Hypereosinophilia
Hypereosinophilia is defined as circulating blood absolute eosinophil counts above 1.5 ×109/L (1,500 eosinophils/μL) recorded on at least two occasions with a minimum time interval of four weeks apart 1. Peripheral blood eosinophil levels vary by age, with upper reference limits being higher in younger children 2. Traditionally, the degree of hypereosinophilia can be classified as mild (absolute eosinophil counts 500 – 1500 eosinophils per microliter), moderate (absolute eosinophil counts 1500 – 5000 eosinophils/μL), or severe (absolute eosinophil counts above 5000 eosinophils per microliter) 3.
An eosinophil is a type of white blood cell that plays an important role in the immune system. White blood cells help protect the body by fighting infections. They are also associated with allergy and asthma.
Patients with hypereosinophilia can have a spectrum of clinical consequences, ranging from relatively benign conditions to diseases associated with organ dysfunction and potentially life-threatening complications 4. Hypereosinophilia is observed in patients with various inflammatory and allergic conditions, as well as diverse hematologic malignancies 5. In hematopoietic stem cell and myeloid neoplasms, eosinophils originate from a malignant clone, whereas in other conditions and disorders, hypereosinophilia is considered a nonneoplastic process triggered by eosinophilopoietic cytokines or by other as yet unknown processes 6. Peripheral blood hypereosinophilia can be transient, episodic, or persistent. In patients with chronic (persistent) hypereosinophilia, tissue infiltration and the effects of eosinophil-derived effector molecules might result in clinically relevant organ pathology or even in (irreversible) organ damage 7. Notably, among a range of effects on multiple organs, endomyocardial fibrosis, thrombosis, or both might be life-threatening consequences in patients with sustained hypereosinophilia. In other patients hypereosinophilia can be persistent but does not lead to measurable organ dysfunction. In these patients the clinical course and outcome remain uncertain; therefore they should be followed for potential disease progression.
Several neoplastic conditions are associated with hypereosinophilia. Myeloid neoplasms variably accompanied by hypereosinophilia are chronic myeloid leukemia (CML), other myeloproliferative neoplasms (MPNs), distinct variants of acute myeloid leukemia, rare forms of myelodysplastic syndromes (MDS), some myelodysplastic syndrome or myeloproliferative neoplasm overlap disorders, and a subset of patients with (advanced) systemic mastocytosis 8. These differential diagnoses have to be considered in cases of unexplained hypereosinophilia, especially when signs of myeloproliferation are present. In such patients a thorough hematologic workup, including bone marrow cytology, histology and immunohistochemistry, cytogenetics, molecular analyses, and staging of potentially affected organ systems, should be initiated. In patients with eosinophilic leukemia, hypereosinophilia is a consistent and predominant feature. Most serious complications of hypereosinophilia (ie, endomyocardial fibrosis, thrombosis, or both) are particularly prone to develop in these patients, especially in the setting of fusion genes involving platelet-derived growth factor receptor α (PDGFRA). This is important clinically because imatinib is usually an effective therapy in patients with PDGFRA fusion genes and leads to complete hematologic and molecular remission in a high proportion of cases 1.
During the past 2 decades, several different classifications of eosinophilic disorders have been proposed in various fields of medicine 9. Although respective criteria and definitions partially overlap, no multidisciplinary global consensus has been developed. In addition, the recent identification of several new molecular and immunologic mechanisms lends greater understanding to the disorders and therefore to logical taxonomy.
Table 1. Definition of hypereosinophilia and hypereosinophilic syndrome
| Proposed term | Proposed abbreviation | Definition and criteria |
| Blood eosinophilia | — | >0.5 Eosinophils × 109/L blood |
| Hypereosinophilia | HE | >1.5 Eosinophils × 109/L blood on 2 examinations (interval ≥1 month*) and/or tissue hypereosinophilia defined by the following†:
|
| Hypereosinophilic syndrome | HES |
|
| Eosinophil-associated single-organ diseases |
|
Footnotes:
*In the case of evolving life-threatening end-organ damage, the diagnosis can be made immediately to avoid delay in therapy.
†Validated quantitative criteria for tissue hypereosinophilia do not exist for most tissues at the present time. Consequently, tissue hypereosinophilic syndrome is defined by a combination of qualitative and semiquantitative findings that will require revision as new information becomes available.
‡Hypereosinophilia-related organ damage (damage attributable to hypereosinophilia): organ dysfunction with marked tissue eosinophil infiltrates and/or extensive deposition of eosinophil-derived proteins (in the presence or absence of marked tissue eosinophils) and 1 or more of the following: (1) fibrosis (lung, heart, digestive tract, skin, and others); (2) thrombosis with or without thromboembolism; (3) cutaneous (including mucosal) erythema, edema/angioedema, ulceration, pruritus, and eczema; and (4) peripheral or central neuropathy with chronic or recurrent neurologic deficit. Less commonly, other organ system involvement (liver, pancreas, kidney, and other organs) and the resulting organ damage can be judged as hypereosinophilia-related pathology, so that the clinician concludes the clinical situation resembles hypereosinophilic syndrome. Note that hypereosinophilic syndrome can manifest in 1 or more organ systems.
Table 2. Classification of hypereosinophilia
| Proposed terminology | Proposed abbreviation | Pathogenesis/definition |
| Hereditary (familial) hypereosinophilia | HEFA | Pathogenesis unknown; familial clustering, no signs or symptoms of hereditary immunodeficiency, and no evidence of a reactive or neoplastic condition/disorder underlying hypereosinophilia |
| HE of undetermined significance | HEUS | No underlying cause of hypereosinophilia, no family history, no evidence of a reactive or neoplastic condition/disorder underlying hypereosinophilia, and no end-organ damage attributable to hypereosinophilia |
| Primary (clonal/neoplastic) hypereosinophilia† | HEN | Underlying stem cell, myeloid, or eosinophilic neoplasm, as classified by World Health Organization (WHO) criteria; eosinophils considered neoplastic cells* |
| Secondary (reactive) hypereosinophilia† | HER | Underlying condition/disease in which eosinophils are considered nonclonal cells*; hypereosinophilia considered cytokine driven in most cases‡ |
Footnotes:
*Clonality of eosinophils is often difficult to demonstrate or is not examined. However, if a myeloid or stem cell neoplasm known to present typically with clonal hypereosinophilia is present or a typical molecular defect is demonstrable (eg, PDGFR or FGFR mutations or BCR/ABL1), eosinophilia should be considered clonal.
†HEN and HER are prediagnostic checkpoints that should guide further diagnostic evaluations but cannot serve as final diagnoses.
‡In a group of patients, HER might be caused/triggered by other as yet unknown processes because no increase in eosinophilopoietic cytokine levels can be documented.
Table 3. Classification of syndromes and conditions accompanied by hypereosinophilia
| Variant | Typical findings |
| Hypereosinophilic syndrome* | |
| Idiopathic hypereosinophilic syndrome | No underlying cause of hypereosinophilia, no evidence of a reactive or neoplastic condition/disorder underlying hypereosinophilia and end-organ damage attributable to hypereosinophilia |
| Primary (neoplastic) hypereosinophilic syndrome (HESN) | |
| Secondary (reactive) hypereosinophilic syndrome (HESR) | Underlying stem cell, myeloid, or eosinophilic neoplasm classified according to World Health Organization (WHO) guidelines and end-organ damage attributable to hypereosinophilia, and eosinophils are considered (or shown) neoplastic (clonal) cells.† |
| Underlying condition/disease in which eosinophils are considered nonclonal cells; hypereosinophilia is considered cytokine driven, and end-organ damage is attributable to hypereosinophilia. | |
| Subvariant: lymphoid variant HES (clonal T cells identified as the only potential cause)‡ | |
| Other conditions and syndromes | |
| Specific syndromes accompanied by hypereosinophilia | Specific syndromes in which the effect of eosinophilia remains unclear but the clinical presentation is distinct and accompanied by hypereosinophilia. |
| Other conditions accompanied by hypereosinophilia | Mostly organ-restricted conditions in which the effect of eosinophilia remains unclear. |
Footnotes:
*Hypereosinophilic syndrome is defined as blood hypereosinophilia with (plus) end-organ damage attributable to tissue hypereosinophilia.
†Clonality of eosinophils is often difficult to demonstrate or is not examined. However, if a myeloid or stem cell neoplasm known to present typically with clonal hypereosinophilia is present or a typical molecular defect is demonstrable (eg, PDGFR or FGFR mutations or BCR/ABL1), eosinophilia should be considered clonal.
‡The lymphoid variant of hypereosinophilic syndrome is regarded as a special form of secondary hypereosinophilic syndrome by several experts, although its exact nature and pathogenesis remain controversial.
Table 4. Syndromes associated with hypereosinophilia

Abbreviation: HE = hypereosinophilia; HES = hypereosinophilic syndrome
[Source 10 ]Table 5. Inflammatory conditions associated with hypereosinophilia

Abbreviation: HE = hypereosinophilia; HES = hypereosinophilic syndrome
[Source 10 ]Table 6. Dermatologic diseases with hypereosinophilia

Hypereosinophilia causes
Hypereosinophilia is associated with a variety of conditions, including allergic, infectious, and neoplastic disorders. Causes of hypereosinophilia may be classified as familial or acquired 11. Familial hypereosinophilia is an autosomal dominant disorder with a stable eosinophil count and a benign clinical course. Acquired hypereosinophilia is further divided into secondary, clonal, and idiopathic eosinophilia 12.
Secondary hypereosinophilia
Secondary hypereosinophilia is a cytokine-derived (interleukin-5 [IL-5]) reactive phenomenon. Worldwide, parasitic diseases are the most common cause, whereas in developed countries, allergic diseases and atopic diseases are the most common cause 13. Other causes include the following:
- Malignancies – Metastatic cancer, T-cell lymphoma 14, colon cancer
- Pulmonary hypereosinophilia – Loffler syndrome 15, Churg-Strauss syndrome, allergic bronchopulmonary aspergillosis
- Connective tissue disorders – Scleroderma, polyarteritis nodosa
- Skin diseases – Dermatitis herpetiformis
- Inflammatory bowel disease
- Sarcoidosis
- Addison disease
- Drug reactions (allergic or toxic)
Clonal hypereosinophilia
Clonal hypereosinophilia is diagnosed by bone marrow histology, cytogenetics, and molecular genetics. Causes include the following:
- Acute leukemia – Pre-B acute lymphoblastic leukemia (ALL), acute myeloid leukemia M4 with bone marrow hypereosinophilia (AML-M4Eo)
- Chronic myeloid disorders
Molecularly defined disorders include the following:
- BCR-ABL chronic myeloid leukemia
- PDGFRA (platelet-derived growth factor receptor, alpha polypeptide)–rearranged hypereosinophilia – Systemic mastocytosis–chronic eosinophilic leukemia (SM-CEL)
- PDGFRβ-rearranged hypereosinophilia
- KIT-mutated systemic mastocytosis
Clinicopathologically assigned disorders include the following:
- Myelodysplastic syndrome (MDS)
- Myeloproliferative disorders (MPDs) – Classic myeloproliferative disorder (polycythemia) and atypical myeloproliferative disorder (chronic eosinophilic leukemia, systemic mastocytosis, chronic myelomonocytic leukemia)
Idiopathic hypereosinophilia
Idiopathic hypereosinophilia is a diagnosis of exclusion when secondary and clonal causes of hypereosinophilia are excluded. Hypereosinophilic syndrome is a subset of idiopathic hypereosinophilia characterized by persistent hypereosinophilia (absolute eosinophil counts above 1,500 eosinophils/μL) of longer than 6 months’ duration associated with organ damage. However, long-term follow-up and X-linked clonality studies indicate that at least some patients with hypereosinophilic syndrome have an underlying clonal myeloid malignancy or a clonal or phenotypically abnormal T-cell population, suggesting a true secondary process.
The literature now favors the view that cases of idiopathic hypereosinophilic syndrome with FIP1L1 indeed represent chronic eosinophilic leukemia, because these patients have a molecular genetic abnormality, specifically an FIP1L1–PDGFRA fusion gene 16. In addition, there are documented cases of acute transformation to either acute myeloid leukemia (AML) or granulocytic sarcoma in some cases of hypereosinophilic syndrome after an interval as long as 24 years. In such cases, a diagnosis of chronic eosinophilic leukemia is made in retrospect when acute transformation provides indirect evidence that the condition was likely to have been a clonal, neoplastic, myeloproliferative disorder from the beginning.
In addition, some patients with hypereosinophilic syndrome present with features typical of myeloproliferative disorders, such as hepatosplenomegaly, the presence of leukocyte precursors in the peripheral blood, increased alkaline phosphatase level, chromosomal abnormalities, and reticulin fibrosis. Cytogenetic studies in such cases may be normal, but molecular genetic studies may show aberrations.
The best-described aberration is the interstitial deletion on chromosome 4q12, resulting in fusion of the 5’ portion of the FIP1L1 gene to the 3’ portion of the PDGFRA gene. This fusion gene encodes for the FIP1L1–PDGFR alpha protein, the constitutively activated tyrosine kinase activity that induces hypereosinophilia. The prevalence of such a mutation is 0.4% in unselected cases of hypereosinophilia, but it can be as high as 12–88% in cohorts that meet the World Health Organization (WHO) criteria for idiopathic hypereosinophilic syndrome, particularly those with features of myeloproliferative disorder (increased levels of tryptase and mast cells in the bone marrow).
Patients with hypereosinophilic syndrome with the PDGFRA mutation have a very high incidence of cardiac involvement and carry a poor prognosis without therapy. Fortunately, the results of imatinib therapy in such cases of hypereosinophilic syndrome are very encouraging.
The other subset of idiopathic hypereosinophilia, hypereosinophilic syndrome with clonal or immunophenotypically aberrant T-cells, is associated with increased secretion of IL-5 and cutaneous manifestations. Simon et al reported immunophenotypic abnormality in 16 of 60 patients with hypereosinophilic syndrome 17. Moreover, nine patients had CD3+CD4+CD8- T cells, three had CD3+CD4-CD8+ cells, three had CD3+CD4-CD8- cells, and two had CD3-CD4+ cells (one patient had two distinct populations). Progression to T-cell lymphoma was observed in this subset of patients with hypereosinophilic syndrome, particularly those with the CD3-CD4+ phenotypes 18.
Chronic eosinophilic leukemia
Chronic eosinophilic leukemia is caused by autonomous proliferation of clonal eosinophilic precursors. Simplified criteria for the diagnosis of chronic eosinophilic leukemia include the following:
- Eosinophil count of at least 1500/µL
- Peripheral blood blast count of >2% and a bone marrow blast cell count that is >5% but < 19% of all nucleated cells
- Criteria for atypical chronic myelogenous leukemia (CML), chronic myelomonocytic leukemia, and chronic granulocytic leukemia (BCR-ABL–positive CML) are not met
- Myeloid cells are demonstrated to be clonal (eg, by detection of clonal cytogenetic abnormality or by demonstration of a very skewed expression of X chromosome genes)
Some of the cytogenetic abnormalities that have been described in chronic eosinophilic leukemia include t(5:12) and t(8:13), and molecular genetic abnormalities include the FIP1L1-PDGFRA fusion gene and ETV6-PDGFRβ.
Hypereosinophilia pathophysiology
Eosinophil production is governed by several cytokines, including IL-3, IL-5, and granulocyte-macrophage colony-stimulating factor (GM-CSF). IL-5 appears to be the most important cytokine that is responsible for differentiation of the eosinophil line 11.
Unlike neutrophils, eosinophils can survive in the tissues for weeks. Their survival in tissues depends on the sustained presence of cytokines. Only eosinophils and basophils and their precursors have receptors for IL-3, IL-5, and GM-CSF. In vitro, eosinophils survive less than 48 hours in the absence of cytokines.
Eosinophil granules contain toxic cationic proteins, which are the primary mediators of tissue damage. These toxins include major basic protein, eosinophil peroxidase, eosinophil-derived neurotoxin, and eosinophil cationic protein. The latter two are ribonucleases. Free radicals produced by the eosinophilic peroxidase and the respiratory burst oxidative pathway of the infiltrating eosinophils further enhance the damage.
Eosinophils amplify the inflammatory cascade by secreting chemoattractants that recruit more eosinophils. Such chemoattractants include the following:
- Eotaxin
- Platelet-activating factor
- The cytokine RANTES (regulated upon activation, normal T cell expressed, and secreted).
Several mechanisms have been proposed for the pathogenesis of hypereosinophilic syndrome, including overproduction of eosinophilopoietic cytokines, their enhanced activity, and defects in the normal suppressive regulation of eosinophilopoiesis. Organ damage induced by hypereosinophilic syndrome is due to the eosinophilic infiltration of the tissues accompanied by the mediator release from the eosinophil granules. Hence, the level of eosinophilia is not a true reflection of organ damage.
The most serious complication of hypereosinophilic syndrome is cardiac involvement, which can result in myocardial fibrosis, chronic heart failure and death. The mechanisms of cardiac damage are not entirely understood, but the damage is marked by severe endocardial fibrotic thickening of either or both ventricles, resulting in restrictive cardiomyopathy due to inflow obstruction.
Hypereosinophilic syndrome
Hypereosinophilic syndrome refers to a group of rare blood disorders characterized by persistent peripheral blood eosinophilia of 1.5 ×109/L (1,500 eosinophils/μL) or higher with evidence of end organ involvement attributable to the eosinophilia and not otherwise explained in the clinical setting 19. In 1975, Chusid et al 20 defined the three features required for a diagnosis of hypereosinophilic syndrome:
- A sustained absolute eosinophil count greater than >1500/µL, which persists for longer than 6 months
- No evidence of parasites, allergy or other known causes of an elevated eosinophil count.
- Signs and symptoms of organ involvement.
Most people have less than 500 eosinophils/microliter (500 eosinophils/μL) in their blood. People with hypereosinophilic syndrome usually have more than 1,500 eosinophils/microliter (1.5 ×109/L) in their blood for 6 months or more and the cause cannot be identified. These eosinophils make their way into various tissues, causing inflammation and eventually organ dysfunction.
Hypereosinophilic syndrome signs and symptoms vary significantly based on which parts of the body are affected. Although any organ system can be involved in hypereosinophilic syndrome, the skin, lungs, digestive tract, heart, blood and central nervous system (brain and spinal cord) are the most commonly affected. Hypereosinophilic syndrome was originally thought to be “idiopathic” or of unknown cause. However, recent advances in diagnostic testing have allowed a cause to be identified in approximately a quarter of cases (e.g., allergic, rheumatologic, infectious, and neoplastic disorders). Moreover, the cause of the eosinophilia in hypereosinophilic syndromes can be primary (myeloid), secondary (lymphocyte-driven), or unknown. Some varieties of hypereosinophilic syndrome tend to run in families. Other types have been associated with certain types of cancers, infections or other health problems.
Hypereosinophilic syndrome can affect anyone. But it occurs more often in men (90%), usually between the ages of 20 and 50.
Hypereosinophilic syndrome usually presents with fever, weight loss, fatigue, and rash. A rash is present in over 50% of patients, but is non-specific in appearance. Most commonly, red swollen itchy nodules (lumps) have been described. It can also resemble urticaria (hives). An enlarged liver and spleen is often present indicating liver and spleen involvement. The lungs, kidneys, heart and nervous system can be affected.
Symptoms of hypereosinophilic syndrome are also common in many other medical problems, making an initial diagnosis more difficult. Some patients have an underlying blood disorder present, most commonly a form of leukemia. Leukemia may also be diagnosed up to 9-12 years after the initial diagnosis of hypereosinophilic syndrome.
Untreated, hypereosinophilic syndrome can become life-threatening. Hypereosinophilic syndrome treatment varies based on the severity of the condition and whether or not an underlying cause has been identified but generally includes imatinib or corticosteroids as an initial treatment 21. Approximately 70% of patients respond to high dose oral corticosteroid therapy with oral prednisone, with the eosinophilia returning to normal levels. Although corticosteroids remain the first-line therapy for most forms of hypereosinophilic syndromes, the availability of an increasing number of novel therapeutic agents, including tyrosine kinase inhibitors and monoclonal antibodies, has necessarily altered the approach to treatment of hypereosinophilic syndromes.
The overall 5-year survival for patients with hypereosinophilic syndrome is 80% and the cause of death is usually heart failure.
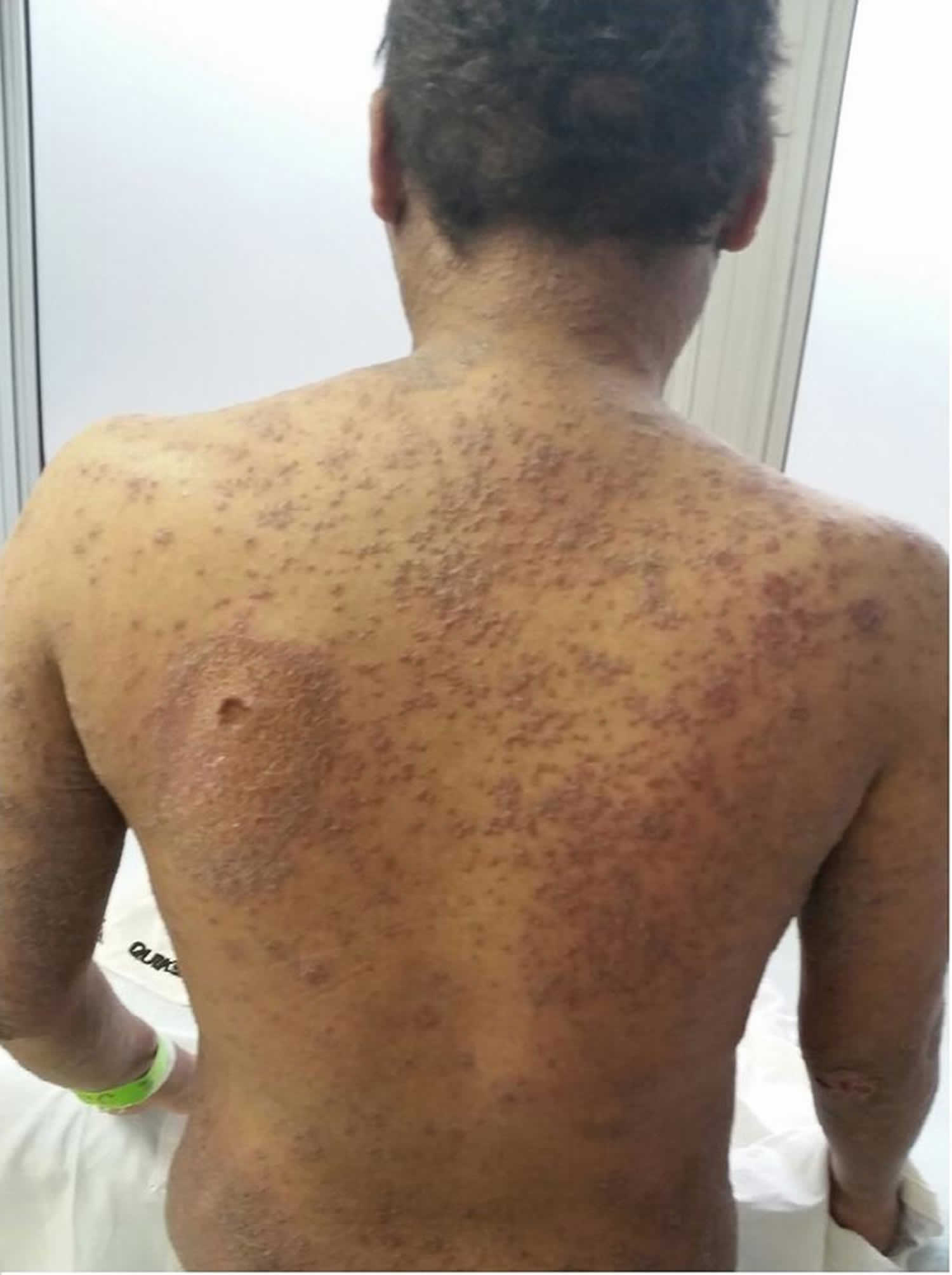
Figure 1. Hypereosinophilic syndrome skin
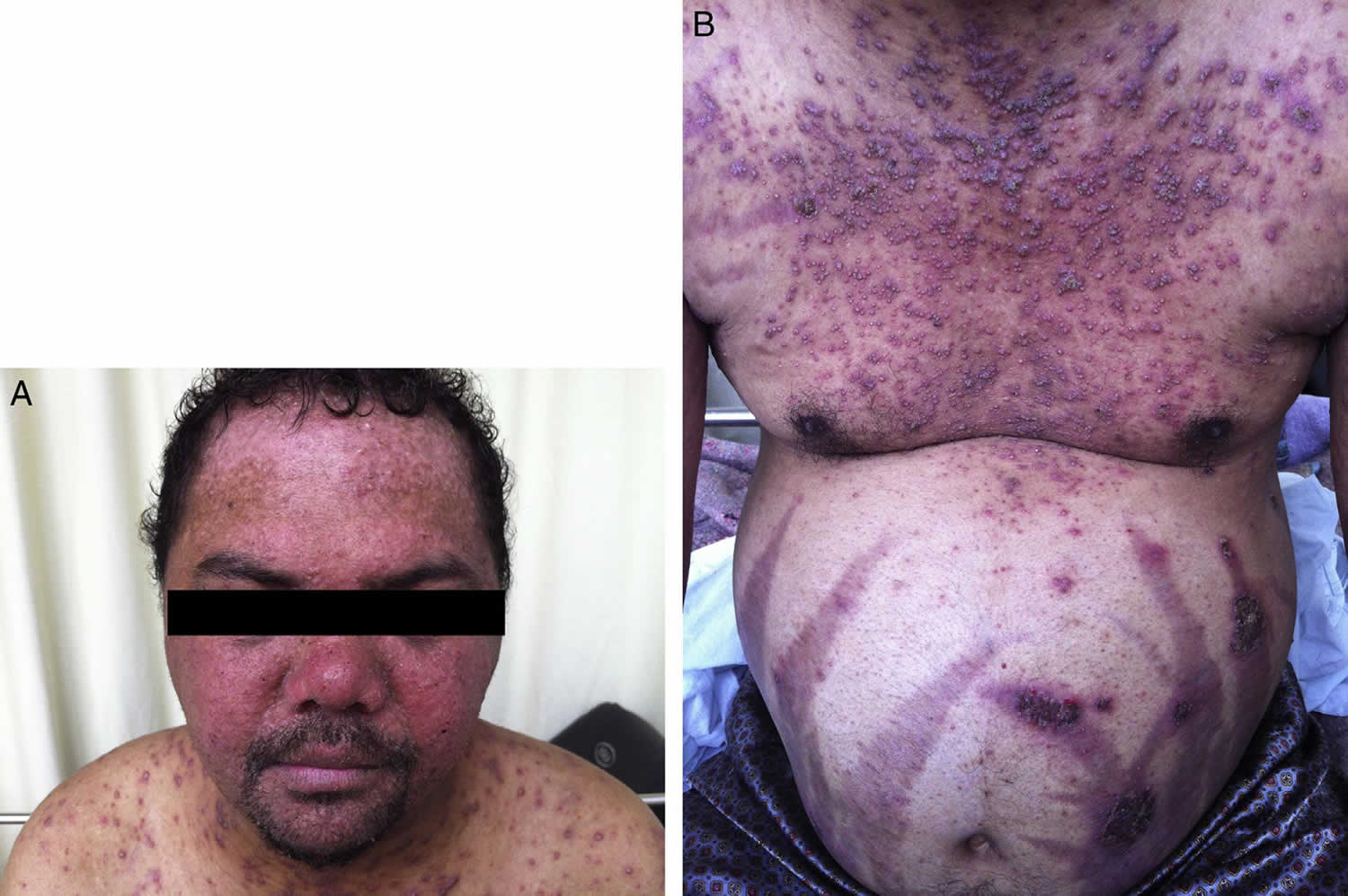
Footnote: A 30-year-old male patient presented to the emergency department with dyspnea, fever and productive cough for three days. He had been taking 20 mg of prednisone for 20 years. On this occasion, the patient was treated for streptococcal glomerulonephritis. After discharge, he did not return and was lost to follow-up. The patient also had pruritic skin lesions since ten years of age, which used to erupt, vanish and reappear spontaneously in two-week intervals. After prednisone was started, the frequency of the lesions was related to its intake, as the frequency of lesion reappearance was much higher without prednisone.
[Source 22 ]Hypereosinophilic syndrome causes
The cause of hypereosinophilic syndrome is not known. Hypereosinophilia is associated with a variety of conditions, including allergic, infectious, and neoplastic disorders. Causes of hypereosinophilia may be classified as familial or acquired 11. Familial hypereosinophilia is an autosomal dominant disorder with a stable eosinophil count and a benign clinical course 21. In autosomal dominant conditions, an affected person only needs a change (mutation) in one copy of the responsible gene in each cell. In some cases, an affected person inherits the mutation from an affected parent. Other cases may result from new (de novo) mutations in the gene. These cases occur in people with no history of the disorder in their family. A person with an autosomal dominant condition has a 50% chance with each pregnancy of passing along the altered gene to his or her child.
Acquired hypereosinophilia is further divided into secondary, clonal, and idiopathic eosinophilia 12.
Secondary hypereosinophilia
Secondary hypereosinophilia is a cytokine-derived (interleukin-5 [IL-5]) reactive phenomenon. Worldwide, parasitic diseases are the most common cause, whereas in developed countries, allergic diseases and atopic diseases are the most common cause 13. Other causes include the following:
- Malignancies – Metastatic cancer, T-cell lymphoma 14, colon cancer
- Pulmonary hypereosinophilia – Loffler syndrome 15, Churg-Strauss syndrome, allergic bronchopulmonary aspergillosis
- Connective tissue disorders – Scleroderma, polyarteritis nodosa
- Skin diseases – Dermatitis herpetiformis
- Inflammatory bowel disease
- Sarcoidosis
- Addison disease
- Drug reactions (allergic or toxic)
Clonal hypereosinophilia
Clonal hypereosinophilia is diagnosed by bone marrow histology, cytogenetics, and molecular genetics. Causes include the following:
- Acute leukemia – Pre-B acute lymphoblastic leukemia (ALL), acute myeloid leukemia M4 with bone marrow hypereosinophilia (AML-M4Eo)
- Chronic myeloid disorders
Molecularly defined disorders include the following:
- BCR-ABL chronic myeloid leukemia
- PDGFRA (platelet-derived growth factor receptor, alpha polypeptide)–rearranged hypereosinophilia – Systemic mastocytosis–chronic eosinophilic leukemia (SM-CEL)
- PDGFRβ-rearranged hypereosinophilia
- KIT-mutated systemic mastocytosis
Clinicopathologically assigned disorders include the following:
- Myelodysplastic syndrome (MDS)
- Myeloproliferative disorders (MPDs) – Classic myeloproliferative disorder (polycythemia) and atypical myeloproliferative disorder (chronic eosinophilic leukemia, systemic mastocytosis, chronic myelomonocytic leukemia)
Idiopathic hypereosinophilia
Idiopathic hypereosinophilia is a diagnosis of exclusion when secondary and clonal causes of hypereosinophilia are excluded. Hypereosinophilic syndrome is a subset of idiopathic hypereosinophilia characterized by persistent hypereosinophilia (absolute eosinophil counts above 1,500 eosinophils/μL) of longer than 6 months’ duration associated with organ damage. However, long-term follow-up and X-linked clonality studies indicate that at least some patients with hypereosinophilic syndrome have an underlying clonal myeloid malignancy or a clonal or phenotypically abnormal T-cell population, suggesting a true secondary process.
The literature now favors the view that cases of idiopathic hypereosinophilic syndrome with FIP1L1 indeed represent chronic eosinophilic leukemia, because these patients have a molecular genetic abnormality, specifically an FIP1L1–PDGFRA fusion gene 16. In addition, there are documented cases of acute transformation to either acute myeloid leukemia (AML) or granulocytic sarcoma in some cases of hypereosinophilic syndrome after an interval as long as 24 years. In such cases, a diagnosis of chronic eosinophilic leukemia is made in retrospect when acute transformation provides indirect evidence that the condition was likely to have been a clonal, neoplastic, myeloproliferative disorder from the beginning.
In addition, some patients with hypereosinophilic syndrome present with features typical of myeloproliferative disorders, such as hepatosplenomegaly, the presence of leukocyte precursors in the peripheral blood, increased alkaline phosphatase level, chromosomal abnormalities, and reticulin fibrosis. Cytogenetic studies in such cases may be normal, but molecular genetic studies may show aberrations.
The best-described aberration is the interstitial deletion on chromosome 4q12, resulting in fusion of the 5’ portion of the FIP1L1 gene to the 3’ portion of the PDGFRA gene. This fusion gene encodes for the FIP1L1–PDGFR alpha protein, the constitutively activated tyrosine kinase activity that induces hypereosinophilia. The prevalence of such a mutation is 0.4% in unselected cases of hypereosinophilia, but it can be as high as 12–88% in cohorts that meet the World Health Organization (WHO) criteria for idiopathic hypereosinophilic syndrome, particularly those with features of myeloproliferative disorder (increased levels of tryptase and mast cells in the bone marrow).
Patients with hypereosinophilic syndrome with the PDGFRA mutation have a very high incidence of cardiac involvement and carry a poor prognosis without therapy. Fortunately, the results of imatinib therapy in such cases of hypereosinophilic syndrome are very encouraging.
The other subset of idiopathic hypereosinophilia, hypereosinophilic syndrome with clonal or immunophenotypically aberrant T-cells, is associated with increased secretion of IL-5 and cutaneous manifestations. Simon et al reported immunophenotypic abnormality in 16 of 60 patients with hypereosinophilic syndrome 17. Moreover, nine patients had CD3+CD4+CD8- T cells, three had CD3+CD4-CD8+ cells, three had CD3+CD4-CD8- cells, and two had CD3-CD4+ cells (one patient had two distinct populations). Progression to T-cell lymphoma was observed in this subset of patients with hypereosinophilic syndrome, particularly those with the CD3-CD4+ phenotypes 18.
Chronic eosinophilic leukemia
Chronic eosinophilic leukemia is caused by autonomous proliferation of clonal eosinophilic precursors. Simplified criteria for the diagnosis of chronic eosinophilic leukemia include the following:
- Eosinophil count of at least 1500/µL
- Peripheral blood blast count of >2% and a bone marrow blast cell count that is >5% but < 19% of all nucleated cells
- Criteria for atypical chronic myelogenous leukemia (CML), chronic myelomonocytic leukemia, and chronic granulocytic leukemia (BCR-ABL–positive CML) are not met
- Myeloid cells are demonstrated to be clonal (eg, by detection of clonal cytogenetic abnormality or by demonstration of a very skewed expression of X chromosome genes)
Some of the cytogenetic abnormalities that have been described in chronic eosinophilic leukemia include t(5:12) and t(8:13), and molecular genetic abnormalities include the FIP1L1-PDGFRA fusion gene and ETV6-PDGFRβ.
Hypereosinophilic syndrome symptoms
Hypereosinophilic syndrome can happen at any age, although it is more common in men (90%), usually between the ages of 20 and 50. People with hypereosinophilic syndrome may suffer from a wide variety of symptoms, depending upon which parts of the body are affected. Early symptoms of hypereosinophilic syndrome may include fatigue, cough, breathlessness, muscle pain, rash and fever.
Hypereosinophilic syndrome symptoms include:
- Skin – rashes, itching, and edema.
- Lung – asthma, cough, difficulty breathing, shortness of breath, recurrent upper respiratory infections, and pleural effusion.
- Gastrointestinal – mouth sores, abdominal pain, vomiting, and diarrhea.
- Musculoskeletal – arthritis, muscle inflammation, muscle aches, and joint pain.
- Nervous system – memory loss, confusion, vertigo, dizziness, paresthesia, speech impairment, and visual disturbances.
- Heart – congestive heart failure, cardiomyopathy, pericardial effusion, and myocarditis.
- Blood – deep venous thrombosis, and anemia.
Affected people can also experience a variety of non-specific symptoms such as fever, weight loss, night sweats and fatigue 21.
Hypereosinophilic syndrome diagnosis
Symptoms of hypereosinophilic syndrome are also common in many other medical problems, making an initial diagnosis more difficult. Many types of disorders can raise your eosinophil level, including parasitic infection, allergic disease, cancers, autoimmune diseases and reactions to medications. When trying to determine whether you have hypereosinophilic syndrome, your doctor is likely to ask about your travel history and any medications you’re taking, to help rule out these other causes.
Testing is individualized according to symptoms and may include stool evaluation to detect parasitic infection, allergy testing to diagnose environmental or food allergies, biopsies of the skin or other organs, blood tests to screen for autoimmunity or CT imaging of affected organs, molecular genetic studies to detect FIP1L1-PDGFRA or other mutations to help determine, diagnosis, prognosis, and treatment.
When diagnosed with hypereosinophilic syndrome, it is important to determine the extent of organ damage. A chest x-ray and echocardiogram are routinely performed to evaluate the heart and lungs. Other tests often performed in hypereosinophilic syndrome patients include liver and kidney function, serum vitamin B12 levels, erythrocyte sedimentation rate (ESR) and serum tryptase levels.
Laboratory tests
Your doctor may also need information from some of the following lab tests:
- Blood tests, to detect autoimmune conditions, parasitic infections, or problems with your liver or kidneys
- Allergy tests, to detect environmental or food allergies
- Stool tests, to detect parasitic infections such as hookworm
- Genetic test, to check for a gene mutation that can cause hypereosinophilic syndrome
Imaging tests
Imaging tests may include:
- X-rays, to check the condition of your lungs
- CT scan, to detect problems in the chest, abdomen and pelvis
- Echocardiogram or MRI, to assess heart function
Hypereosinophilic syndrome treatment
Treatment for hypereosinophilic syndrome is aimed at reducing your eosinophil count to prevent tissue damage, especially to your heart. Specific treatment depends on your symptoms, the severity of your condition and the cause of your hypereosinophilic syndrome.
If you have no symptoms and your eosinophil count is low enough, you might require no treatment other than close monitoring for any changes related to hypereosinophilic syndrome.
Standard hypereosinophilic syndrome treatment includes corticosteroid medications such as prednisone, and chemotherapeutic agents such as hydroxyurea, chlorambucil and vincristine. Interferon-alpha may also be used as a treatment. This medication must be administered by frequent injections.
Research is uncovering new therapies for hypereosinophilic syndrome. One new approach for controlling malignant cell growth is the use of tyrosine kinase inhibitors such as Gleevec (imatinib). Monoclonal antibody therapy, such as alemtuzumab (anti-CD52) has also shown promise for treatment of hypereosinophilic syndrome. In fact, based on a recent phase III randomized, placebo-controlled trial 23, the monocloncal antibody Nucala or mepolizumab (anti-IL-5) has just received approval by the U.S. Food and Drug Administration (FDA) for treatment of adults and children aged 12 years and older with hypereosinophilic syndrome for six months or longer without another identifiable non-blood related cause of the disease.
Medications
Systemic corticosteroids, such as prednisone, are the first line treatment. Other treatment options include:
- Hydroxyurea (Droxia, Hydrea, Siklos)
- Imatinib (Gleevec)
- Vincristine
Because hypereosinophilic syndrome can increase your risk of blood clots, you might also be prescribed blood-thinning medications such as warfarin (Coumadin).
Surgery and other procedures
If nothing else has worked, your doctor might suggest a stem cell or bone marrow transplant.
Hypereosinophilic syndrome prognosis
The prognosis in hypereosinophilic syndrome depends on the organ systems involved, disease severity and response to therapy. Outcomes can vary greatly from one person to the next. There is no cure. In very severe cases, hypereosinophilic syndrome may be fatal, but there is hope. Survival rates have improved greatly. In 1975, only 12% of hypereosinophilic syndrome patients survived three years 20. Today management of cardiovascular disease by early echocardiographic monitoring and advances in medical and surgical therapies have improved the overall survival with more than 80% of hypereosinophilic syndrome patients survive five years or more 24. A study of 40 cases by Lefebcve et al 25 showed a 5-year survival of 80% and a 15-year survival of 42%.
The availability of tyrosine kinase inhibitors such as imatinib, which prevent progression of cardiac disease and other organ damage—particularly in FIP1L1/PGDFRA–positive cases—will likely further improve the prognosis of hypereosinophilic syndrome. However, FIP1L1/PGDFRA– negative cases of hypereosinophilic syndrome that are resistant to corticosteroids have not been shown to have a durable response to imatinib.
Lastly, additional insight into the molecular pathogenesis of such cases of hypereosinophilic syndrome is required to develop effective targeted therapies.
Features that indicate a favorable prognosis in hypereosinophilic syndrome include the following:
- Angioedema
- Urticaria
- Elevated serum IgE level
- Sustained response to corticosteroids
- Early diagnosis and intensive management
The presence of features that are suggestive of myeloproliferative disorder and leukocytosis greater than 90,000/μ L carry a worse prognosis in hypereosinophilic syndrome.
Hypereosinophilic syndrome life expectancy
Hypereosinophilic syndrome is a chronic and progressive disorder that is potentially fatal. Blast transformation could occur after many years. True idiopathic hypereosinophilic syndrome is generally indolent, but patients with characteristics suggestive of a myeloproliferative/neoplastic disorder and those who develop chronic heart failure have a worse prognosis.
An older review of 57 patients with advanced hypereosinophilic syndrome reported a mean survival of 9 months and a 3-year survival rate of 12% 20. A later analysis from France noted an 80% survival at 5 years and a 42% survival at 15 years among 40 patients with hypereosinophilic syndrome 25.
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e9. doi:10.1016/j.jaci.2012.02.019 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4091810[↩][↩][↩][↩][↩]
- Bellamy GJ, Hinchliffe RF, Crawshaw KC, Finn A, Bell F. Total and differential leucocyte counts in infants at 2, 5 and 13 months of age. Clin Lab Haematol. 2000 Apr;22(2):81-7. doi: 10.1046/j.1365-2257.2000.00288.x[↩]
- Burris D, Rosenberg CE, Schwartz JT, et al. Pediatric Hypereosinophilia: Characteristics, Clinical Manifestations, and Diagnoses. J Allergy Clin Immunol Pract. 2019;7(8):2750-2758.e2. doi:10.1016/j.jaip.2019.05.011 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6842676[↩]
- Schwartz JT, Fulkerson PC. An Approach to the Evaluation of Persistent Hypereosinophilia in Pediatric Patients. Front Immunol. 2018;9:1944. Published 2018 Sep 3. doi:10.3389/fimmu.2018.01944 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6130221[↩]
- Bain BJ, Fletcher SH. Chronic eosinophilic leukemias and the myeloproliferative variant of the hypereosinophilic syndrome. Immunol Allergy Clin North Am. 2007 Aug;27(3):377-88. doi: 10.1016/j.iac.2007.06.001[↩]
- Tefferi A, Patnaik MM, Pardanani A. Eosinophilia: secondary, clonal and idiopathic. Br J Haematol. 2006 Jun;133(5):468-92. doi: 10.1111/j.1365-2141.2006.06038.x[↩]
- Hogan SP, Rosenberg HF, Moqbel R, Phipps S, Foster PS, Lacy P, Kay AB, Rothenberg ME. Eosinophils: biological properties and role in health and disease. Clin Exp Allergy. 2008 May;38(5):709-50. doi: 10.1111/j.1365-2222.2008.02958.x[↩]
- Tefferi A, Skoda R, Vardiman JW. Myeloproliferative neoplasms: contemporary diagnosis using histology and genetics. Nat Rev Clin Oncol. 2009 Nov;6(11):627-37. doi: 10.1038/nrclinonc.2009.149[↩]
- Gotlib J. Eosinophilic myeloid disorders: new classification and novel therapeutic strategies. Curr Opin Hematol. 2010 Mar;17(2):117-24. doi: 10.1097/MOH.0b013e3283366c70[↩]
- Valent P, Klion AD, Horny HP, et al. Contemporary consensus proposal on criteria and classification of eosinophilic disorders and related syndromes. J Allergy Clin Immunol. 2012;130(3):607-612.e9. doi:10.1016/j.jaci.2012.02.019 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4091810/bin/NIHMS598504-supplement-supplement_1.pdf[↩][↩][↩]
- Wardlaw AJ. Eosinophils and Related Disorders. Kaushansky K, Lichtman MA, Prchal JT, Levi MM, Press OW, Burns LJ, Caligiuri MA, eds. Williams Hematology. 9th ed. New York, NY: McGraw-Hill Education; 2016. 947-64.[↩][↩][↩]
- Gleich GJ, Leiferman KM. The hypereosinophilic syndromes: current concepts and treatments. Br J Haematol. 2009 May. 145(3):271-85.[↩][↩]
- Curtis C, Ogbogu P. Hypereosinophilic Syndrome. Clin Rev Allergy Immunol. 2016 Apr. 50 (2):240-51.[↩][↩]
- Helbig G, Wieczorkiewicz A, Dziaczkowska-Suszek J, Majewski M, Kyrcz-Krzemien S. T-cell abnormalities are present at high frequencies in patients with hypereosinophilic syndrome. Haematologica. 2009 Sep. 94(9):1236-41.[↩][↩]
- Cincin AA, Ozben B, Tanrikulu MA, Baskan O, Agirbasli M. Large apical thrombus in a patient with persistent heart failure and hypereosinophilia: Löffler endocarditis. J Gen Intern Med. 2008 Oct. 23(10):1713-8.[↩][↩]
- Yamada Y, Sanchez-Aguilera A, Brandt EB, et al. FIP1L1/PDGFRalpha synergizes with SCF to induce systemic mastocytosis in a murine model of chronic eosinophilic leukemia/hypereosinophilic syndrome. Blood. 2008 Sep 15. 112(6):2500-7.[↩][↩]
- Simon HU, Plötz SG, Dummer R, Blaser K. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. 1999 Oct 7. 341(15):1112-20.[↩][↩]
- Brugnoni D, Airó P, Rossi G, et al. A case of hypereosinophilic syndrome is associated with the expansion of a CD3-CD4+ T-cell population able to secrete large amounts of interleukin-5. Blood. 1996 Feb 15. 87(4):1416-22.[↩][↩]
- Amy D. Klion; How I treat hypereosinophilic syndromes. Blood 2015; 126 (9): 1069–1077. doi: https://doi.org/10.1182/blood-2014-11-551614[↩]
- Chusid MJ, Dale DC, West BC, Wolff SM. The hypereosinophilic syndrome: analysis of fourteen cases with review of the literature. Medicine (Baltimore). 1975 Jan. 54(1):1-27.[↩][↩][↩]
- Noh HR, Magpantay GG. Hypereosinophilic syndrome. Allergy Asthma Proc. January 2017; 38(1):78-81.[↩][↩][↩]
- Queiroz Neto, Miguel Pedro de, & Gondim Filho, Fernando Antônio Galvão. (2017). Idiopathic hypereosinophilic syndrome with 20 years of diagnostic delay. Revista Brasileira de Hematologia e Hemoterapia, 39(2), 170-174. https://doi.org/10.1016/j.bjhh.2016.11.008[↩]
- Roufosse F, Kahn J-E, Rothenberg ME, Wardlaw AJ, Klion AD, Kirby SY, Gilson MJ, Bentley JH, Bradford ES, Yancey SW, Steinfeld J, Gleich GJ, Efficacy and safety of mepolizumab in hypereosinophilic syndrome: a Phase III, randomized, placebo-controlled trial, Journal of Allergy and Clinical Immunology (2020), doi: https://doi.org/10.1016/j.jaci.2020.08.037[↩]
- Hypereosinophilic Syndrome (HES). https://www.aaaai.org/conditions-and-treatments/related-conditions/hypereosinophilic-syndrome[↩]
- Lefebvre C, Bletry O, Degoulet P, et al. [Prognostic factors of hypereosinophilic syndrome. Study of 40 cases] [French]. Ann Med Interne (Paris). 1989. 140(4):253-7.[↩][↩]


{kind=link}