in the blood). The production of heme (haem) is a multi-step process that requires 8 different enzymes. Uroporphyrinogen decarboxylase (UROD) is responsible for the fifth step in heme biosynthesis process, in which carbon and oxygen atoms are removed from uroporphyrinogen III (the product of the fourth step) to form coproporphyrinogen III. In subsequent steps, three other enzymes produce and modify compounds that ultimately lead to heme.
Scientists have determined that the UROD gene is located on the short arm (p) of chromosome 1 (1p34.1) 20. Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 also known as autosomes and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosome 1p34.1” refers to band 34.1 on the short arm of chromosome 1. The numbered bands specify the location of the thousands of genes that are present on each chromosome. At least 30 different mutations of the UROD gene have been identified in patients with hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT), with 1 predominant missense mutation (glycine–to–glutamic acid substitution at codon 281) in Spanish patients with hepatoerythropoietic porphyria (HEP) 9.
In hepatoerythropoietic porphyria (HEP), the uroporphyrinogen decarboxylase (UROD) enzyme activity is usually less than 10% its normal levels 20. Such low enzyme activity results in the abnormal accumulation of specific porphyrins and related chemicals in the body, especially within the bone marrow, red blood cells, liver and skin. Symptoms develop because of this abnormal accumulation of porphyrins and related chemicals. When porphyrins accumulate in the skin, they absorb sunlight and enter an excited state (photoactivation). This abnormal activation results in the characteristic damage to the skin found in individuals with hepatoerythropoietic porphyria (HEP) 20. The liver removes porphyrins from the blood plasma and secretes it into the bile. When porphyrins accumulate in the liver, they can cause toxic damage to the liver.
The rarity of hepatoerythropoietic porphyria (HEP) makes identification of additional risk factors difficult to assess 1. However, the existence of instances of late-onset disease suggest that risk factors may play a role in some hepatoerythropoietic porphyria (HEP) individuals 26. Excess iron may contribute to UROD inhibition by providing an oxidative environment that is apparently required for generating a UROD inhibitor 27. Hepatic hepcidin expression has been shown to regulate iron homeostasis and likely plays a role in development of porphyria cutanea tarda (PCT); however, the role that
hepcidin plays in porphyria cutanea tarda (PCT) development has not been clearly defined 28.
Alcohol and its metabolites may induce the enzymes ALAS1 and CYP2E1, generate reactive oxygen species (ROS) that contribute to oxidative damage, cause mitochondrial injury, deplete reduced glutathione and other antioxidant defenses, increase endotoxin production, and activate Kupffer cells leading to liver inflammation. In addition, alcohol has been found to impair iron-mediated expression of hepatic hepcidin and to decrease hepatic expression of hepcidin, which may help lead to increased iron in hepatocytes 29, 30.
Smoking may increase oxidative stress in hepatocytes and induce hepatic CYP1A2 which is important in the development of uroporphyria in rodent models. Hepatitis C is associated with excess fat, some iron accumulation, mitochondrial dysfunction, and oxidative stress in hepatocytes – all of which may contribute to the development of porphyria cutanea tarda (PCT). Dysregulation of hepcidin may contribute to iron accumulation in hepatitis C 31, 32.
Estrogens can generate reactive oxygen species (ROS) in some experimental systems; however, the mechanism by which they are a susceptibility factor has not been established. Estrogen mimetics (e.g., tamoxifen) have been shown to be associated with porphyria cutanea tarda (PCT) in several cases. The liver is the site of estrogen metabolism; first pass kinetics leads to a much higher hepatic concentration of estrogen and may also contribute to an increased oxidative environment in some individuals 33.
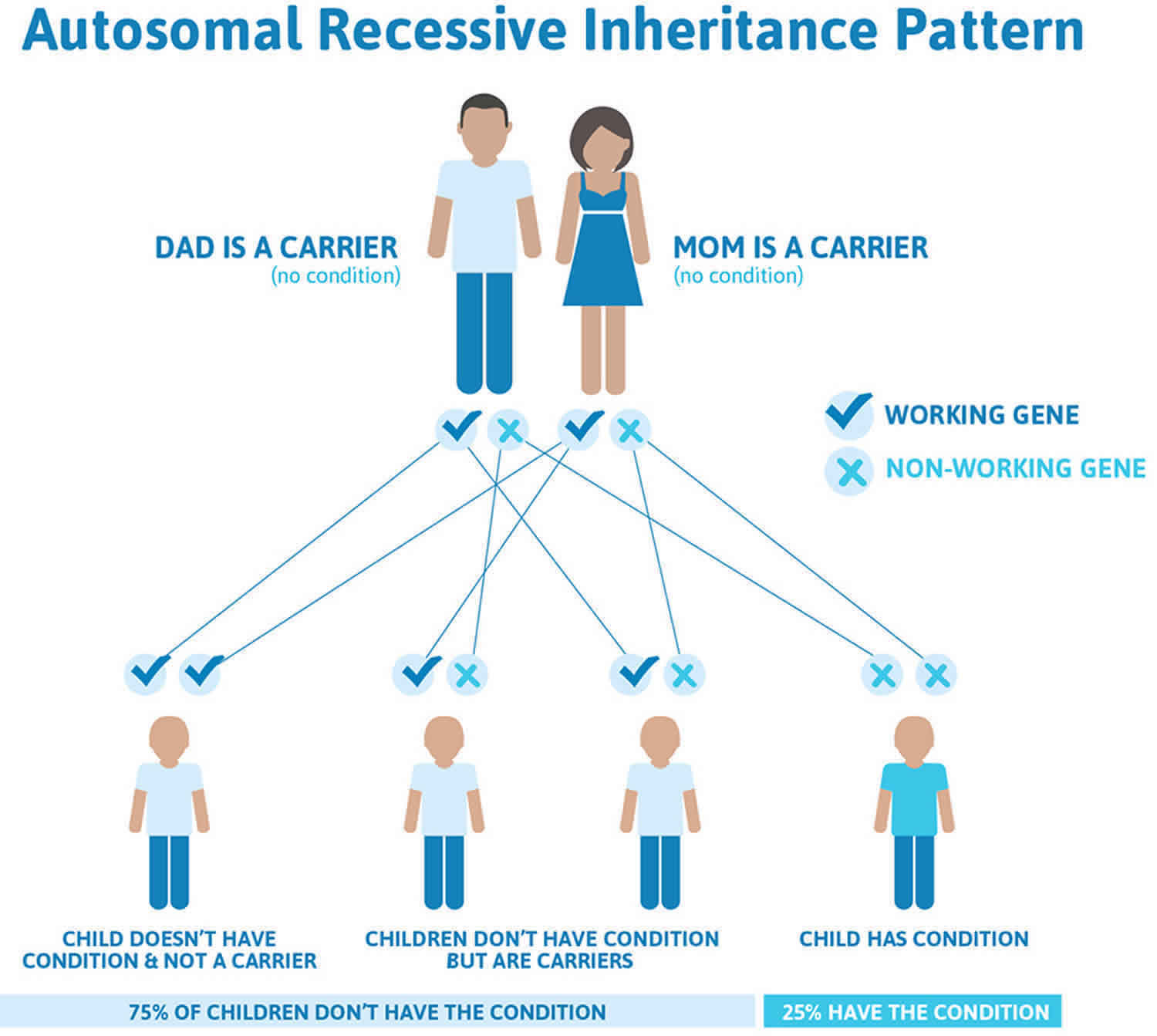
Hepatoerythropoietic Porphyria inheritance pattern
Hepatoerythropoietic porphyria (HEP) is inherited as an autosomal recessive trait. Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Genetic counseling is recommended for affected individuals and their families.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 8. Hepatoerythropoietic porphyria autosomal recessive inheritance pattern
Hepatoerythropoietic Porphyria Pathophysiology
Reduced activity of the liver enzyme uroporphyrinogen decarboxylase (UROD) to 15%-20% of normal in all tissues leads to the accumulation of substrate, uroporphyrinogen, and the intermediate products of the reaction in all cells. Cells with a high demand for heme production include red blood cells (erythrocytes) and liver cells (hepatocytes); therefore, accumulation is more pronounced in these cell types predominantly in the liver.
The substrates uroporphyrinogen and intermediates accumulate in cells in the form of oxidized porphyrins mostly uroporphyrin and heptacarboxylporphyrin that are then transported into the plasma, where they are deposited in the skin and other tissues. In the skin, these porphyrins interact with blue light (the Soret band, ~410 nm) to produce skin damage.
Excess plasma porphyrins (i.e., uroporphyrin and heptacarboxylporphyrin) are excreted via the urine. Intermediates further along in the pathway (i.e., protoporphyrin and zinc protoporphyrin) are eliminated in the bile.
Hepatoerythropoietic Porphyria Signs and Symptoms
Hepatoerythropoietic porphyria (HEP) symptoms and severity can vary from one person to another. Onset is usually within the first two years of life, but mild cases that go undiagnosed until adulthood have been reported. Although hepatoerythropoietic porphyria (HEP) is associated with specific, characteristic symptoms, several factors, including the small number of identified cases, make it difficult to establish the full range of associated symptoms of hepatoerythropoietic porphyria (HEP).
Severe skin photosensitivity is usually the first sign. Affected infants may have extremely fragile skin that can peel or blister on minimal impact is common. Reddening of the skin is also common (erythema). Blistering skin lesions can develop on sun-exposed skin such as the hands and face. Photosensitivity can be severe and can cause scarring, erosion, and disfigurement. Bacterial infection of skin lesions can occur. Abnormal, excessive hair growth (hypertrichosis) may also occur on sun-exposed skin. Affected skin may darken or lose color (hyper- or hypopigmentation). Small bumps with a distinct white head (milia) may also develop. Some affected individuals have teeth that are reddish-brown colored (erythrodontia) as a result of the deposition of porphyrins in the enamel layer of the developing tooth 1.
Low levels of circulating red blood cells (anemia) may also occur. Anemia may be due to the premature destruction of red blood cells (hemolysis). Anemia associated with hepatoerythropoietic porphyria (HEP) may be mild or severe. Severe anemia may be associated with fatigue, pale skin, irregular heartbeat, chest pain, dizziness, and abnormally cold hands and feet. Some individuals may have an abnormally enlarged liver and/or spleen (hepatosplenomegaly).
Mild cases of hepatoerythropoietic porphyria (HEP) can go undiagnosed until adulthood. Overt photosensitivity may not be seen, and mild skin damage can be mistaken for other conditions during childhood.
Note: The clinical features of hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT) are indistinguishable. However, in striking contrast to hepatoerythropoietic porphyria (HEP), familial porphyria cutanea tarda (F-PCT) rarely if ever manifests in infancy or childhood. Rather, as is the case for porphyria cutanea tarda type 1 (nonfamilial, acquired), familial porphyria cutanea tarda (F-PCT) is a disease of middle-aged persons, usually with several risk factors for development of overt disease.
Hepatoerythropoietic Porphyria Diagnosis
A diagnosis of hepatoerythropoietic porphyria (HEP) is based upon identification of characteristic symptoms, a detailed medical history, a thorough clinical evaluation and a variety of specialized tests.
Hepatoerythropoietic porphyria (HEP) should be suspected in infants or children with the following clinical findings, suggestive laboratory findings, and family history.
Hepatoerythropoietic porphyria (HEP) may be considered in infants and children with these clinical features:
- Blistering skin lesions/vesicles/bullae
- Abnormal, excessive hair (hypertrichosis)
- Scarring
- Passage of red or dark urine
Note: The features of hepatoerythropoietic porphyria (HEP) generally resemble those of congenital erythropoietic porphyria (CEP).
Diagnosis of hepatoerythropoietic porphyria (HEP) can be made by demonstrating significant elevations of specific porphyrins in your urine and stool, as well as identification of a specific fluorescence emission peak in plasma. DNA genetic testing to identify the specific mutations in an individual’s UROD genes is the most specific and sensitive test to confirm the diagnosis of hepatoerythropoietic porphyria (HEP).
Clinical Testing and Workup
Screening tests can help diagnose hepatoerythropoietic porphyria (HEP) by measuring the levels of certain porphyrins in blood plasma, urine and red blood cells. Urine and plasma porphyrins show an increase predominantly of uroporphyrin and heptacarboxylporphyrin. Consider erythrocyte zinc protoporphyrin levels, which are significantly increased in hepatoerythropoietic porphyria (HEP), to differentiate hepatoerythropoietic porphyria (HEP) from congenital erythropoietic porphyria (CEP).
Biochemical Characteristics of Hepatoerythropoietic Porphyria 1
- Plasma: Increased uroporphyrin, heptacarboxylporphyrin (~620 nm) (Fluorescence emission peak of diluted plasma at neutral pH, following excitation at 400-410 nm)
- Urine: Increased uroporphyrin, heptacarboxylporphyrin
- Erythrocytes (red blood cells): Increased zinc protoporphyrin
Porphyrin patterns in hepatoerythropoietic porphyria (HEP) are similar to those seen in porphyria cutanea tarda (PCT) with elevation of highly carboxylated porphyrins and isocoproporphyrins. In contrast to porphyria cutanea tarda (PCT), there are markedly increased levels of zinc protoporphyrin in red blood cells in hepatoerythropoietic porphyria (HEP) patients which is due to accumulation of pathway intermediates being metabolized to protoporphyrins.
Hepatoerythropoietic Porphyria Differential Diagnosis
Symptoms of the following disorders can be similar to those of hepatoerythropoietic porphyria (HEP).
Congenital erythropoietic porphyria (CEP) is a rare inherited metabolic disorder resulting from the deficient function of the enzyme uroporphyrinogen III cosynthase (UROS), the fourth enzyme in the heme biosynthetic pathway. Due to the impaired function of UROS (uroporphyrinogen III cosynthase) enzyme, excessive amounts of particular porphyrins accumulate, particularly in the bone marrow, plasma, red blood cells, urine, teeth, and bones. The major symptom of congenital erythropoietic porphyria (CEP) is hypersensitivity of the skin to sunlight and some types of artificial light, such as fluorescent lights (photosensitivity). After exposure to light, the photo-activated porphyrins in the skin cause bullae (blistering) and the fluid-filled sacs rupture, and the lesions often get infected. These infected lesions can lead to scarring, bone loss, and deformities. The hands, arms, and face are the most commonly affected areas. Congenital erythropoietic porphyria (CEP) is inherited as an autosomal recessive genetic disorder. Typically, there is no family history of the disease. Both parents are usually healthy, but each carries a defective gene that they can pass to their children. Affected offspring have two copies of the defective gene, one inherited from each parent.
There are other conditions that may cause signs and symptoms that are similar to those seen in hepatoerythropoietic porphyria (HEP). Such conditions include other cutaneous porphyrias, sporadic porphyria cutanea tarda (i.e., porphyria cutanea tarda type 1 that is not associated with a UROD pathogenic variant), drug-induced photosensitivity, epidermolysis bullosa, various forms of lupus, and solar urticarial.
Sporadic porphyria cutanea tarda also called porphyria cutanea tarda type 1 that is not associated with a UROD genetic mutation is clinically indistinguishable from hepatoerythropoietic porphyria (HEP) and familial porphyria cutanea tarda (F-PCT) and is highly influenced by susceptibility factors associated with porphyria cutanea tarda (PCT). In these cases, the excess porphyrins are produced only in the liver.
The skin histopathologic findings of pseudoporphyria are similar to those of hepatoerythropoietic porphyria (HEP), however, pseudoporphyria is not associated with porphyrin biochemical abnormalities. Medications, chronic kidney failure, and excessive sun exposure or UV radiation have been reported to cause pseudoporphyria 34.
Table 1. Other Types of Porphyria in Hepatoerythropoietic Porphyria Differential Diagnosis
| Disorder | Gene | Mode of inheritance | Skin Lesions | Distinguishing Features / Comment |
|---|---|---|---|---|
| Hereditary coproporphyria (HCP) | CPOX (coproporphyrinogen oxidase) | Autosomal Dominant | Blistering skin lesions closely resembling lesions of congenital erythropoietic porphyria (CEP) |
|
| Variegate porphyria (VP) | PPOX (protoporphyrinogen oxidase) | Autosomal Dominant | Blistering skin lesions are nearly identical to those in hepatoerythropoietic porphyria (HEP). Cutaneous manifestations in hepatoerythropoietic porphyria (HEP) are chronic & blistering like in variegate porphyria (VP) but are usually more severe than those of variegate porphyria (VP), because circulating porphyrin levels in hepatoerythropoietic porphyria (HEP) are usually much higher than in variegate porphyria (VP). |
|
| Familial porphyria cutanea tarda (F-PCT) | UROD (uroporphyrinogen decarboxylase) | Autosomal Dominant | Skin lesions resemble those of hepatoerythropoietic porphyria (HEP) but are less severe & typically begin later, in the 5th or 6th decade of life. |
|
| Congenital erythropoietic porphyria (CEP) |
|
| The skin lesions of congenital erythropoietic porphyria (CEP), like those seen in hepatoerythropoietic porphyria (HEP), appear early in life (i.e., in infancy or childhood) and are severe & mutilating. | In both congenital erythropoietic porphyria (CEP) and hepatoerythropoietic porphyria (HEP), ↑ severity is attributed to plasma concentration of porphyrin. Although congenital erythropoietic porphyria (CEP) can be mistaken for hepatoerythropoietic porphyria (HEP), urine porphyrin analysis which shows marked ↑ in uroporphyrin and coproporphyrin type 1 in congenital erythropoietic porphyria (CEP) helps exclude other cutaneous porphyrias. Fecal analysis may be necessary, particularly for persons with late onset. |
Hepatoerythropoietic Porphyria Treatment
The treatment of hepatoerythropoietic porphyria (HEP) is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, hematologists, dermatologists, hepatologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may benefit affected individuals and their families.
There is no specific, FDA-approved therapy for individuals with hepatoerythropoietic porphyria (HEP). Because the disorder is so rare, most treatment information is based other forms of porphyria.
Avoidance and/or protection from sunlight will benefit individuals with hepatoerythropoietic porphyria (HEP) and can include the use of clothing styles with long sleeves and pant legs, made with double layers of fabric or of light-exclusive fabrics, wide brimmed hats, gloves, and sunglasses. Topical sunscreens are generally ineffective, but certain tanning products with ingredients that increase pigmentation may be helpful. Affected individuals may also benefit from window tinting and the use of vinyl or films to cover the windows of their homes and cars.
Phlebotomies (removal of blood) to lower the amount of porphyrins in the liver, which are used to treat individuals with porphyria cutanea tarda (PCT), are generally ineffective in individuals with hepatoerythropoietic porphyria (HEP) since elevated iron levels are not a feature of the disorder 35, 15. Another treatment for porphyria cutanea tarda (PCT), the antimalarial drug hydroxychloroquine, was effective in at least one case reported in the medical literature.
Anemia may require treatment in some cases. Blood transfusions have been used to treat some individuals. Recombinant erythropoietin (rEPO) a synthetic version of the naturally occurring hormone erythropoietin, which helps the body produce more red blood cells, was successfully used to treat severe anemia in an individual with hepatoerythropoietic porphyria (HEP) whose anemia was not associated with increased red cell destruction.
Light Avoidance
Avoiding exposure to sunlight is the most important way to manage symptoms for people with hepatoerythropoietic porphyria (HEP). People with hepatoerythropoietic porphyria (HEP) need to wear protective clothing, and have windows tinted in their cars and homes. Most sunscreens are not effective because they do not block light in the blue-violet range, which is the type of light that triggers reactions in porphyria.
Agents to avoid
Older individuals should avoid known triggering factors: alcohol, oral estrogen, iron overload, smoking, and drugs that induce the cytochrome P450s.
Blister and Wound Care
There is a risk of developing infections in blisters, particularly if they rupture. Prescription antibiotic ointments may be required to manage infections. Care from a dermatologist is recommended.
Hepatoerythropoietic Porphyria Prognosis
Due to the extreme rarity of hepatoerythropoietic porphyria (HEP) with less than 100 reported cases, little is known about the prognosis of hepatoerythropoietic porphyria (HEP) patients 17, 36, 19. The prognosis for hepatoerythropoietic porphyria (HEP) can be complicated by liver involvement, and liver function test must be monitored regularly. However, with proper treatment and management, many patients with hepatoerythropoietic porphyria (HEP) can maintain a good quality of life 37, 38. No increased risk for liver cancer (hepatocellular carcinoma) has been documented in hepatoerythropoietic porphyria (HEP) 1.
Hepatoerythropoietic porphyria (HEP) typically presents early in infancy or early childhood with signs and symptoms such as extreme photosensitivity, recurrent skin blisters (bullae) and skin erosions on sun exposed skin with secondary bacterial infections that may result in severe skin scarring, sclerodermatous change, and deformities of the hand 4. Sun-induced skin redness (erythema) and blistering occurred by age 2 years in 75% of reported cases 4. Sclerodactyly (a condition where the skin on the fingers and/or toes thickens and tightens, making them feel hard and less flexible), osteolysis (active resorption of bone tissue) and shortening of the phalanges, and progressive joint deformities can occur as well 4, 15.
Spontaneous improvement of acute photosensitivity during later childhood, but persistent skin fragility, has been described 39, 40, 41, 42. Other patients have presented in the second or third decade of life with mild skin fragility or photodistributed annular plaques 43, 44, 41, 45.
Other signs and symptoms inlcude abnormal excessive hair (hypertrichosis), red-brown discoloration of teeth (erythrodontia) and pink-to-red urine. There may be bone fragility due to expansion of the bone marrow and vitamin deficiencies, especially vitamin D deficiency. Red blood cells have a shortened life-span with mild or severe hemolytic anemia. Synthesis of heme and hemoglobin is actually increased to compensate for the shortened red blood cell survival and is associated with abnormal enlargement of the liver and/or spleen (hepatosplenomegaly) 17, 4, 15. Current treatment recommendations resemble those for familial porphyria cutanea tarda (F-PCT) 1.
Mild cases of hepatoerythropoietic porphyria (HEP) may go unrecognized until adulthood and can be clinically indistinguishable from porphyria cutanea tarda (PCT), the most common form of porphyria in humans where the porphyria cutanea tarda (PCT) that may be acquired in 75% to 80% of cases (also known as porphyria cutanea tarda type 1 or sporadic porphyria cutanea tarda) or occur in individuals with a mutation of one UROD gene (autosomal dominant inheritance or porphyria cutanea tarda type 2 or familial porphyria cutanea tarda). Hepatoerythropoietic Porphyria (HEP) is the autosomal recessive form of familial porphyria cutanea tarda (F-PCT). Skin photosensitivity is generally much more severe in hepatoerythropoietic porphyria (HEP) than in porphyria cutanea tarda (PCT).
Compared to porphyria cutanea tarda (PCT), the skin features of hepatoerythropoietic porphyria (HEP) typically have earlier onset, increased severity leading to disfigurement, and closer resemblance to those in congenital erythropoietic porphyria (CEP) 8, 15. However, extracutaneous findings including hemolytic anemia are more frequent and severe in congenital erythropoietic porphyria (CEP) than hepatoerythropoietic porphyria (HEP) 8, 15, 44.
In contrast to the autosomal recessive form of variegate porphyria (VP), which is characterized by developmental delay and seizures 46, neurologic abnormalities are not typically associated with hepatoerythropoietic porphyria (HEP) or congenital erythropoietic porphyria (CEP) 8, 47, 48. Nevertheless, developmental delay and seizures have been previously reported in hepatoerythropoietic porphyria (HEP) 49, 11. A 4-year-old boy had delayed speech and language skills then presented with focal seizures and acute left hemiparesis 49. Two young adults, ages 21 and 23 years, with severe hepatoerythropoietic porphyria (HEP) developed generalized seizures and had neuroimaging evidence of cerebral cortical atrophy and punctate calcifications in the frontal lobes, presumably related to hypoxic injury as in other porphyrias 11. These observations, together with recent affected siblings’ developmental delay, support neurologic assessment of hepatoerythropoietic porphyria (HEP) patients in order to better define this possible complication 4.
Cantatore-Francis et al 4 reported 2 sisters with painful polyarticular arthritis, represents a typical (but heretofore unrecognized) inflammatory precedent of joint deformity in hepatoerythropoietic porphyria (HEP) or an idiosyncratic inflammatory process, perhaps triggered by porphyrin deposition together with exposure to ultraviolet light or another environmental insult, remains to be determined.
Anemia was present in more than 50% hepatoerythropoietic porphyria (HEP) patients for whom hematologic status was reported (15/27) 4, but severe anemia requiring transfusions or administration of erythropoietin (EPO) has only been observed in a few individuals 43.
- Rudnick S, Phillips J, Bonkovsky H; Porphyrias Consortium of the Rare Diseases Clinical Research Network. Hepatoerythropoietic Porphyria. 2013 Oct 31 [Updated 2022 Dec 22]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK169003[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Kaya Ç, Heydari A, Kızılay O, Kızılay O, Sezgin G. Hepatoerythropoietic Porphyria with Coexisting BTD And CNGB1 Genetic Mutations: A First Case Report. Eur J Case Rep Intern Med. 2025 Jan 25;12(3):005198. doi: 10.12890/2025_005198[↩]
- Balogun O, Nejak-Bowen K. Understanding Hepatic Porphyrias: Symptoms, Treatments, and Unmet Needs. Semin Liver Dis. 2024 May;44(2):209-225. doi: 10.1055/s-0044-1787076[↩][↩]
- Cantatore-Francis JL, Cohen-Pfeffer J, Balwani M, Kahn P, Lazarus HM, Desnick RJ, Schaffer JV. Hepatoerythropoietic porphyria misdiagnosed as child abuse: cutaneous, arthritic, and hematologic manifestations in siblings with a novel UROD mutation. Arch Dermatol. 2010 May;146(5):529-33. doi: 10.1001/archdermatol.2010.89[↩][↩][↩][↩][↩][↩][↩][↩]
- Hepatoerythropoietic Porphyria (HEP). https://porphyriafoundation.org/for-patients/types-of-porphyria/hep/[↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ. Disorders of heme biosynthesis: X-linked sideroblastic anemia and the porphyrias. In: Scriver CS, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th. New York, NY: McGraw-Hill; 2001. pp. 2961–3062.[↩]
- de Verneuil H, Grandchamp B, Beaumont C, Picat C, Nordmann Y. Uroporphyrinogen decarboxylase structural mutant (Gly281—-Glu) in a case of porphyria. Science. 1986 Nov 7;234(4777):732-4. doi: 10.1126/science.3775362[↩]
- Smith SG. Hepatoerythropoietic porphyria. Semin Dermatol. 1986;5(2):125–137.[↩][↩][↩][↩]
- Ged C, Ozalla D, Herrero C, et al. Description of a New Mutation in Hepatoerythropoietic Porphyria and Prenatal Exclusion of a Homozygous Fetus. Arch Dermatol. 2002;138(7):957–960. doi:10.1001/archderm.138.7.957[↩][↩][↩]
- 92 – Congenital and Hereditary Disorders of the Skin. Avery’s Diseases of the Newborn (Eleventh Edition) 2024, Pages 1332-1346, 1346.e1-1346.e2 https://doi.org/10.1016/B978-0-323-82823-9.00092-1[↩]
- Berenguer J, Blasco J, Cardenal C, Pujol T, Cruces Prado MJ, Herrero C, Mascaró JM, de la Torre C, Mercader JM. Hepatoerythropoietic porphyria: neuroimaging findings. AJNR Am J Neuroradiol. 1997 Sep;18(8):1557-60. https://pmc.ncbi.nlm.nih.gov/articles/instance/8338136/pdf/9296199.pdf[↩][↩][↩]
- Kaya Ç, Heydari A, Kızılay O, Kızılay O, Sezgin G. Hepatoerythropoietic Porphyria with Coexisting BTD And CNGB1 Genetic Mutations: A First Case Report. Eur J Case Rep Intern Med. 2025 Jan 25;12(3):005198. doi: 10.12890/2025_005198 [↩][↩]
- Anderson KE, Sassa S, Bishop DF, Desnick RJ, Disorders of Heme Biosynthesis: X-Linked Sideroblastic Anemia and the Porphyrias, in: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G (Eds.), The Online Metabolic and Molecular Bases of Inherited Disease, The McGraw-Hill Companies, Inc., New York, NY, 2014.[↩]
- Pin˜ol J, Herrero C, Almeida J, et al. Porphyrie he´ pato-erythro-cytaire: une nouvelle forme de porphyrie. Ann Dermatol Venereol 1975;102:129 –136[↩]
- Elder GH. Hepatic porphyrias in children. J Inherit Metab Dis. 1997 Jun;20(2):237-46. doi: 10.1023/a:1005313024076[↩][↩][↩][↩][↩][↩][↩]
- Biesecker LG, Adam MP, Alkuraya FS, et al. A dyadic approach to the delineation of diagnostic entities in clinical genomics. Am J Hum Genet. 2021 Jan 7;108(1):8-15. doi: 10.1016/j.ajhg.2020.11.013[↩]
- de Verneuil H, Beaumont C, Deybach JC, Nordmann Y, Sfar Z, Kastally R. Enzymatic and immunological studies of uroporphyrinogen decarboxylase in familial porphyria cutanea tarda and hepatoerythropoietic porphyria. Am J Hum Genet. 1984 May;36(3):613-22. https://pmc.ncbi.nlm.nih.gov/articles/instance/1684444/pdf/ajhg00165-0121.pdf[↩][↩][↩]
- Phillips JD, Whitby FG, Stadtmueller BM, Edwards CQ, Hill CP, Kushner JP. Two novel uroporphyrinogen decarboxylase (URO-D) mutations causing hepatoerythropoietic porphyria (HEP) Transl Res. 2007;149(2):85–91. doi: 10.1016/j.trsl.2006.08.006[↩]
- Granata BX, Parera VE, Melito VA, Teijo MJ, Batlle AM, Rossetti MV. The very first description of a patient with hepatoerythropoietic porphyria in Argentina. Biochemical and molecular studies. Cell Mol Biol (Noisy-le-grand). 2009 Feb 16;55(1):61-5.[↩][↩]
- Hepatoerythropoietic Porphyria. https://rarediseases.org/rare-diseases/hepatoerythropoietic-porphyria/[↩][↩][↩][↩][↩]
- Weiss Y, Chen B, Yasuda M, Nazarenko I, Anderson KE, Desnick RJ. Porphyria cutanea tarda and hepatoerythropoietic porphyria: Identification of 19 novel uroporphyrinogen III decarboxylase mutations. Mol Genet Metab. 2019 Nov;128(3):363-366. doi: 10.1016/j.ymgme.2018.11.013[↩]
- Aarsand AK, Boman H, Sandberg S. Familial and sporadic porphyria cutanea tarda: characterization and diagnostic strategies. Clin Chem. 2009 Apr;55(4):795-803. doi: 10.1373/clinchem.2008.117432[↩]
- Lin, Jou & Shi, Donglu. (2021). Photothermal and photovoltaic properties of transparent thin films of porphyrin compounds for energy applications. Applied Physics Reviews. 8. 011302. https://doi.org/10.1063/5.0036961[↩]
- Panawala, Lakna. (2017). What is the Function of Hemoglobin in the Human Body. https://www.researchgate.net/publication/313841668_What_is_the_Function_of_Hemoglobin_in_the_Human_Body[↩]
- Edel Y, Mamet R. Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. 2018 Apr 19;9(2):e0013. doi: 10.5041/RMMJ.10333[↩]
- Triviboonvanich S, Junnu S, Srisawat C, Silpa-archa N. Late-onset hepatoerythropoietic porphyria presenting with facial deformities and erythrodontia. Thai J Dermatol. 2019;35:73–80.[↩]
- Ryan Caballes F, Sendi H, Bonkovsky HL. Hepatitis C, porphyria cutanea tarda and liver iron: an update. Liver Int. 2012 Jul;32(6):880-93. doi: 10.1111/j.1478-3231.2012.02794.x[↩]
- Ajioka RS, Phillips JD, Weiss RB, Dunn DM, Smit MW, Proll SC, Katze MG, Kushner JP. Down-regulation of hepcidin in porphyria cutanea tarda. Blood. 2008 Dec 1;112(12):4723-8. doi: 10.1182/blood-2008-02-138222[↩]
- Harrison-Findik DD, Klein E, Crist C, Evans J, Timchenko N, Gollan J. Iron-mediated regulation of liver hepcidin expression in rats and mice is abolished by alcohol. Hepatology. 2007 Dec;46(6):1979-85. doi: 10.1002/hep.21895[↩]
- Harrison-Findik DD, Schafer D, Klein E, Timchenko NA, Kulaksiz H, Clemens D, Fein E, Andriopoulos B, Pantopoulos K, Gollan J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J Biol Chem. 2006 Aug 11;281(32):22974-82. doi: 10.1074/jbc.M602098200[↩]
- Fujita N, Sugimoto R, Motonishi S, Tomosugi N, Tanaka H, Takeo M, Iwasa M, Kobayashi Y, Hayashi H, Kaito M, Takei Y. Patients with chronic hepatitis C achieving a sustained virological response to peginterferon and ribavirin therapy recover from impaired hepcidin secretion. J Hepatol. 2008 Nov;49(5):702-10. doi: 10.1016/j.jhep.2008.05.014[↩]
- Nishina S, Hino K, Korenaga M, Vecchi C, Pietrangelo A, Mizukami Y, Furutani T, Sakai A, Okuda M, Hidaka I, Okita K, Sakaida I. Hepatitis C virus-induced reactive oxygen species raise hepatic iron level in mice by reducing hepcidin transcription. Gastroenterology. 2008 Jan;134(1):226-38. doi: 10.1053/j.gastro.2007.10.011[↩]
- Bulaj ZJ, Franklin MR, Phillips JD, Miller KL, Bergonia HA, Ajioka RS, Griffen LM, Guinee DJ, Edwards CQ, Kushner JP. Transdermal estrogen replacement therapy in postmenopausal women previously treated for porphyria cutanea tarda. J Lab Clin Med. 2000 Dec;136(6):482-8. doi: 10.1067/mlc.2000.111024[↩]
- Beer K, Applebaum D, Nousari C. Pseudoporphyria: discussion of etiologic agents. J Drugs Dermatol. 2014 Aug;13(8):990-2. https://jddonline.com/articles/pseudoporphyria-discussion-of-etiologic-agents-S1545961614P0990X/[↩]
- Sassa S. Modern diagnosis and management of the porphyrias. Br J Haematol. 2006;135(3):281–292. doi: 10.1111/j.1365-2141.2006.06289.x[↩]
- Piñol Aguadé J, Herrero C, Almeida J, Castells Mas A, Ferrando J, De Asprer J, Palou A, Giménez A. Porphyrie hépato-érythrocytaire. Une nouvelle forme de porphyrie [Hepato-erythrocytic porphyria. A new type of porphyria]. Ann Dermatol Syphiligr (Paris). 1975;102(2):129-36. French.[↩]
- Herrick AL, McColl KE, Moore MR, Cook A, Goldberg A. Controlled trial of haem arginate in acute hepatic porphyria. Lancet. 1989;1:1295–1297. doi: 10.1016/s0140-6736(89)92688-3[↩]
- Petersen CS, Thomsen K. High-dose hydroxychloroquine treatment of porphyria cutanea tarda. J Am Acad Dermatol. 1992;26:614–619. doi: 10.1016/0190-9622(92)70090-3[↩]
- Gunther WW. The porphyrias and erythropoietic protoporphyria: an unusual case. Australas J Dermatol. 1967;9(1):23–30. doi: 10.1111/j.1440-0960.1967.tb01147.x[↩]
- Ged C, Ozalla D, Herrero C, et al. Description of a new mutation in hepatoerythropoietic porphyria and prenatal exclusion of a homozygous fetus. Arch Dermatol. 2002;138(7):957–960. doi: 10.1001/archderm.138.7.957[↩]
- Remenyik É, Lecha M, Badenas C, et al. Childhood-onset mild cutaneous porphyria with compound heterozygotic mutations in the uroporphyrinogen decarboxylase gene. Clin Exp Dermatol. 2008;33(5):602–605. doi: 10.1111/j.1365-2230.2008.02734.x[↩][↩]
- Czarnecki DB. Hepatoerythropoietic Porphyria. Arch Dermatol. 1980;116(3):307–311. doi:10.1001/archderm.1980.01640270067017[↩]
- Horina JH, Wolf P. Epoetin for severe anemia in hepatoerythropoietic porphyria. N Engl J Med. 2000;342(17):1294–1295. doi: 10.1056/NEJM200004273421717[↩][↩]
- Armstrong DK, Sharpe PC, Chambers CR, Whatley SD, Roberts AG, Elder GH. Hepatoerythropoietic porphyria: a missense mutation in the UROD gene is associated with mild disease and an unusual porphyrin excretion pattern. Br J Dermatol. 2004;151(4):920–923. doi: 10.1111/j.1365-2133.2004.06101.x[↩][↩]
- Moran-Jimenez MJ, Ged C, Romana M, Enriquez De Salamanca R, Taïeb A, Topi G, D’Alessandro L, de Verneuil H. Uroporphyrinogen decarboxylase: complete human gene sequence and molecular study of three families with hepatoerythropoietic porphyria. Am J Hum Genet. 1996 Apr;58(4):712-21. https://pmc.ncbi.nlm.nih.gov/articles/instance/1914669/pdf/ajhg00017-0069.pdf[↩]
- Palmer RA, Elder GH, Barrett DF, Keohane SG. Homozygous variegate porphyria: a compound heterozygote with novel mutations in the protoporphyrinogen oxidase gene. Br J Dermatol. 2001;144(4):866–869. doi: 10.1046/j.1365-2133.2001.04147.x[↩]
- Elder GH. Hepatic porphyrias in children. J Inher Metab Dis. 1997;20(2):237–246. doi: 10.1023/a:1005313024076[↩]
- Murphy GM. The cutaneous porphyrias: a review. The British Photodermatology Group. Br J Dermatol. 1999;140(4):573–581. doi: 10.1046/j.1365-2133.1999.02754.x[↩]
- Parsons JL, Sahn EE, Holden KR, Pai GS. Neurologic disease in a child with hepatoerythropoietic porphyria. Pediatr Dermatol. 1994;11(3):216–221. doi: 10.1111/j.1525-1470.1994.tb00589.x[↩][↩]





{kind=link}