Joubert syndrome
Joubert syndrome, also called familial aplasia of the vermis, molar tooth midbrain-hindbrain malformation, agenesis of cerebellar vermis or cerebello-oculo-renal syndrome, is a rare autosomal recessive genetic neurodevelopmental disorder that affects the brain’s cerebellum and brainstem, leading to issues with balance and coordination such as ataxia (loss of muscle control), as well as other neurological problems such as low muscle tone (hypotonia), developmental delays and intellectual disability, abnormal eye movements (nystagmus), abnormal breathing patterns 1, 2, 3, 4, 5, 6, 7, 8. The key feature of Joubert syndrome is a specific brain malformation called the “molar tooth sign” on brain scans such as magnetic resonance imaging (MRI), indicating a malformed cerebellum and brainstem – consisting of cerebellar hypoplasia with vermian dysplasia, thick and horizontally oriented superior cerebellar peduncles, and an abnormally deep interpeduncular fossa (see Figure 3 below) 3, 9. The “molar tooth sign” results from the abnormal development of structures near the back of the brain, including the cerebellar vermis and the brainstem 9. The “molar tooth sign” got its name because the characteristic brain abnormalities resemble the cross-section of a molar tooth when seen on an MRI 6.
Joubert syndrome is a multisystem neurodevelopmental disorder that affects many parts of the body. The signs and symptoms of Joubert syndrome vary among affected individuals, even among members of the same family. Most infants with Joubert syndrome have low muscle tone (hypotonia) in infancy, which contributes to difficulty coordinating movements (ataxia) in early childhood. Other characteristic features of Joubert syndrome include episodes of unusually fast (hyperpnea) or slow (apnea) breathing in infancy, and abnormal eye movements (ocular motor apraxia) 9. Most affected individuals have delayed development and intellectual disability, which can range from mild to severe 9. Distinctive facial features can also occur in Joubert syndrome; these include a broad forehead, arched eyebrows, droopy eyelids (ptosis), widely spaced eyes (hypertelorism), low-set ears, and a triangle-shaped mouth 9.
Joubert syndrome can include a broad range of additional signs and symptoms. Joubert syndrome is sometimes associated with other eye abnormalities such as retinal dystrophy, which can cause vision loss, and coloboma, which is a gap or split in a structure of the eye, kidney disease including polycystic kidney disease and nephronophthisis, liver disease, skeletal abnormalities such as the presence of extra fingers and toes, or hormone (endocrine) problems 9. A combination of the characteristic features of Joubert syndrome and one or more of these additional signs and symptoms were previously referred to as Joubert syndrome and related disorders (JSRD). Now, however, any instances that involve the molar tooth sign on brain scans, including those with these additional signs and symptoms, are usually considered Joubert syndrome 9.
Joubert syndrome signs and symptoms:
- Low muscle tone (hypotonia) in infancy.
- Difficulty to coordinate voluntary muscle movements (ataxia).
- Breathing problems (hyperpnea, apnea).
- Abnormal eye movements (nystagmus, strabismus).
- Developmental delays and intellectual disabilities.
- Possible facial differences.
- Physical deformities (polydactyly, cleft lip or palate, and tongue abnormalities).
- Other symptoms may include kidney and liver disease, and skeletal abnormalities.
- Seizures.
Reliable epidemiological data on Joubert syndrome are lacking 10. Joubert syndrome is estimated to affect between 1 in 80,000 and 1 in 100,000 newborns 10. However, this estimate may be too low because Joubert syndrome has such a large range of possible features and is likely underdiagnosed 11, 3, 9.
About 94 percent of Joubert syndrome cases is caused by mutations in more than 40 genes 12, 13, 14, 15, 16, 17. These genetic mutations that cause Joubert syndrome are more common in certain ethnic groups, such as Ashkenazi Jewish, French-Canadian, and Hutterite populations. In the remaining cases, the genetic cause is unknown. In the Askhenazi Jews, the predicted prevalence of Joubert syndrome is increased up to 1 in 34,000 and 1 in 40,000 newborns due to the presence of the founder mutation p.R73L in the TMEM216 gene, with a carrier frequency of 1:90 to 1:100 18, 19. Similarly, in the Hutterite population, about 1:17 healthy subjects carry the founder mutation p.R18X in the TMEM237 gene, leading to a Joubert syndrome prevalence of about 1 in 1150 20.
Mutations in the genes associated with Joubert syndrome lead to problems with the structure and function of cell structures called primary cilia 21, 22, 23, 24. Primary cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in sensing the physical environment and in chemical signaling. Primary cilia are important for the structure and function of many types of cells, including brain cells (neurons) and certain cells in the kidneys and liver. Primary cilia are also necessary for the perception of sensory input, which is interpreted by the brain for sight, hearing, and smell. Defects in these cell structures can disrupt important chemical signaling pathways during development. Although researchers believe that defective primary cilia are responsible for most of the features of Joubert syndrome, it is not completely understood how they lead to specific developmental abnormalities.
The diagnosis of Joubert syndrome is based on physical symptoms and the “molar tooth sign” as seen on an MRI. Diagnostic criteria for Joubert syndrome continue to evolve but most experts agree that the neuroradiologic finding of the “molar tooth sign” is necessary to make the diagnosis. The diagnosis of Joubert syndrome is based on the presence of the following three primary criteria 25, 6, 26:
- The “molar tooth sign” based on MRI findings (hypoplasia of the cerebellar vermis)
- Hypotonia (weak muscle tone) in infancy with later development of ataxia
- Developmental delays/intellectual disability
A genetic diagnosis of Joubert syndrome can be confirmed via molecular genetic testing, which is available for the many genes that have been shown to cause Joubert syndrome. A molecular genetic diagnosis can be established in about 60% to 90% of patients. Carrier molecular genetic testing and prenatal diagnosis are available if one of these gene mutations has been identified in an affected family member.
The treatment for Joubert syndrome is symptomatic and supportive. Developmental delays are usually treated with physical therapy, occupational therapy, speech therapy and infant stimulation. Children with Joubert syndrome should be evaluated by appropriate specialists including nephrologists, ophthalmologists, geneticists and neurologists. Annual screening is recommended for liver, kidney and retinal abnormalities.
Infants and children with abnormal breathing patterns should be considered for apnea monitoring and supportive therapy may require stimulatory medications or supplemental oxygen; mechanical support; or tracheostomy in rare cases. Other interventions may include speech therapy for oromotor dysfunction; occupational and physical therapy; educational support, including special programs for the visually impaired; and feedings by gastrostomy tube. Surgery may be required for polydactyly and symptomatic ptosis and/or strabismus. Corrective lenses may be required for optic refractive errors. Nephronophthisis, end-stage renal disease, liver failure and/or fibrosis are treated with standard approaches, which may include dialysis and/or kidney transplantation.
Genetic counseling is recommended for individuals with Joubert syndrome and their families.
Figure 1. Joubert syndrome
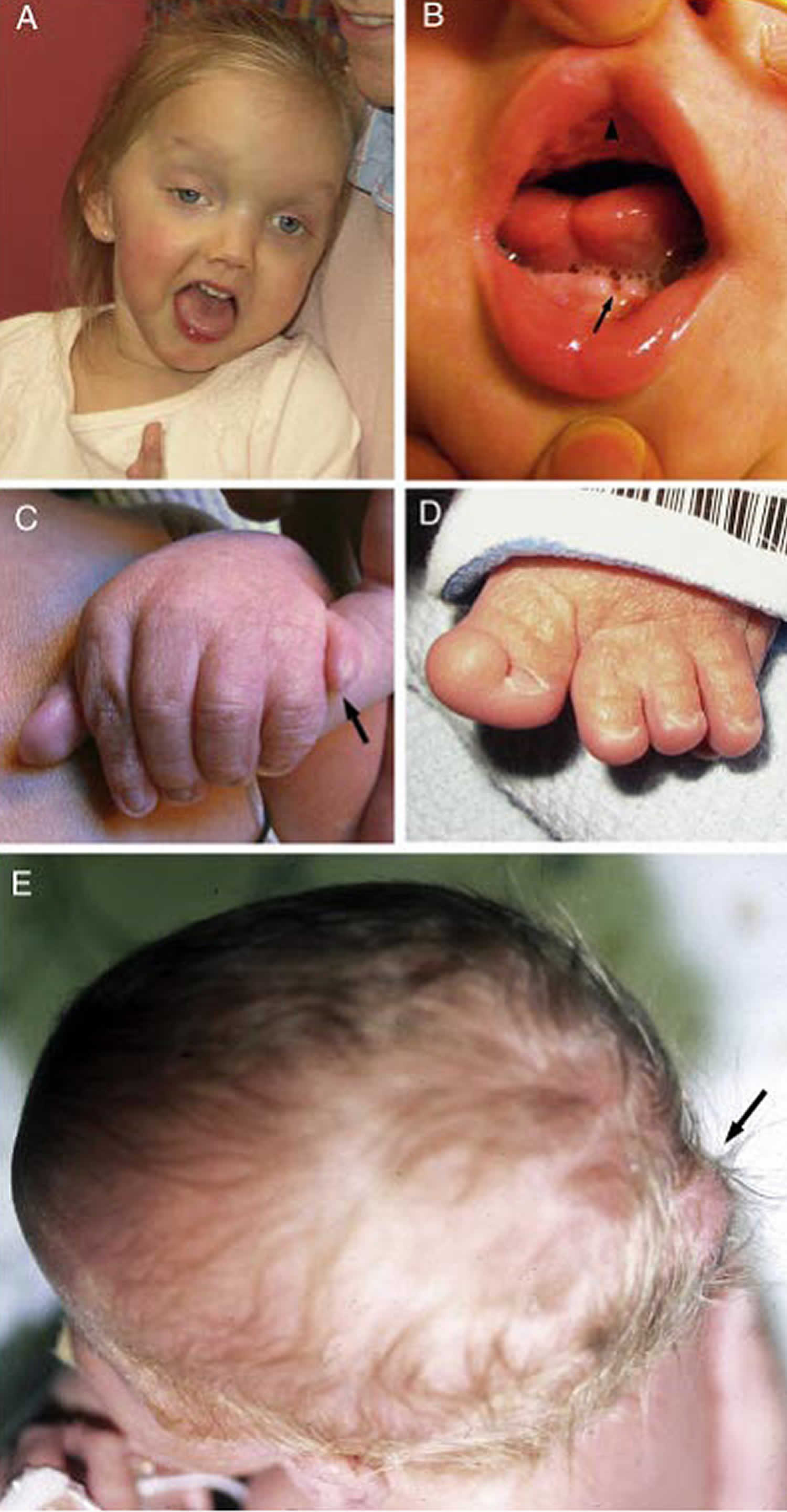
Footnotes: Joubert syndrome clinical features.
(A) Facial features in a girl with Joubert syndrome at age 27 months showing broad forehead, arched eyebrows, strabismus, eyelid ptosis (on right eye), and open mouth configuration indicating reduced facial tone
(B) Oral findings in a child with oral-facial-digital features of Joubert syndrome showing midline upper lip cleft (arrowhead), midline groove of tongue, and bumps of the lower alveolar ridge (arrow)
(C) Left hand of an infant with Joubert syndrome and postaxial polydactyly (arrow)
(D) Left foot of an infant with Joubert syndrome and preaxial polydactyly of the hallux (extra big toe)
(E) View from above of an infant with a small occipital encephalocele with protrusion of the occiput of the skull (arrow)
[Source 3 ]Figure 2. Joubert syndrome brain MRI
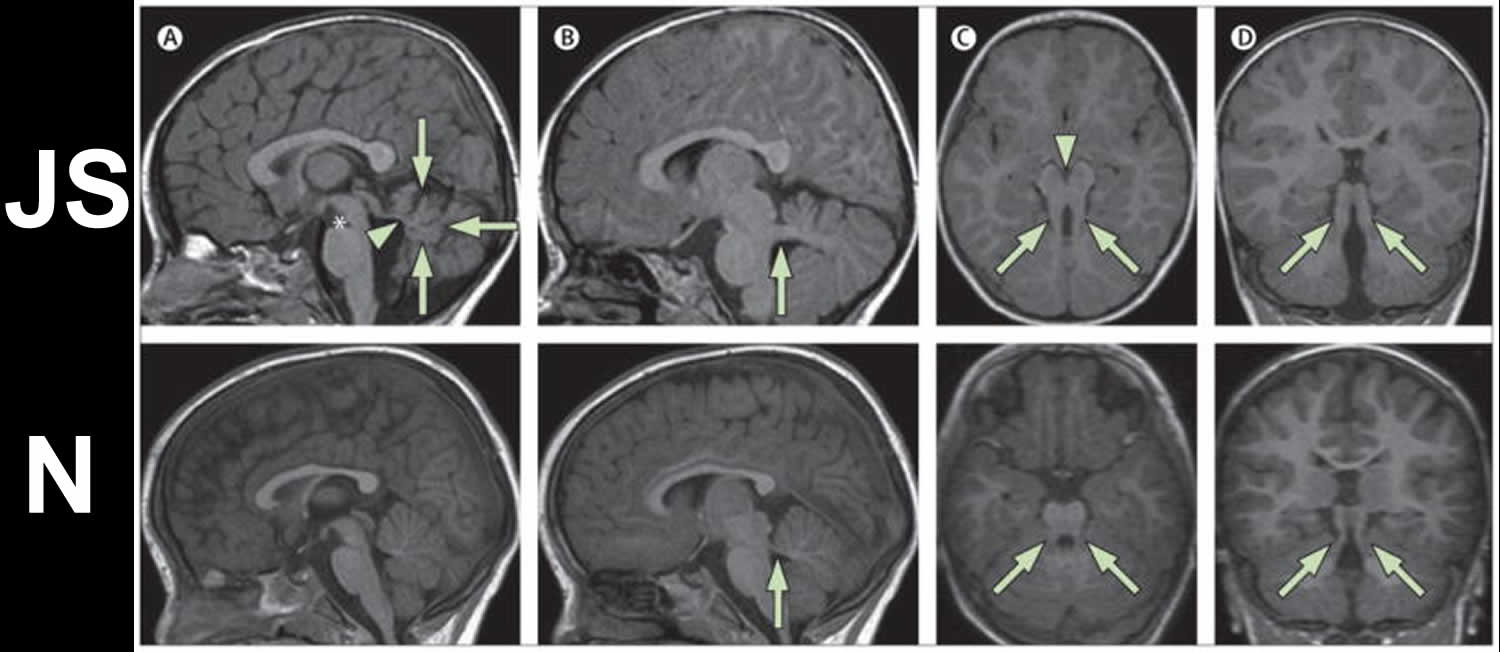
Footnotes: Neuroimaging findings in a 2-year-old child with Joubert Syndrome (JS upper panels) compared to a healthy control (N lower panels). Molar tooth sign in Joubert syndrome.
JS upper panels:
(a) Parasagittal T1-weighted image shows the thickened, elongated and horizontally orientated superior cerebellar peduncles (white arrow).
(b) Midsagittal T1-weighted image demonstrates a moderate hypoplasia and dysplasia of the cerebellar vermis (white arrows) with secondary distortion and enlargement of the fourth ventricle with rostral shifting of the fastigium (white arrow head). A deepened interpeduncular fossa is also noted.
(c) Axial T1-weighted image at the level of the pontomesencephalic junction shows the “molar tooth sign” with a deepened interpeduncular fossa (white arrowhead) and elongated, thickened and horizontally orientated superior cerebellar peduncles (white arrows). Additionally, the cerebellar vermis appears to be hypoplastic and its remnants dysplastic.
(d) Coronal T1-weighted image reveals the thickened superior cerebellar peduncles (white arrows).
N lower panels: Healthy control MRI image.
[Source 10 ]Figure 3. Joubert syndrome brain MRI
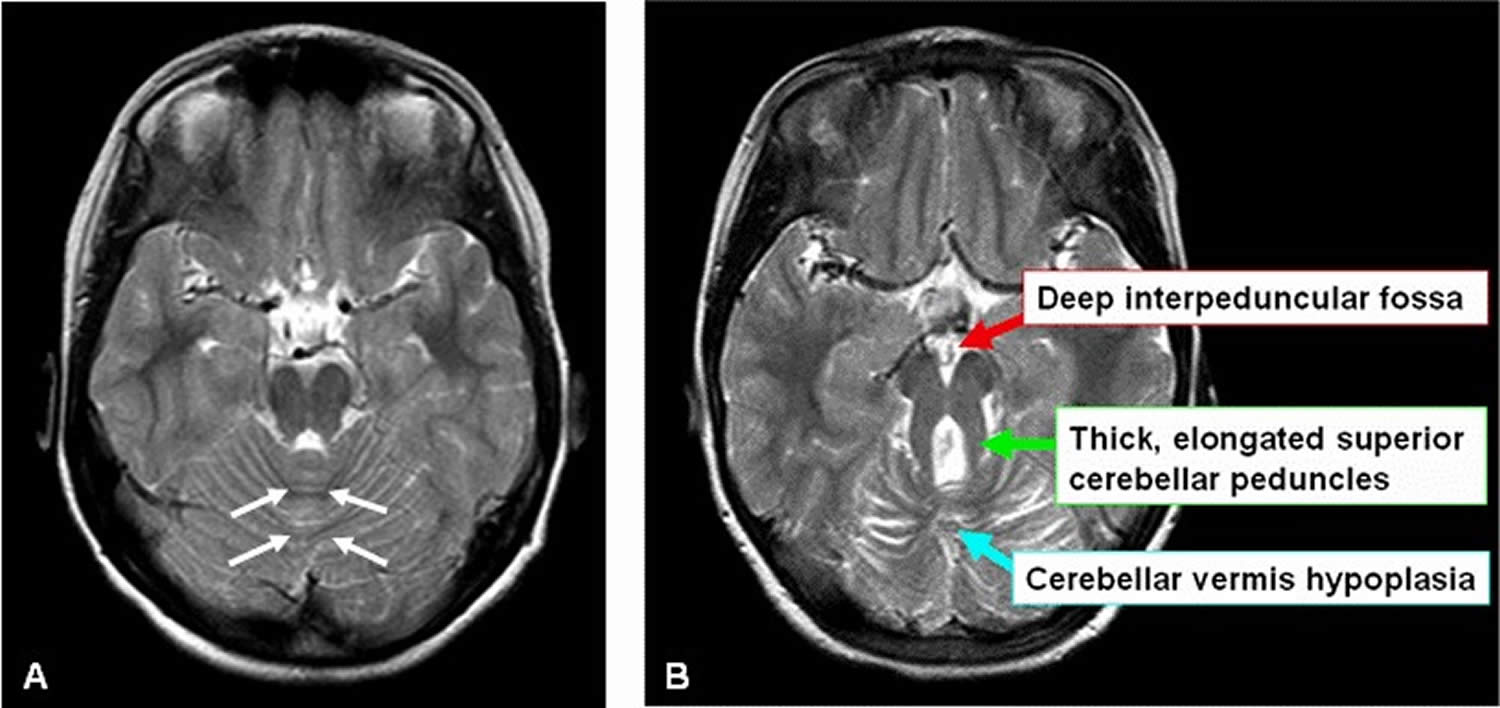
Footnotes: Molar tooth sign (MTS) in Joubert syndrome.
(A) Healthy control brain scan. Axial MRI image through the cerebellum and brain stem showing intact cerebellar vermis (outlined by white arrows)
(B) Axial MRI image through the cerebellum and brain stem of a child with Joubert syndrome. Arrows indicate the three key components of the molar tooth sign.
- An abnormally deep interpeduncular fossa
- Prominent, straight, and thickened superior cerebellar peduncles
- Hypoplasia of the vermis (the midline portion of the cerebellum)
Figure 4. Iris coloboma
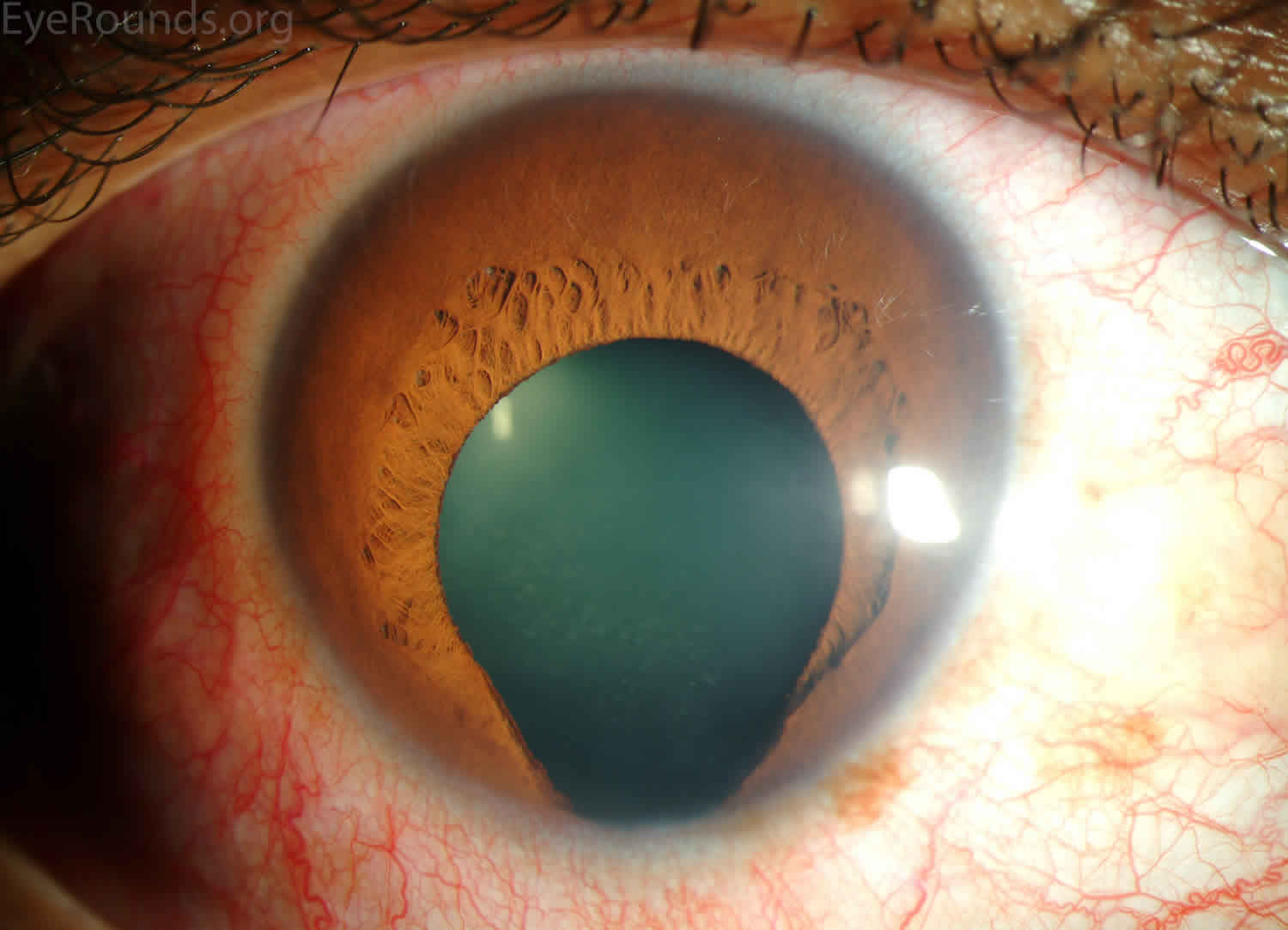
Footnotes: A coloboma is a term that is used to describe missing tissue in your eye – the eyelids, iris, lens, ciliary body, zonules, choroid, retina or optic nerve. Colobomas are present in a person’s eye when they’re born. A coloboma can affect one or both eyes. The most recognizable and common colobomas affect your iris (the colored part of your eye) and cause your pupil (the dark center of your eye) to have a keyhole shape. But there are many types of colobomas, most of which you can’t see externally (on the outside). Depending on the location of the coloboma, symptoms may include unusual appearance of the eye or eyelid, blurry vision, blind spots, and sensitivity to light.
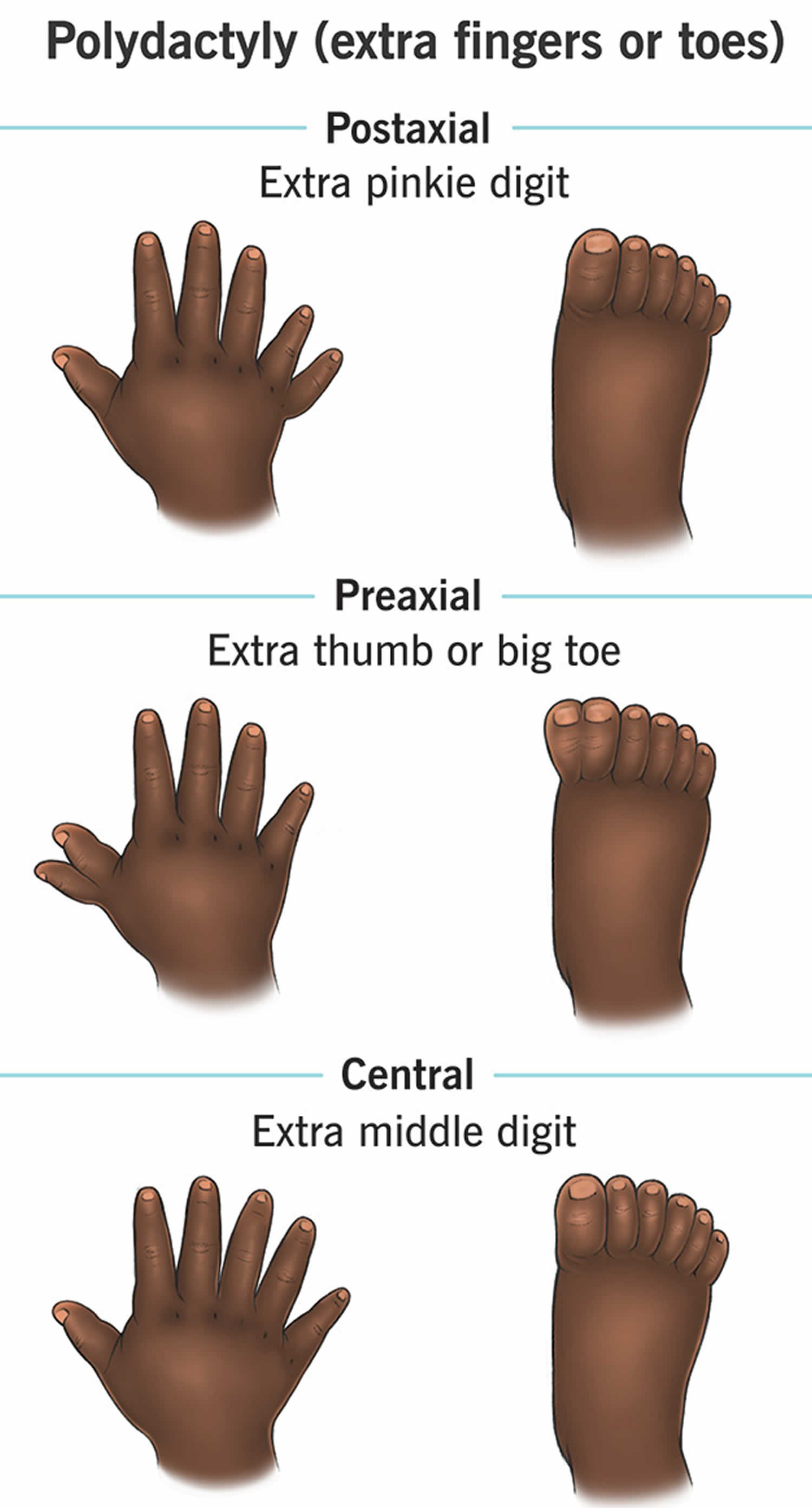
[Source 27 ]Figure 5. Polydactyly (extra fingers or toes)
Is Joubert syndrome more common in people of French Canadian descent?
Yes. There is a relatively high prevalence of Joubert syndrome and related disorders in the French Canadian population, with several founder effects noted 28. A founder effect is the effect on a gene pool that occurs when a new population is formed (founded) by a small number of individuals from a larger population, with limited genetic variation. Founder effects are typically associated with an increase in the frequency of a specific autosomal recessive allele (version of a gene) 29.
The family first described by Joubert et al in 1969 has been traced to a founder who immigrated to Quebec from France in the 1600s. However, it reportedly appears that there are other founder groups in the French Canadian population 28. Scientists are not aware of reports stating the prevalence or carrier frequency of Joubert syndrome in this population.
The exact incidence or prevalence of Joubert syndrome and related disorders in the general population has not been determined, but many authors use a range between 1 in 80,000 and 1 in 100,000. This may be an underestimate 28.
Is there a connection between Joubert syndrome and autism?
There is a connection, but it remains somewhat unclear. Autism is a relatively common condition, occurring in 1 in 500 children, and is more likely in boys with a 4:1 male:female ratio. There are likely many different causes for this increasingly common diagnosis. Features of classical autism include poor eye contact and limited communication skills as well as repetitive or self-stimulatory behaviors. Autism has been reported in a number of children with Joubert syndrome in several publications. However, more recent surveys suggest that these behavioral disturbances do not represent classic autism, but are more likely to be related to the underlying cerebellar disorder, eye movement problems, and associated developmental disabilities 30.
Can steroid use increase the chance to have a child with Joubert syndrome?
No. A person with a history of steroid use does not have a higher chance to have a child with Joubert syndrome. Joubert syndrome is a genetic condition caused by changes (mutations) in genes that can be passed from parent to child. These mutations are present from birth and are not acquired during a person’s lifetime.
Joubert syndrome types
Joubert syndrome have also been described in any related condition showing the “molar tooth sign” (MTS) in brain imaging. Joubert syndrome (JS) was originally described in 1968 in four siblings with agenesis of the cerebellar vermis presenting episodic hyperpnoea, abnormal eye movements, ataxia and intellectual disability 31. Several years later, a pathognomonic midbrain-hindbrain malformation, the “molar tooth sign” (MTS), was detected first in Joubert syndrome (JS) 32 and then in several other conditions previously considered as distinct entities 33:
The term “Joubert Syndrome and Related Disorders” (JSRD) was used as a collective term to represent all conditions sharing the “molar tooth sign” (MTS) in brain imaging with additional specific organ involvement 34, 35, 36, 10:
- Purely neurological Joubert syndrome (pure JS)
- Joubert syndrome with retinal disease (JS-Ret). Joubert syndrome with retinal disease (JS-Ret) is characterized by a pigmentary retinopathy that may be indistinguishable from classic retinitis pigmentosa. It can occasionally be severe with neonatal onset of congenital blindness; however, the retinal disease may not be progressive and is not always present in infancy or early childhood.
- Joubert syndrome with renal disease (JS-Ren). Joubert syndrome with renal disease (JS-Ren) has been described traditionally in two forms – nephronophthisis (kidney inflammation and scarring) and polycystic kidney disease; however, these now appear to be part of a continuum with the specific renal manifestation varying by stage of kidney disease.
- Arima syndrome or Joubert syndrome with oculorenal disease (JS-OR). In Joubert syndrome with oculorenal disease (JS-OR), retinal disease and kidney impairment often occur together in the same individual.
- Joubert syndrome with hepatic disease (JS-H). Joubert syndrome with hepatic disease (JS-H) usually presents with hepatic (liver) fibrosis that is usually progressive but rarely symptomatic at birth.
- COACH syndrome (colobomas, cognitive impairment [“oligophrenia”], ataxia, cerebellar vermis hypoplasia, and hepatic fibrosis)
- Varadi-Papp syndrome or Joubert syndrome with oral-facial-digital features (JS-OFD) (Joubert syndrome with oral-facial-digital features [tongue hamartomas, oral frenulae, and polydactyly with a Y-shaped metacarpal]). In Joubert syndrome with oral-facial-digital features (JS-OFD), oral findings can include midline upper-lip cleft, midline groove of tongue, hamartomas of the alveolar ridge and cleft palate. Craniofacial features often include wide-spaced eyes, low-set ears and small jaw (micrognathia).
- Other less common clinical Joubert syndrome subtypes, e.g., Joubert syndrome with acrocallosal features, Joubert syndrome with Jeune asphyxiating thoracic dystrophy.
Occasional features observed in all subgroups include pre‐, meso‐, or post‐axial polydactyly, which can variably involve hands and feet, and other central nervous system (brain and spinal cord) abnormalities, such as corpus callosum abnormalities, hydrocephalus, encephalocele, or polymicrogyria.
While these complex signs and symptoms had initially been termed “Joubert Syndrome and Related Disorders” (JSRD) 37, 38, nowadays the unifying term “Joubert syndrome” (JS) applies to all patients with the “molar tooth sign” (MTS) in brain imaging, including those with and without any extra‐neurological involvement listed above 10.
Joubert syndrome causes
About 94 percent of Joubert syndrome is caused by mutations in more than 40 genes 12, 13, 14, 15, 16, 17. These genetic mutations that cause Joubert syndrome are more common in certain ethnic groups, such as Ashkenazi Jewish, French-Canadian, and Hutterite populations. In the remaining cases, the genetic cause is unknown. Mutations in the genes associated with Joubert syndrome lead to problems with the structure and function of cell structures called primary cilia 21, 22, 23, 24. Primary cilia are microscopic, finger-like projections that stick out from the surface of cells and are involved in sensing the physical environment and in chemical signaling 39, 40, 41. Primary cilia are important for the structure and function of many types of cells, including brain cells (neurons) and certain cells in the kidneys and liver. Primary cilia are also necessary for the perception of sensory input, which is interpreted by the brain for sight, hearing, and smell. Defects in these cell structures can disrupt important chemical signaling pathways during development. Although researchers believe that defective primary cilia are responsible for most of the features of Joubert syndrome, it is not completely understood how they lead to specific developmental abnormalities.
Of note, almost all Joubert syndrome genes have also been implicated in other ciliopathies, such as Meckel syndrome (MKS), isolated nephronophthisis (NPHP), Leber congenital amaurosis (LCA), oral‐facial‐digital syndromes (OFDS), Bardet‐Biedl syndrome (BBS), and others 12.
Depending on the gene involved, various clinical presentations of Joubert syndrome can occur in an affected individual. A mutation in the AHI1 (JBTS3) gene is responsible for Joubert syndrome in approximately 7% to 10% of families 8. Affected individuals with AHI1 (JBTS3) gene mutation often have impaired vision due to retinal dystrophy. A mutation in the NPHP1 (JBTS4) gene causes approximately 1-2% of Joubert syndrome 8. Affected individuals with NPHP1 (JBTS4) gene mutation often develop a progressive kidney disease called nephronophthisis 8. A mutation in the CEP290 (JBTS5) gene causes about 7 to 10% of Joubert syndrome. Mutations in the CPLANE1, CC2D2A, INPP5E, KIAA0586, MKS1, RPGRIP1L TCTN2, TMEM67 and TMEM216 genes, along with other less common genetic causes are also associated with Joubert syndrome 8.
Joubert syndrome inheritance pattern
Joubert syndrome typically has an autosomal recessive pattern of inheritance, which means an individual inherits an abnormal gene from each parent 42, 8. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they usually do not show signs and symptoms of the condition. If an individual receives one working gene and one abnormal gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the abnormal gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier, like the parents, is 50% with each pregnancy. The chance for a child to receive working genes from both parents is 25%. The risk is the same for males and females.
Parents who are close relatives (consanguineous) have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
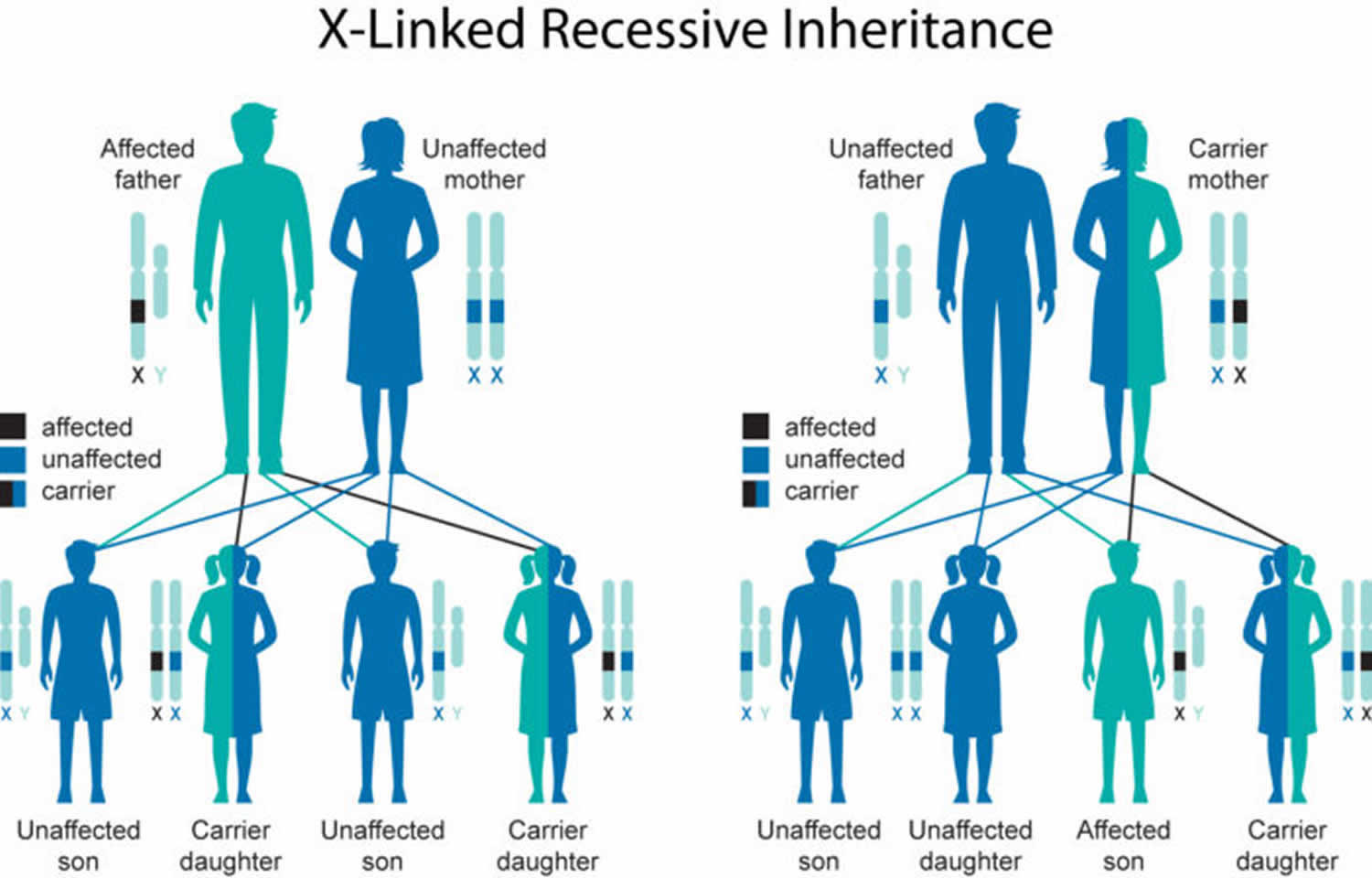
Rare cases of Joubert syndrome caused by mutations in the OFD1 gene are inherited in an X-linked recessive pattern 43, 8. In these cases, the abnormal gene is located on the X chromosome, which is one of the two sex chromosomes. In males (who have only one X chromosome [XY]), one altered copy of the gene in each cell is sufficient to cause Joubert syndrome. In females (who have two X chromosomes [XX]), a mutation would have to occur in both copies of the gene to cause Joubert syndrome. Because it is unlikely that females (who have two X chromosomes [XX]) will have two altered copies of the abnormal gene, males (who have only one X chromosome [XY]) are affected by X-linked recessive disorders much more frequently than females. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
Female carriers of an X-linked disorder have a 25% chance with each pregnancy to have a carrier daughter like themselves, a 25% chance to have a non-carrier daughter, a 25% chance to have a son affected with the disease and a 25% chance to have an unaffected son.
If a male with an X-linked disorder is able to reproduce, he will pass the non-working gene to all of his daughters who will be carriers. A male cannot pass an X-linked gene to his sons because males always pass their Y chromosome instead of their X chromosome to male offspring.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 6. Joubert syndrome autosomal recessive inheritance pattern
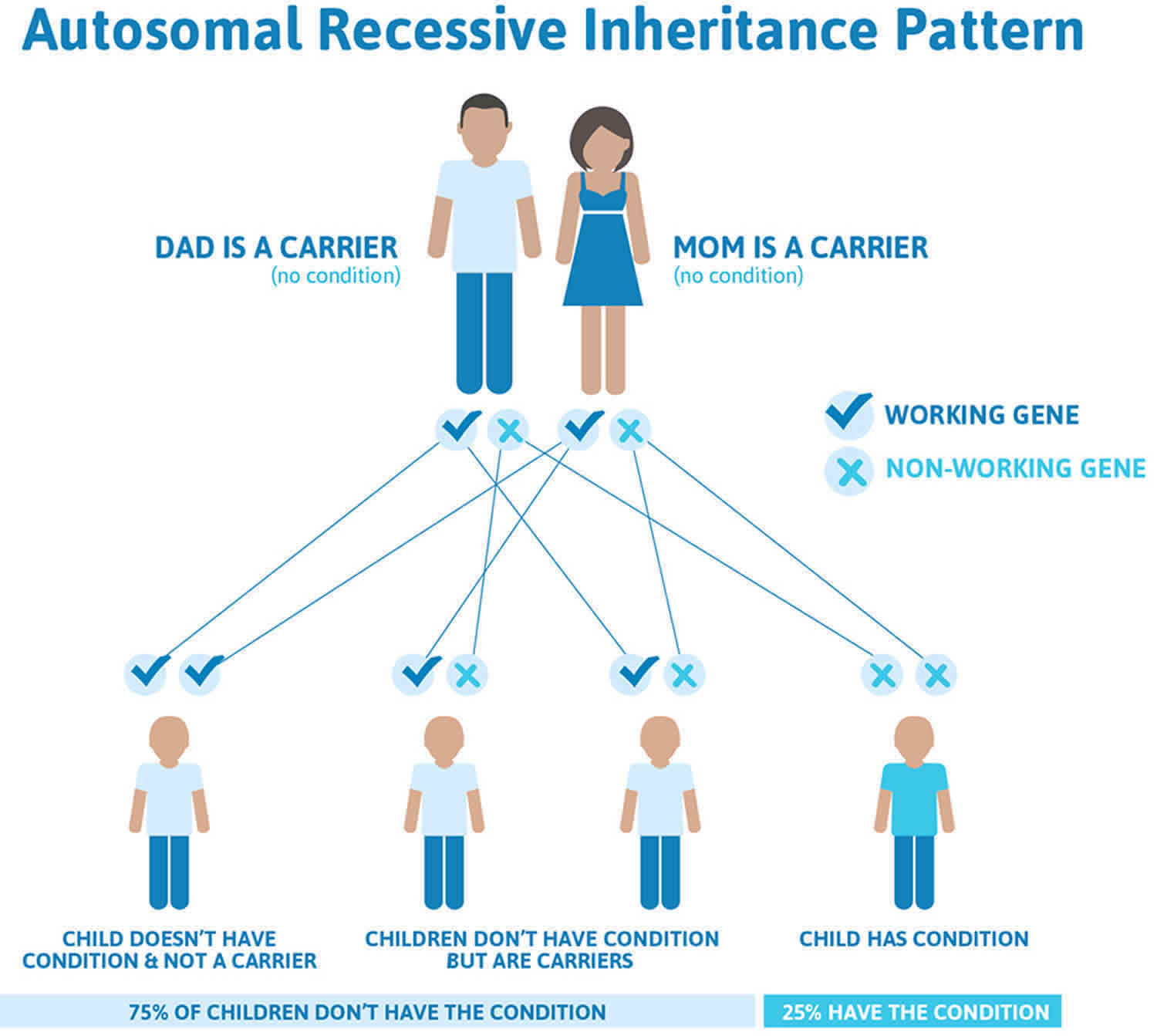
Figure 7. Joubert syndrome X-linked recessive inheritance pattern
Should I have carrier testing if my male partner has a child with Joubert syndrome?
Joubert syndrome is predominantly inherited in an autosomal recessive manner, in which case both parents must be carriers to have an affected child. In rare cases, Joubert syndrome is inherited in an X-linked recessive manner 28.
- If a male child has autosomal recessive Joubert syndrome, it is assumed his father is a carrier. To have another affected child with a different partner, the partner must also be a carrier of autosomal recessive Joubert syndrome. Many genes can be responsible for Joubert syndrome (some of which are unknown), and digenic inheritance is possible 28. This means that children may be at risk if one parent carries a mutation in one responsible gene, and the other parent carries a mutation in a different responsible gene.
- If a male child has X-linked recessive Joubert syndrome, the most likely reason is that his mother is a carrier of X-linked Joubert syndrome, and the father is not a carrier. Males cannot be “carriers” of X-linked recessive conditions; if they have a mutation in the responsible X-linked gene, they will be affected.
Because the genetics of Joubert syndrome can be complex, people with questions about carrier testing are strongly encouraged to speak with a genetic counselor or other genetics professional. Whether to have genetic testing is a personal decision. A genetic counselor can provide more detailed information about the genetic risks to specific family members. They also discuss the risks, benefits, and limitations of genetic testing, and can facilitate the testing process for people who decide to have testing.
Joubert syndrome symptoms
Joubert syndrome is a multisystem neurodevelopmental disorder that affects many parts of the body where most individuals with Joubert syndrome typically present as infants with low muscle tone (hypotonia), abnormal eye movements (ocular motor apraxia), respiratory control disturbances with unusually fast (hyperpnea) or slow (apnea) breathing, and as children or adults with difficulty coordinating movements (ataxia) and/or mild to severe delayed development and intellectual disability 44, 10, 3.
Many of the clinical symptoms of Joubert syndrome are apparent in infancy and most affected children have delays in gross motor developments. The most common features are lack of muscle control (ataxia), abnormal breathing patterns with unusually fast (hyperpnea) or slow (apnea) breathing, sleep apnea, abnormal eye movements (ocular motor apraxia) and tongue movements and low muscle tone (hypotonia). Intellect ranges from normal to severe intellectual disability. Joubert syndrome is characterized by a specific finding on an MRI called a “molar tooth sign” in which the cerebellar vermis of the brain is absent or underdeveloped and the brain stem is abnormal.
In addition to these main features, the majority of individuals with Joubert syndrome also have involvement of other body systems including the eye, kidney, liver, and skeleton 45. Joubert syndrome is sometimes associated with other eye abnormalities such as retinal dystrophy (degenerative disorders of the retina), and coloboma (a gap or split in a structure of the eye), which can cause vision loss or blindness and sensitivity to light, abnormal eye movements (nystagmus) and/or oculomotor apraxia (difficulty in smooth ocular pursuit with jerkiness in gaze and tracking), crossed eyes (strabismus) and droopy eyelids (ptosis). Other problems sometimes associated with Joubert syndrome include kidney disease such as polycystic kidney disease and nephronophthisis (a rare genetic disorder that leads to end-stage kidney disease that is characterized by kidney scarring (fibrosis), inflammation, and cyst formation, impairing kidney function that progress to terminal renal failure during the second decade (juvenile form) or before the age of 5 years (infantile form)) and/or liver disease, skeletal abnormalities such as the presence of extra fingers and toes (polydactyly), scoliosis (abnormal sideways curve of the spine), a gap in the skull with protrusion of the membranes that cover the brain (encephalocele) and hormone (endocrine) problems 9. Distinctive facial features can also occur in Joubert syndrome, these include a broad forehead, arched eyebrows, widely spaced eyes (hypertelorism), low-set ears, and a triangle-shaped mouth 9.
The signs and symptoms of Joubert syndrome vary among affected individuals, even among members of the same family and several of the additional features may be progressive, thereby substantially complicating medical management.
Neonatal period and early infancy
Clinical findings in the neonatal period and early infancy include low muscle tone (hypotonia), abnormal breathing pattern such as unusually fast (hyperpnea) or slow (apnea) breathing that may alternate, and abnormal eye movements including nystagmus and/or oculomotor apraxia (i.e., difficulty in smooth ocular pursuit with jerkiness in gaze and tracking), crossed eyes (strabismus) and droopy eyelids (ptosis). Oculomotor apraxia can sometimes manifest as head thrusting (as a compensatory mechanism for the inability to initiate saccades) or as horizontal head titubation (i.e., a “no-no” head tremor) 3.
Children and adults
Clinical findings that are identified or emerge with time include cognitive impairment (problems with a person’s ability to think, learn, remember, use judgement, and make decisions) and neurologic findings, as well as involvement of the eyes, kidney, liver, and skeleton.
Developmental delay and intellectual disability
Almost all individuals with Joubert syndrome manifest hypotonia and motor delays during infancy and early childhood, frequently evolving to ataxia (impaired balance or coordination) with age. While most reports indicate that patients with Joubert syndrome have substantial cognitive impairment (problems with a person’s ability to think, learn, remember, use judgement, and make decisions), the range of ability is quite broad with a minority of individuals having cognition in the normal range 46, 47, 48, 3. A sample of 32 patients aged 1 to 17 years (mean 6 years) evaluated with the Child Development Inventory displayed a mean developmental age equivalent of 19 months 49. IQ (intelligence quotient) between 30 and 80 have been reported in patients with Joubert syndrome 50, 47. However, difficulties measuring IQ in the presence of significant motor, speech, and visual impairments may be encountered. Ideally, neuropsychological and school evaluations should examine both full-scale intelligence quotient (FSIQ) and the generalized ability index (GAI) or similar measures to best reflect the individual’s intellectual and functional abilities 3.
The range of neurodevelopmental outcomes is very broad. Speech production is impacted out of proportion to language comprehension due to oral motor apraxia. Monitoring of development coupled with standard therapies (physical, occupational, and speech-language) and educational interventions aim to build on individual strengths and address specific weaknesses. Impaired balance or coordination (ataxia) and oculomotor apraxia (difficulty in smooth ocular pursuit with jerkiness in gaze and tracking) often improve as children mature. Most children with Joubert syndrome eventually walk independently, although a subset require assistance such as walkers and wheelchairs for ambulation. Adults with Joubert syndrome are much less well studied, but cognitive and functional impacts are lifelong 51.
Autism has been reported in children with Joubert syndrome, but the severe oral-motor dyspraxia and oculomotor issues in Joubert syndrome cause speech delay and abnormal eye contact, complicating the assessment of two core features of the diagnosis (communication and social interaction) 52, 53, 54. Takahashi et al 55 reported that none of 31 patients with Joubert syndrome exceeded the Autism Behavior Checklist cutoff for autism, and they did not find increased load of neuropsychiatric issues seen in families of individuals with autism. Furthermore, many patients with Joubert syndrome are very interested in social interaction, engage in pretend play and exhibit theory of mind, features inconsistent with autism 10.
Eye and vision disorders
Abnormal eye movements are uniformly present in Joubert syndrome though highly variable in severity and may be subtle 56, 57, 58, 59, 50, 60, 61, 62. The presence of nystagmus, saccades instead of smooth pursuit and tracking/acquiring targets with head movements rather than eye movements are the predominant clinical signs 10. Quantitative eye movement recordings demonstrate that the gains (target velocity/eye velocity) for smooth pursuit, saccades, optokinetic nystagmus and vestibuloocular reflex are variably reduced 57, 63, 64. Each of these eye movement deficits reflect abnormalities of specific neural ensembles in the cerebellar vermis (oculomotor, nodulus, uvula), flocculus/paraflocculus, deep cerebellar nuclei, vestibular nuclei, pontine nuclei and/or inferior olives 10.
Retinal dystrophy (30% of individuals) ranging from congenital retinal blindness diagnosed in infancy with an attenuated or extinguished electroretinogram (ERG) (i.e., Leber congenital amaurosis) to retinitis pigmentosa, a slowly progressive degeneration that can result in nyctalopia (night blindness) or eventual blindness after the second decade 10, 51, 43. Early onset of the retinal dystrophy shares overlapping clinical features with Leber congenital amaurosis, and preservation of vision despite markedly abnormal electroretinogram (ERG) findings supports more severe involvement of rod versus cone photoreceptors in some cases 65, 56.
Chorioretinal coloboma, optic nerve coloboma (17% of individuals) and optic atrophy is associated with significant visual impairment if the macula or optic nerve are involved 51.
Both chorioretinal coloboma and retinal dystrophy (2%-3% of individuals) 66, 67.
Microphthalmia (a developmental disorder of the eye in which one or both eyes are abnormally small and have anatomic malformations) and/or anophthalmia (a rare genetic birth defect characterized by the complete absence of one or both eyes), frequently accompanying colobomas, have rarely been identified in few patients with biallelic variants in CELSR2, CEP120, CSPP1, PDE6D, TMEM67, TMEM138, and TMEM216 68, 69, 70, 71, 72, 73, 74, 75.
Strabismus (crossed eye) and droopy eyelid (ptosis) (19% of individuals) are additional ocular findings that may be corrected with surgical intervention 66, 10. Amblyopia and refractive errors are also common 67.
Kidney disease
Kidney disease is present in 23% to 38% of individuals with Joubert syndrome and was traditionally described as “cystic dysplasia” and “nephronophthisis” with onset in late childhood or later 76, 77. However, these two findings now appear to be part of a continuum in which the manifestations vary by the stage of kidney disease 78, 79, 12, 51. Nephronophthisis is a rare genetic disorder that leads to end-stage kidney disease that is characterized by kidney scarring (fibrosis), inflammation, and cyst formation, impairing kidney function that progress to terminal renal failure during the second decade (juvenile form) or before the age of 5 years (infantile form) 80, 81. The first sign of later onset kidney disease is often failure to concentrate urine (salt-losing renal insufficiency) followed by echogenic kidneys on ultrasound and eventual renal failure. In patients with AHI1 mutations, onset of kidney disease has been reported in young adults 82, 83. Saraiva and Baraitser 84 reported kidney disease in 30% of cases in the literature.
In early-onset kidney disease, findings may be consistent with cystic dysplasia (i.e., multiple variably sized cysts in immature kidneys with fetal lobulations). In the first or second decade of life, kidney disease often presents as a urine-concentrating defect that results in polyuria and polydipsia (i.e., juvenile nephronophthisis) that may go undetected until manifestations of end-stage kidney disease (ESKD) become evident, such as fatigue, growth restriction, and/or anemia. Ultrasound examination typically shows small, scarred kidneys with increased echogenicity and occasional cysts at the corticomedullary junction.
Progression to end-stage kidney disease, occurring on average by age 13 years, is the leading cause of death in individuals with Joubert syndrome after age one year 78.
Liver disease
Liver disease is typically congenital hepatic fibrosis characterized by developmental anomalies of biliary ductal plate remodeling with elevated transaminases and gamma-glutamyl transferase (GGT) with preserved hepatocellular (synthetic) function 3. COACH syndrome (Cerebellar vermis hypoplasia, Oligophrenia – developmental delay/mental retardation, Ataxia, Colobomas, and Hepatic fibrosis) is considered a sub-type of Joubert syndrome 85, 86, 38. Clinically, the liver disease can be asymptomatic or present with mildly elevated serum transaminases, but more often, it is identified by liver imaging or signs of portal hypertension (gastroesophageal varices, hepatomegaly, splenomegaly, hypersplenism, ascites and rarely, upper gastrointestinal bleeding) in up to 13% of individuals 87, 51, 12. Liver biopsy findings are in the spectrum of the ductal plate malformation and congenital liver fibrosis, and the fibrosis is progressive at least in some cases 88. Severe liver disease requires transjugular intrahepatic portosystemic shunt (TIPS) or liver transplantation and can result in death 87, 51, 12.
Skeletal disorder
Extra fingers and toes (polydactyly) is seen in 8% to 19% of Joubert syndrome cases and is a feature of many ciliopathies (a group of genetically inherited disorders caused by defects in cilia, hairlike structures that protrude from the surface of cells) 44, 36. Most frequently, it is post-axial polydactyly with (little finger) side of the hand or fibular (little toe) side of the foot, although it can be pre-axial polydactyly with having an extra digit (thumb or big toe) located on the thumb or big toe side (radial or tibial side) of the hand or foot; or very rarely, mesoaxial polydactyly with the presence of an extra finger or toe between the thumb/hallux and little finger/toe. In general, extra fingers and toes (polydactyly) is not functionally significant, and surgical correction is at the discretion of the patient/family.
Although various skeletal features and related short-rib thoracic dysplasias have been reported in Joubert syndrome, it is rare for this to be the primary presentation 89, 90, 91, 92.
Neuromuscular scoliosis (abnormal sideways curve of the spine) with minimal prevalence of 5% requires close monitoring, especially during puberty and may require further intervention 51.
Breathing disorder
Children and adults with Joubert syndrome are at increased risk for sleep-related apnea and central sleep apnea and obstructive sleep apnea, especially if obese 51. Sleep-disordered breathing can worsen neurobehavioral/psychiatric manifestations and if untreated can result in pulmonary hypertension 93.
Oral motor dysfunction
Oral motor dysfunction may include the following 3:
- Difficulty swallowing (dysphagia) that may manifest as excess drooling and feeding difficulties predisposing to aspiration and respiratory complications. A substantial minority of individuals require gastrostomy tube placement.
- Speech apraxia a motor speech disorder that makes it difficult to plan and coordinate the movements needed to produce speech, a nearly universal finding that may account (at least in part) for the discrepancy between expressive and receptive communication abilities 94, 95.
- Dystonic movements are involuntary, abnormal muscle contractions that cause twisting, repetitive movements, and abnormal postures, which often include facial grimacing and/or biting the tongue and cheeks (perhaps self-injurious), for which there are no established treatments 51.
Other neurologic signs and symptoms
Other neurologic findings can include the following 3:
- Early hypotonia often evolves with time to become cerebellar ataxia that frequently improves with age 51
- Neurobehavioral/psychiatric manifestations include inattention, hyperactivity, stereotypies, emotional lability, anxiety, self-injury, aggression, and autism 51
- Seizures of all types (10% of individuals), which require standard neurologic evaluation and treatment 51, 12
- Temperature dysregulation (prevalence unknown), including episodic unexplained fevers and heat intolerance that can be associated with decreased activity during hot weather.
Infections
A small subset of individuals typically those with OFD1-related Joubert syndrome experience frequent sinopulmonary infections consistent with a primary ciliary dyskinesia (i.e., motile ciliopathy) 43, 96. These infections may be severe and require hospitalization.
Other signs and symptoms
While many patients with Joubert syndrome display dysmorphic facial features, no easily recognizable facial appearance has been described 97, 98. Specific craniofacial findings of oral-facial-digital syndrome type 1 (OFD1) (cleft palate, oral frenulae, and tongue lobulations or hamartomas) may be present in OFD1-related Joubert syndrome (Figure 1B) 12.
Micropenis and pituitary dysfunction have also been reported in small numbers of patients with Joubert syndrome 84, 37.
Endocrine abnormalities seen in <5% of individuals with Joubert syndrome include panhypopituitarism, isolated growth hormone deficiency, or thyroid hormone deficiency 66, 99, 51.
Laterality defects sseen in <1% of individuals with Joubert syndrome include situs inversus (a rare congenital condition in which the organs in your chest and abdomen are positioned in a mirror image of their normal location) 66.
Hirschsprung’s disease, a congenital disorder where nerve cells are missing in the large intestine, preventing the bowel from properly pushing stool through, manifested by intractable constipation has been described so far in few Joubert syndrome individuals 36, while this condition is significantly associated with another ciliopathy, Bardet–Biedl syndrome 100.
Joubert syndrome diagnosis
The diagnosis of Joubert syndrome is based on physical symptoms and the “molar tooth sign” as seen on an MRI. Diagnostic criteria for Joubert syndrome continue to evolve but most experts agree that the neuroradiologic finding of the “molar tooth sign” is necessary to make the diagnosis. The diagnosis of Joubert syndrome is based on the presence of the following three primary criteria 25, 6, 26:
- The “molar tooth sign” based on MRI findings (hypoplasia of the cerebellar vermis)
- Hypotonia (weak muscle tone) in infancy with later development of ataxia
- Developmental delays/intellectual disability
To establish the extent of disease in an individual diagnosed with Joubert syndrome, the following baseline evaluations to identify the extent of disease in affected infants/children are recommended 101. Recommendations were developed by a consensus panel and are outlined on the Joubert Syndrome and Related Disorders Foundation website (https://jsrdf.org).
- Examination of high-quality MRI scan to assess for cerebral malformations, neuronal migration disorders, or cephaloceles that could portend a poorer prognosis or seizures, if not done at the time of diagnosis
- A baseline neurologic evaluation with particular attention to tone, respiratory pattern (tachypnea and apnea), eye movements, development, and cerebellar function
- Sleep history with polysomnogram as baseline evaluation and particularly if symptomatic apnea is present
- Assessment of oromotor function by a speech therapist and/or by fluoroscopic swallowing studies
- Developmental assessment with age-appropriate tools
- Evaluation by a pediatric ophthalmologist via dilated eye examination for colobomas and retinal changes, as well as strabismus and ptosis, with consideration of specialized testing such as visual-evoked potentials, electroretinogram, and ocular motility testing
- Abdominal ultrasound examination to evaluate for hepatic fibrosis or renal cysts and/or findings consistent with nephronophthisis (e.g., loss of corticomedullary differentiation)
- Tests of renal function, including blood pressure, blood urea nitrogen (BUN), serum creatinine concentration, complete blood count (CBC), and urinalysis from first-morning void for specific gravity to test concentrating ability (if feasible)
- Liver function tests including serum concentrations of transaminases, albumin, bilirubin, and prothrombin time
- For males with micropenis or any child with signs of growth hormone deficiency, endocrine evaluation for other pituitary abnormalities
- Skeletal survey and/or limb radiographs if there is suspicion of a skeletal dysplasia such as short-rib polydactyly or Jeune asphyxiating thoracic dystrophy
- Consultation with a clinical geneticist to document family history, to evaluate growth and head size, and to evaluate for other anomalies including polydactyly, dysmorphic facial features, tongue tumors/lobulations, and micropenis
- Siblings or relatives who have clinical features similar to those of an individual with Joubert syndrome warrant genetic consultation. If the pathogenic variant(s) have been identified in a proband, testing symptomatic relatives for these pathogenic variants is appropriate.
A genetic diagnosis of Joubert syndrome can be confirmed via molecular genetic testing, which is available for the many genes that have been shown to cause Joubert syndrome. A molecular genetic diagnosis can be established in about 60% to 90% of patients. Carrier molecular genetic testing and prenatal diagnosis are available if one of these gene mutations has been identified in an affected family member.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Clinical Testing and Work-Up
Because Joubert syndrome is a multisystem neurodevelopmental disorder, and the diagnosis is often made initially in the setting of a neurology, genetics or developmental pediatrics clinic, the frequent involvement of other organ systems means that monitoring for complications is required, with referral to the corresponding subspecialists when specific organ involvement is suspected.
The following baseline evaluations to identify the extent of disease are recommended by a consensus panel:
- Examination of high-quality MRI scan (if not done at the time of diagnosis) to assess for cerebral malformations and other brain abnormalities that may indicate a poorer prognosis or seizures.
- A baseline neurologic evaluation with particular attention to tone, respiratory pattern, eye movements, development and cerebellar function.
- Neuropsychological and developmental evaluation.
- Evaluation by a pediatric ophthalmologist via dilated eye examination for eye abnormalities such as colobomas, retinal changes, strabismus and ptosis.
- Abdominal ultrasound examination for evaluation of possible liver and kidney abnormalities.
- Tests of renal function, including blood pressure, blood urea nitrogen (BUN), serum creatinine concentration, complete blood count (CBC) and urine analysis.
- Liver function tests including serum concentrations of transaminases, albumin, bilirubin and prothrombin time.
- Skeletal survey and/or limb radiographs if there is suspicion of a skeletal dysplasia such as short-rib polydactyly.
- Consultation with a clinical geneticist to document family history, evaluate growth and head size and to evaluate for other physical anomalies such as polydactyly or dysmorphic facial features.
Brain abnormalities
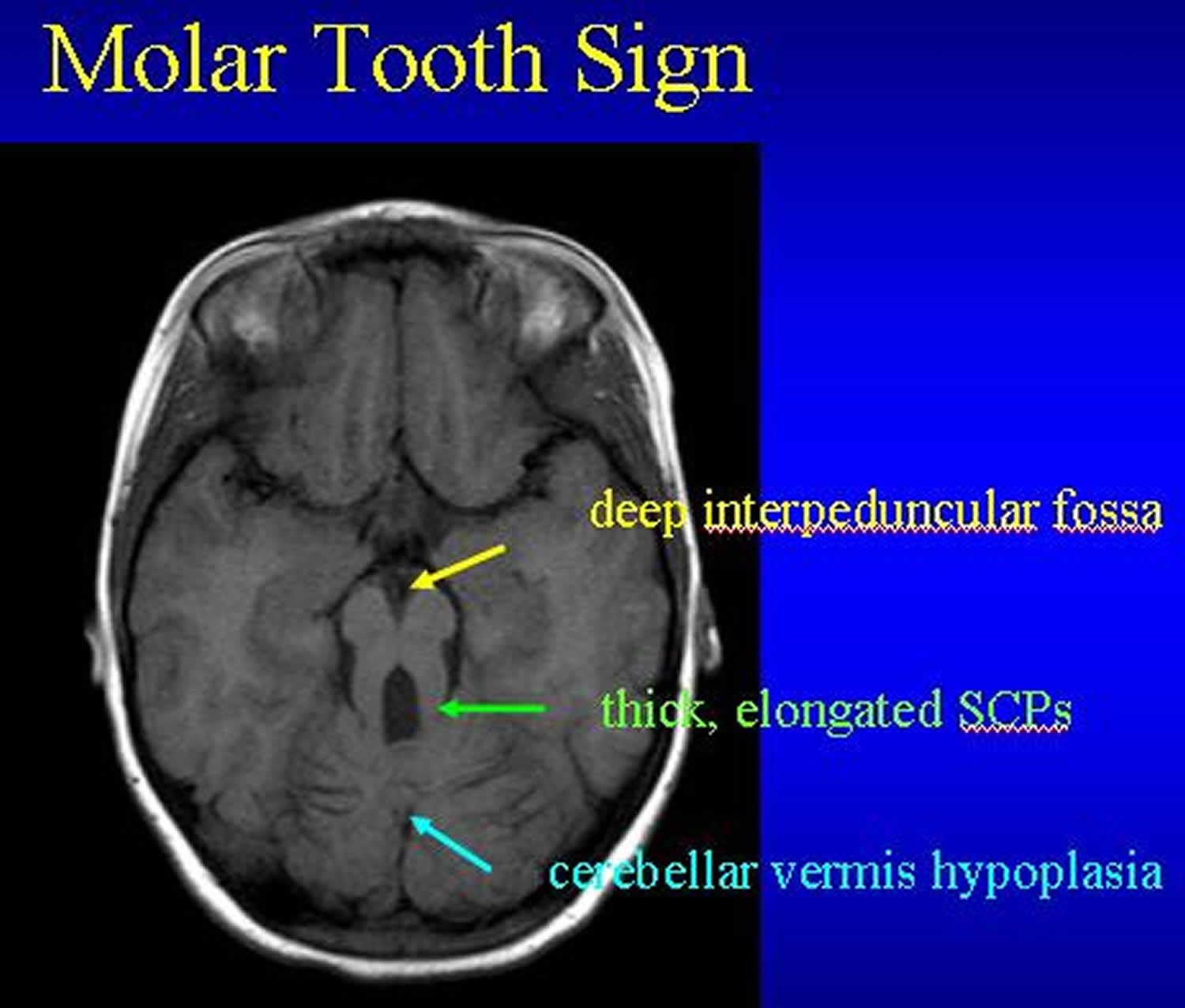
Molar tooth sign (MTS). MRI findings comprise the following (see Figure 3 above) 3, 25, 6, 26:
- An abnormally deep interpeduncular fossa
- Prominent, straight, and thickened superior cerebellar peduncles
- Hypoplasia of the vermis (the midline portion of the cerebellum)
An additional proposed near-universal feature is superior cerebellar dysplasia characterized by absence of the superior cerebellar peduncle decussation, which can be identified on fractional anisotropy imaging 51, 12.
To ensure the most accurate imaging to establish the diagnosis of molar tooth sign (MTS), both high-quality MRI with thin (≤3 mm) axial cuts through the posterior fossa from the midbrain to the pons and standard axial, coronal, and sagittal cuts are recommended 51, 12.
Note:
- Because a subset of individuals with pathogenic variants in Joubert syndrome-associated genes have MRI findings without obvious molar tooth sign (MTS), the spectrum of brain findings in Joubert syndrome likely extends beyond molar tooth sign (MTS).
- Conversely, cerebellar and brain stem malformations identified on brain MRI may be interpreted as “mild molar tooth sign” leading to an erroneous diagnosis of Joubert syndrome 12.
Less common brain anomalies observed with molar tooth sign (MTS) include the following 51, 12:
- Cerebellar hemisphere enlargement
- Malrotation of the hippocampi
- Ventriculomegaly
- Dandy-Walker malformation
- Dysgenesis of the corpus callosum
- Polymicrogyria
- Heterotopia
- Occipital encephalocele (see Figure 1E above)
- Abnormal brain stem and/or hypothalamic hamartomas, particularly in those with oral-facial-digital features 102.
Pre-implantation genetic testing
Pre-implantation Genetic Testing is an early form of prenatal genetic diagnosis where abnormal embryos with specific known single gene condition or chromosome variation are identified, thereby allowing transfer of genetically healthy embryos during an in vitro fertilization (IVF), maximising the chance of a healthy baby. In vitro fertilization (IVF) is a fertility treatment where eggs are fertilized with sperm outside the body in a laboratory, and then a resulting embryo is transferred to the uterus. Pre-implantation Genetic Testing has become an integral part of Assisted Reproductive Technology (ART) procedures 103.
The option of in vitro fertilization (IVF) and preimplantation genetic diagnosis allows couples to reduce the chance of having a child with a specific genetic condition without having to face the difficult decision of whether to stop a naturally conceived pregnancy that has been shown to be affected by testing during pregnancy. IVF and preimplantation genetic diagnosis can also reduce the chance of repeated miscarriages for couples where for example one partner carries a chromosome translocation.
As with any IVF procedure, stress and sometimes disappointment can arise when undertaking the preimplantation genetic diagnosis process. Success rates for having a child from an IVF cycle followed by preimplantation genetic diagnosis vary from IVF center to center (these are available from the individual IVF provider) but tend to follow standard IVF success rates.
Couples may balance the financial and emotional cost of the IVF procedure followed by preimplantation genetic diagnosis, with that of considering termination of pregnancy or continuing a pregnancy for a child with a genetic condition conceived naturally.
In preparation for pregnancy, reproductive genetic carrier screening is also available for women and their partners. This test screens for whether there is an increased chance for other recessive genetic conditions in pregnancy.
Who is suitable for Pre-implantation Genetic Testing?
You may wish to consider pre-implantation genetic testing if you are concerned about any of the following issues:
- Either or both partners are carriers of single gene mutations that you want to avoid passing on to future children
- Either partner has a chromosome rearrangement (called a translocation) that can result in chromosomal variations in the eggs or sperm that could lead to either miscarriage or health problems in the child
- A previous pregnancy has been affected by a chromosomal variation
- Advanced maternal age (usually to test for Down syndrome where the mother is over 38 years old)
- Recurrent miscarriage
- Repeated IVF failure (where 5 or more embryos have been transferred without pregnancy).
How is Pre-implantation Genetic Testing performed?
In pre-implantation genetic testing, the woman goes through a standard in vitro fertilization (IVF) cycle. Hormones are given to the woman to stimulate her ovaries and allow the collection of a number of her eggs called oocytes. After the eggs are removed, they are fertilized in the laboratory with sperm. Those eggs that are successfully fertilized divide and multiply to form a developing embryo. When the embryo is at the blastocyst stage in the IVF laboratory, a few cells are removed to test for the specific genetic condition in question. In general, the removal of these cells does not appear to harm the developing embryo. Only those embryos that do not have the genetic condition tested for, will be transferred into the woman’s uterus.
Joubert syndrome treatment
Guidelines for the evaluation and management of patients with Joubert syndrome were developed by a consensus panel convened by the Joubert syndrome Foundation and Related Cerebellar Disorders 104. The treatment for Joubert syndrome is symptomatic and supportive. Developmental delays are usually treated with physical therapy, occupational therapy, speech therapy and infant stimulation. Children with Joubert syndrome should be evaluated by appropriate specialists including nephrologists, ophthalmologists, geneticists and neurologists. Annual screening is recommended for liver, kidney and retinal abnormalities include yearly ophthalmologic evaluation, urinalysis, renal and liver ultrasounds, as well as serum transaminases, blood urea nitrogen (BUN), and creatinine to monitor for and allow early treatment of the medical complications described above. Lifelong monitoring for obstructive and central sleep apnea is also warranted. Specific developmental and behavioral supports for Joubert syndrome do not exist, and interventions are tailored to the needs of each individual 44.
Infants and children with abnormal breathing patterns should be considered for apnea monitoring and supportive therapy may require stimulatory medications or supplemental oxygen; mechanical support; or tracheostomy in rare cases. Other interventions may include speech therapy for oromotor dysfunction; occupational and physical therapy; educational support, including special programs for the visually impaired; and feedings by gastrostomy tube. Surgery may be required for polydactyly and symptomatic ptosis and/or strabismus. Corrective lenses may be required for optic refractive errors. Nephronophthisis, end-stage renal disease, liver failure and/or fibrosis are treated with standard approaches, which may include dialysis and/or kidney transplantation.
Genetic counseling is recommended for individuals with Joubert syndrome and their families.
Table 1. Joubert Syndrome recommended evaluations following diagnosis
| System/Concern | Evaluation | Comment |
|---|---|---|
| Constitutional | Measure height, weight, occipitofrontal circumference [OFC] | |
| Neurologic | By neurologist familiar w/neurodevelopmental disorders |
|
| Respiratory | By pulmonologist and/or sleep medicine provider | Include assessment for sleep apnea |
| Dysphagia / Poor feeding | By gastroenterologist / nutritionist / feeding team |
|
| Ophthalmologic | By ophthalmologist | Assess for:
|
| Dysarthria / Speech apraxia | By speech-language pathologist | Focus on speech apraxia when warranted |
| Ataxia/activities of daily living (ADL) | Physical medicine & rehab / physical therapy (PT) & occupational therapy (OT) eval | Include assessment of:
|
| Skeletal | By primary care provider / orthopedist |
|
| Development (infants & young children) | Developmental assessment |
|
| Cognitive abilities (school-age children & adults) | School-based standardized assessments | Provision of services & resources as directed by educational assessments |
| Neurobehavioral/ psychiatric manifestations | By developmental pediatrician | For infants and young children: screening for concerns incl sleep disturbances, attention-deficit/hyperactivity disorder (ADHD), anxiety, &/or findings suggestive of autism spectrum disorder |
| By mental health specialist | For school-age children & adults | |
| Kidney disease | By nephrologist / primary care provider |
|
| Liver disease | By primary care provider |
|
| Recurrent sinopulmonary infections | Pulmonary medicine consultation | Only in persons with OFD1-related Joubert syndrome |
| Genetic counseling | By genetics professionals 2 | Obtain a pedigree & inform affected persons & their families re nature, mode of inheritance, and implications of Joubert syndrome to facilitate medical and personal decision making & reproductive options |
| Family support & resources | By clinicians, wider care team, & family support organizations | Assessment of family & social structure to determine need for:
|
Footnotes:
- Now commonly confirmed by ocular coherence tomography (OCT) rather than electroretinogram (ERG)
- Clinical geneticist, certified genetic counselor, certified genetic nurse, genetics advanced practice provider (nurse practitioner or physician assistant)
Table 2. Joubert Syndrome Treatment Options
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Seizures | Standardized treatment with anti-seizure medication by experienced neurologist | Many anti-seizure medications may be effective; none has been demonstrated effective specifically for this disorder. Education for parents/caregivers 1 |
| Respiratory |
| By pulmonologist |
| Dysphagia/poor feeding |
| By nutritionist / speech-language pathologist / gastroenterologist |
| Ophthalmologic | Surgery for symptomatic ptosis and/or strabismus | By ophthalmologist |
| Low vision services |
| |
| Dysarthria / Speech apraxia | By speech-language pathologist |
|
| Ataxia/Spasticity/activities of daily living (ADL) | Physical therapy (PT)/occupational therapy (OT) |
|
| Skeletal involvement |
| By orthopedist or primary care provider |
| Development (infants & young children) | Age-related individualized educational support |
|
| Cognitive abilities (school-age children & adults) | ||
| Neurobehavioral and psychiatric problems |
| By mental health specialist |
| Kidney disease | Standard practices for complications of cystic kidney disease and end-stage kidney disease (ESKD) |
|
| Liver disease | Standard practices for liver failure &/or hepatic fibrosis | By gastroenterologist familiar with congenital hepatic fibrosis |
| Family/Community |
|
|
Footnotes:
- Education of parents/caregivers regarding common seizure presentations is appropriate.
Table 3. Joubert Syndrome recommended monitoring
| System/Concern | Evaluation | Frequency | |
|---|---|---|---|
| Feeding/Growth | Measure growth parameters. Assess nutritional status & safety of oral intake. | At each visit | |
| Respiratory | Clinical surveillance for apnea | ||
| Involve sleep medicine if new manifestations emerge. | As needed | ||
| Neurologic | Monitor those w/seizures as clinically indicated. Assess for new manifestations such as seizures, changes in tone, & ataxia. | Per treating neurologist | |
| Development | Monitor developmental & educational progress. | At each visit | |
| Neurobehavioral/psychiatric manifestations | Assessment for anxiety, ADHD, autism spectrum disorder, aggression, & self-injury | ||
| Musculoskeletal/ADL | OT/PT assessment of mobility, self-help skills | ||
| Scoliosis | For those under care of orthopedist or for children during period of rapid growth | Per treating clinician | |
| Ophthalmologic involvement | Eye eval for known ophthalmologic involvement & those w/molecular diagnosis that places them at ↑ risk for retinal dystrophy (See Table 1.) | Per treating ophthalmologist | |
| Low vision services | Per treating clinician | ||
| Dysarthria / Speech apraxia | Per treating speech-language pathologist | Per treating speech-language pathologist | |
| Kidney disease | For those w/molecular diagnosis that places them at ↑ risk for kidney disease (see Table 1) who are not manifesting kidney disease: | Clinical monitoring for polyuria, polydipsia, & fatigue | Yearly until at least age 20 yrs |
| Estimate creatine & hematocrit; consider urine osmolarity & cystatin C | Every 1-2 yrs | ||
| For those w/kidney disease: per treating nephrologist | Per treating nephrologist | ||
| Liver involvement | For those w/molecular diagnosis that places them at ↑ risk for age-related liver disease (see Table 1): | Monitor for hepatic disease (abdominal ultrasound for spleen size). | Yearly during childhood (until at least age 20 yrs) |
| Monitor platelets, GGT, & ALT. | Every 1-2 yrs | ||
| For those w/liver disease: per treating hepatologist | Per treating hepatologist | ||
| Endocrine | Assess pubertal development. | When age appropriate | |
| Family/Community | Assess family need for social work support (e.g., palliative/respite care, home nursing, other local resources), care coordination, or follow-up genetic counseling if new questions arise (e.g., family planning). 1 | At each visit | |
Abbreviations: ADHD = attention-deficit/hyperactivity disorder; ADL = activities of daily living; ALT = alanine transaminase; ASD = autism spectrum disorder; GGT = gamma-glutamyl transferase; OT = occupational therapy; PT = physical therapy; SLP = speech-language pathologist
Respiratory
- Infants and children with abnormal breathing patterns should be considered for apnea monitoring if the abnormality is severe. Supportive therapy may include stimulatory medications such as caffeine or supplementary oxygen, particularly in the newborn period.
- Anesthetic management during surgical procedures for infants with significant respiratory disturbance may be accomplished in some cases by the use of:
- In rare cases, mechanical support and/or tracheostomy may be considered in a child with severe respiratory dysfunction.
Aggressive treatment of middle ear infections is indicated to avoid conductive hearing loss.
Hypotonia and therapeutic interventions
- Appropriate management and therapy of oromotor dysfunction by a speech therapist
- Nasogastric feeding tubes or gastrostomy tube placement for feeding in children with severe dysphagia
- Occupational, physical, and speech therapy through early intervention programs
- Individualized educational assessment and support for school-aged children to maximize school performance
- Periodic neuropsychologic and developmental testing at appropriate ages
Other central nervous system malformations
- Neurosurgical consultation is indicated for those with evidence of hydrocephalus (rapidly increasing head circumference and/or bulging fontanelle). Note: When hydrocephalus occurs in JS, it rarely requires shunting.
- Posterior fossa cysts and fluid collections rarely require intervention.
- Encephalocele may require primary surgical closure.
- Seizures should be evaluated and treated by a neurologist using standard antiepileptic drugs.
- A variety of psychotropic medications have been used to treat the behavioral complications in Joubert syndrome; no single medication has been uniformly effective for all children.
Eye problems
- Surgery as needed for symptomatic ptosis, strabismus, or amblyopia
- Corrective lenses for refractive errors
- Possible vision therapies for oculomotor apraxia, although specific studies are lacking in this disorder
- Interventions for the visually impaired when congenital blindness or progressive retinal dystrophy are present
Renal disease
- Consultation with a nephrologist is indicated.
- End-stage renal disease (ESRD) resulting from nephronophthisis frequently requires dialysis and/or kidney transplantation during the teenage years or later.
- Hypertension, anemia, and other complications of ESRD require specific treatment.
Hepatic fibrosis
- Consultation with a gastroenterologist is indicated.
- Liver failure and/or fibrosis should be managed by a gastroenterologist with arrangements for surgical intervention such as portal shunting for esophageal varices and portal hypertension, as appropriate.
- Some individuals have needed orthotopic liver transplantation.
Skeletal
- Surgical treatment for polydactyly
- Appropriate medical management by an orthopedic specialist for scoliosis
Other
- Orofacial clefting is treated by standard surgical interventions.
- Tongue tumors that impair normal swallowing or cause respiratory obstruction may require surgical resection.
- Symptoms of obstructive sleep apnea and/or tongue hypertrophy in older individuals may require evaluation with a polysomnogram and/or by an otolaryngologist for consideration of adenoidectomy, tonsillectomy, or surgical tongue reduction. Some children have used Bilevel Positive Airway Pressure (BiPAP) or continuous positive airway pressure/power (CPAP) at night.
- Consultation with an endocrinologist for menstrual irregularities and for pituitary hormone deficiency (with hormone replacement as indicated) is appropriate.
- Obesity should be managed with appropriate measures, including diet, exercise, and behavioral therapies
- Congenital heart defects and situs abnormalities should be treated by conventional therapies.
- Surgical correction of Hirschsprung disease (if present) is indicated.
Prevention of secondary complications
Antibiotic prophylaxis for surgical and dental procedures is indicated for individuals with structural cardiac anomalies.
Surveillance
Because no uniformly reliable distinguishing characteristics allow prediction of the complications that may develop in an infant or young child with Joubert syndrome, a number of annual evaluations are recommended:
- Pediatric and neurologic evaluation and monitoring of growth, sexual maturation, breathing (including apnea symptoms), and motor function
- Neuropsychological and developmental evaluation and testing, as appropriate
- Ophthalmologic evaluation for visual acuity, tracking ability, and development of retinal dystrophy
- Abdominal ultrasound examination for evaluation of possible liver and kidney abnormalities
- Liver function tests
- Evaluation of renal function: measurement of blood pressure, serum concentrations of BUN and creatinine, CBC, and assessment of first-morning void urinalysis
Agents and circumstances to avoid
Individuals with renal impairment should avoid nephrotoxic medications such as nonsteroidal anti-inflammatory drugs.
Individuals with liver impairment should avoid hepatotoxic medications.
Joubert syndrome prognosis
The prognosis for infants with Joubert syndrome depends on whether or not the cerebellar vermis is partially developed or entirely absent, as well as on the extent and severity of other organ involvement, such as the kidneys and liver. Some children have a mild form of the disorder, with minimal motor disability and good mental development, while others may have severe motor disability, moderate impaired mental development, and multi-organ impairments. Some patients may have a shortened lifespan due to complications of the disease, including kidney or liver abnormalities.
Little is known about mortality in Joubert syndrome. After the first months of life, Joubert syndrome prognosis varies considerably among Joubert syndrome subgroups, depending on the extent and severity of organ involvement 36. A wide range of central nervous system (brain and spinal cord) malformations are described in association to the molar tooth sign (MTS) that can dramatically affect the clinical outcome and prognosis of patients with Joubert syndrome 36. These include hydrocephalus, cystic enlargement of the posterior fossa, abnormalities of the corpus callosum, white matter cysts, hypothalamic hamartoma, and absence of the pituitary gland 107, 108, 109, 110. Abnormal migration defects, mainly periventricular nodular heterotopia, and cortical organization defects such as polymicrogyria have also been reported 111, 112. Patients with these brain malformations present a higher incidence of epilepsy, which is otherwise a rare feature of Joubert syndrome 111, 109. A small number of cases present with occipital (meningo) encephalocele of variable severity 113, 114, 31.
Poretti et al 115 reported that neuroimaging may predict the neurodevelopmental outcome, as a high degree of vermis hypoplasia was found to correlate with worse prognosis. In the University of Washington cohort, 40 of 565 patients with Joubert syndrome were deceased, mainly due to extra‐neurological involvement, such as kidney disease or liver fibrosis 116. The underlying genetic defects had been identified in 80% (32/40) of the deceased cohort. Although the numbers were too small for any statistical analyses, six of nine individuals with biallelic variants in CEP290 had died from complications of kidney disease, while two of the three with molecular defects in TMEM67 had died from liver fibrosis, and all three individuals with TCTN2‐related Joubert syndrome died from respiratory complications. Therefore, a close monitoring of these issues should be considered in patients with these additional risk factors 78.
- What is Joubert Syndrome? https://jsrdf.org/what-is-js[↩]
- Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, et al. Healthcare recommendations for Joubert syndrome. Am J Med Genet Part A. 2020; 182: 229–249. https://doi.org/10.1002/ajmg.a.61399[↩]
- Glass IA, Dempsey JC, Parisi M, et al. Joubert Syndrome. 2003 Jul 9 [Updated 2025 Mar 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Nuovo, S. , Bacigalupo, I. , Ginevrino, M. , Battini, R. , Bertini, E. , Borgatti, R. , … Vanacore, N. (2020). Age and sex prevalence estimate of Joubert syndrome in Italy. Neurology, 94(8), e797–e801. 10.1212/wnl.0000000000008996[↩]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969 Sep;19(9):813-25. doi: 10.1212/wnl.19.9.813[↩]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997 Oct;12(7):423-30. doi: 10.1177/088307389701200703[↩][↩][↩][↩][↩]
- Joubert Syndrome. https://www.ninds.nih.gov/health-information/disorders/joubert-syndrome[↩]
- Joubert Syndrome. https://rarediseases.org/rare-diseases/joubert-syndrome[↩][↩][↩][↩][↩][↩][↩]
- Joubert syndrome. https://medlineplus.gov/genetics/condition/joubert-syndrome[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol. 2013 Sep;12(9):894-905. doi: 10.1016/S1474-4422(13)70136-4[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Juric-Sekhar G, Adkins J, Doherty D, Hevner RF. Joubert syndrome: brain and spinal cord malformations in genotyped cases and implications for neurodevelopmental functions of primary cilia. Acta Neuropathol. 2012;123:695–709. doi: 10.1007/s00401-012-0951-2[↩]
- Gana S, Serpieri V, Valente EM. Genotype-phenotype correlates in Joubert syndrome: A review. Am J Med Genet C Semin Med Genet. 2022 Mar;190(1):72-88. doi: 10.1002/ajmg.c.31963[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Reiter, J. F. , & Leroux, M. R. (2017). Genes and molecular pathways underpinning ciliopathies. Nature Reviews. Molecular Cell Biology, 18(9), 533–547. 10.1038/nrm.2017.60[↩][↩]
- Bachmann‐Gagescu, R. , Dempsey, J. C. , Phelps, I. G. , O’Roak, B. J. , Knutzen, D. M. , Rue, T. C. , … Doherty, D. (2015). Joubert syndrome: A model for untangling recessive disorders with extreme genetic heterogeneity. Journal of Medical Genetics, 52(8), 514–522. 10.1136/jmedgenet-2015-103087[↩][↩]
- Parisi, M. A. (2019). The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity. Transl Sci Rare Dis, 4(1–2), 25–49. 10.3233/trd-190041[↩][↩]
- Phelps, I. G. , Dempsey, J. C. , Grout, M. E. , Isabella, C. R. , Tully, H. M. , Doherty, D. , & Bachmann‐Gagescu, R. (2018). Interpreting the clinical significance of combined variants in multiple recessive disease genes: Systematic investigation of Joubert syndrome yields little support for oligogenicity. Genetics in Medicine, 20(2), 223–233. 10.1038/gim.2017.94[↩][↩]
- Shaheen, R. , Szymanska, K. , Basu, B. , Patel, N. , Ewida, N. , Faqeih, E. , … Alkuraya, F. S. (2016). Characterizing the morbid genome of ciliopathies. Genome Biology, 17(1), 242. 10.1186/s13059-016-1099-5[↩][↩]
- Edvardson S, Shaag A, Zenvirt S, et al. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010;86:93–7. doi: 10.1016/j.ajhg.2009.12.007[↩]
- Valente EM, Logan CV, Mougou-Zerelli S, et al. Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat Genet. 2010 Jul;42(7):619-25. doi: 10.1038/ng.594[↩]
- Huang L, Szymanska K, Jensen VL, et al. TMEM237 Is Mutated in Individuals with a Joubert Syndrome Related Disorder and Expands the Role of the TMEM Family at the Ciliary Transition Zone. Am J Hum Genet. 2011;89:713–30. doi: 10.1016/j.ajhg.2011.11.005[↩]
- Brancati F., Dallapiccola B., Valente E.M. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010;5:20. doi: 10.1186/1750-1172-5-20[↩][↩]
- Baker K., Beales P.L. Making sense of cilia in disease: The human ciliopathies. Am. J. Med. Genet. C. Semin. Med. Genet. 2009;151C:281–295. doi: 10.1002/ajmg.c.30231[↩][↩]
- Sharma N., Berbari N.F., Yoder B.K. Ciliary dysfunction in developmental abnormalities and diseases. Curr. Top. Dev. Biol. 2008;85:371–427. doi: 10.1016/S0070-2153(08)00813-2[↩][↩]
- Lee J.E., Gleeson J.G. Cilia in the nervous system: Linking cilia function and neurodevelopmental disorders. Curr. Opin. Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05[↩][↩]
- D’Abrusco, F. , Arrigoni, F. , Serpieri, V. , Romaniello, R. , Caputi, C. , Manti, F. , et al. (2021). Get your molar tooth right: Joubert syndrome misdiagnosis unmasked by whole‐exome sequencing. Cerebellum. 10.1007/s12311-021-01350-8[↩][↩][↩]
- Maria BL, Quisling RG, Rosainz LC, Yachnis AT, Gitten J, Dede D, Fennell E. Molar tooth sign in Joubert syndrome: clinical, radiologic, and pathologic significance. J Child Neurol. 1999 Jun;14(6):368-76. doi: 10.1177/088307389901400605[↩][↩][↩]
- Iris coloboma. https://www.eyerounds.org/atlas/pages/iris-coloboma/index.htm#gsc.tab=0[↩]
- Parisi M, Glass I. Joubert Syndrome. 2003 Jul 9 [Updated 2017 Jun 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325[↩][↩][↩][↩][↩]
- Francesco Brancati, Bruno Dallapiccola and Enza Maria Valente. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010; 5:20:https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-20[↩]
- Takahashi TN, Farmer JE, Deidrick KK, Hsu BS, Miles JH, Maria BL. Joubert syndrome is not a cause of classical autism. Am J Med Genet A. 2005;132A:347–51.[↩]
- Joubert M, Eisenring JJ, Andermann F. Familial dysgenesis of the vermis: a syndrome of hyperventilation, abnormal eye movements and retardation. Neurology. 1968 Mar;18(3):302-3.[↩][↩]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703[↩]
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima Senior-Loken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999;86:459–469. doi: 10.1002/(SICI)1096-8628(19991029)86:5<459::AID-AJMG12>3.0.CO;2-C[↩]
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, Graham JM Jr, Maria BL, Barkovich AJ, Dobyns WB. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004;125:125–134. doi: 10.1002/ajmg.a.20437[↩]
- Poretti, A. , Huisman, T. A. , Scheer, I. , & Boltshauser, E. (2011). Joubert syndrome and related disorders: Spectrum of neuroimaging findings in 75 patients. AJNR. American Journal of Neuroradiology, 32(8), 1459–1463. 10.3174/ajnr.A2517[↩]
- Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis. 2010 Jul 8;5:20. doi: 10.1186/1750-1172-5-20[↩][↩][↩][↩][↩]
- Gleeson JG, Keeler LC, Parisi MA, Marsh SE, Chance PF, Glass IA, Graham JM Jr, Maria BL, Barkovich AJ, Dobyns WB. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet A. 2004 Mar 1;125A(2):125-34; discussion 117. doi: 10.1002/ajmg.a.20437[↩][↩]
- Satran D, Pierpont ME, Dobyns WB. Cerebello-oculo-renal syndromes including Arima, Senior-Löken and COACH syndromes: more than just variants of Joubert syndrome. Am J Med Genet. 1999 Oct 29;86(5):459-69.[↩][↩]
- Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364:1533–43. doi: 10.1056/NEJMra1010172[↩]
- Arts HH, Knoers NV. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2012;28:863–74. doi: 10.1007/s00467-012-2259-9[↩]
- Lee JE, Gleeson JG. Cilia in the nervous system: linking cilia function and neurodevelopmental disorders. Curr Opin Neurol. 2011;24:98–105. doi: 10.1097/WCO.0b013e3283444d05[↩]
- Serpieri V, D’Abrusco F, Dempsey JC, et al. University of Washington Center for Mendelian Genomics (UW-CMG) group. SUFU haploinsufficiency causes a recognisable neurodevelopmental phenotype at the mild end of the Joubert syndrome spectrum. J Med Genet. 2022 Sep;59(9):888-894. doi: 10.1136/jmedgenet-2021-108114[↩]
- Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B, van Wijk E, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FP, van Bokhoven H, de Brouwer AP. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009 Oct;85(4):465-81. doi: 10.1016/j.ajhg.2009.09.002[↩][↩][↩]
- Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol. 2009 Sep;16(3):143-54. doi: 10.1016/j.spen.2009.06.002[↩][↩][↩]
- Bachmann-Gagescu R, Dempsey JC, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015 Aug;52(8):514-22. doi: 10.1136/jmedgenet-2015-103087[↩]
- Casaer P, et al. Variability of outcome in Joubert syndrome. Neuropediatrics. 1985;16:43–5. doi: 10.1055/s-2008-1052543[↩]
- Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-Up in Children with Joubert Syndrome. Neuropediatr. 1997;28:204–211. doi: 10.1055/s-2007-973701[↩][↩]
- Ziegler AL, Deonna T, Calame A. Hidden intelligence of a multiply handicapped child with Joubert syndrome. Dev Med Child Neurol. 1990;32:261–6. doi: 10.1111/j.1469-8749.1990.tb16933.x[↩]
- Gitten J, Dede D, Fennell E, Quisling R, Maria BL. Neurobehavioral development in Joubert syndrome. J Child Neurol. 1998;13:391–7. doi: 10.1177/088307389801300806[↩]
- Hodgkins PR, et al. Joubert syndrome: long-term follow-up. Dev Med Child Neurol. 2004;46:694–9. doi: 10.1017/s0012162204001161[↩][↩]
- Bachmann-Gagescu R, Dempsey JC, Bulgheroni S, et al. Healthcare recommendations for Joubert syndrome. Am J Med Genet A. 2020 Jan;182(1):229-249. doi: 10.1002/ajmg.a.61399[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Holroyd S, Reiss AL, Bryan RN. Autistic features in Joubert syndrome: a genetic disorder with agenesis of the cerebellar vermis. Biol Psychiatry. 1991;29:287–294. doi: 10.1016/0006-3223(91)91291-x[↩]
- Ozonoff S, Williams BJ, Gale S, Miller JN. Autism and autistic behavior in Joubert syndrome. J Child Neurol. 1999;14:636–641. doi: 10.1177/088307389901401003[↩]
- Braddock BA, Farmer JE, Deidrick KM, Iverson JM, Maria BL. Oromotor and communication findings in joubert syndrome: further evidence of multisystem apraxia. J Child Neurol. 2006;21:160–3. doi: 10.1177/08830738060210020501[↩]
- Takahashi TN, et al. Joubert syndrome is not a cause of classical autism. Am J Med Genet A. 2005;132:347–51. doi: 10.1002/ajmg.a.30500[↩]
- Lambert SR, Kriss A, Gresty M, Benton S, Taylor D. Joubert syndrome. Arch Ophthalmol. 1989;107:709–713. doi: 10.1001/archopht.1989.01070010727035[↩][↩]
- Tusa RJ, Hove MT. Ocular and oculomotor signs in Joubert syndrome. J Child Neurol. 1999;14:621–627. doi: 10.1177/088307389901401001[↩][↩]
- Moore AT, Taylor DS. A syndrome of congenital retinal dystrophy and saccade palsy–a subset of Leber’s amaurosis. Br J Ophthalmol. 1984;68:421–31. doi: 10.1136/bjo.68.6.421[↩]
- Sztriha L, Al-Gazali LI, Aithala GR, Nork M. Joubert’s syndrome: new cases and review of clinicopathologic correlation. Pediatr Neurol. 1999;20:274–81. doi: 10.1016/s0887-8994(98)00154-4[↩]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813[↩]
- Boltshauser E, Isler W. Joubert syndrome: episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57–66. doi: 10.1055/s-0028-1091505[↩]
- Maria BL, et al. “Joubert syndrome” revisited: key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703[↩]
- Weiss A, et al. Eye Movement Abnormalities in Joubert Syndrome. Investigative Ophthalmology & Visual Science. 2009 doi: 10.1167/iovs.08-3299[↩]
- Khan AO, et al. Ophthalmic features of Joubert syndrome. Ophthalmology. 2008;115:2286–9. doi: 10.1016/j.ophtha.2008.08.005[↩]
- Lambert SR, Kriss A, Taylor D, Coffey R, Pembrey M. Follow-up and diagnostic reappraisal of 75 patients with Leber’s congenital amaurosis. Am J Ophthalmol. 1989;107:624–31. doi: 10.1016/0002-9394(89)90259-6[↩]
- Bachmann-Gagescu R, Phelps IG, Dempsey JC, Sharma VA, Ishak GE, Boyle EA, Wilson M, Marques Lourenço C, Arslan M; University of Washington Center for Mendelian Genomics; Shendure J, Doherty D. KIAA0586 is Mutated in Joubert Syndrome. Hum Mutat. 2015 Sep;36(9):831-5. doi: 10.1002/humu.22821[↩][↩][↩][↩]
- Brooks BP, Zein WM, Thompson AH, Mokhtarzadeh M, Doherty DA, Parisi M, Glass IA, Malicdan MC, Vilboux T, Vemulapalli M, Mullikin JC, Gahl WA, Gunay-Aygun M. Joubert Syndrome: Ophthalmological Findings in Correlation with Genotype and Hepatorenal Disease in 99 Patients Prospectively Evaluated at a Single Center. Ophthalmology. 2018 Dec;125(12):1937-1952. doi: 10.1016/j.ophtha.2018.05.026[↩][↩]
- Ben-Salem S, Al-Shamsi AM, Gleeson JG, Ali BR, Al-Gazali L. Mutation spectrum of Joubert syndrome and related disorders among Arabs. Hum Genome Var. 2014 Nov 6;1:14020. doi: 10.1038/hgv.2014.20. Erratum in: Hum Genome Var. 2015 Mar 26;2:15001. doi: 10.1038/hgv.2015.1[↩]
- Edvardson S, Shaag A, Zenvirt S, Erlich Y, Hannon GJ, Shanske AL, Gomori JM, Ekstein J, Elpeleg O. Joubert syndrome 2 (JBTS2) in Ashkenazi Jews is associated with a TMEM216 mutation. Am J Hum Genet. 2010 Jan;86(1):93-7. doi: 10.1016/j.ajhg.2009.12.007 Erratum in: Am J Hum Genet. 2010 Feb;86(2):294. Shanske, Alan L [added].[↩]
- Kroes HY, Monroe GR, van der Zwaag B, Duran KJ, de Kovel CG, van Roosmalen MJ, Harakalova M, Nijman IJ, Kloosterman WP, Giles RH, Knoers NV, van Haaften G. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet. 2016 Feb;24(2):214-20. doi: 10.1038/ejhg.2015.84[↩]
- Lee JH, Silhavy JL, Lee JE, Al-Gazali L, Thomas S, Davis EE, Bielas SL, Hill KJ, Iannicelli M, Brancati F, Gabriel SB, Russ C, Logan CV, Sharif SM, Bennett CP, Abe M, Hildebrandt F, Diplas BH, Attié-Bitach T, Katsanis N, Rajab A, Koul R, Sztriha L, Waters ER, Ferro-Novick S, Woods CG, Johnson CA, Valente EM, Zaki MS, Gleeson JG. Evolutionarily assembled cis-regulatory module at a human ciliopathy locus. Science. 2012 Feb 24;335(6071):966-9. doi: 10.1126/science.1213506[↩]
- Powell L, Barroso-Gil M, Clowry GJ, Devlin LA, Molinari E, Ramsbottom SA, Miles CG, Sayer JA. Expression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development. BMC Dev Biol. 2020 Dec 9;20(1):26. doi: 10.1186/s12861-020-00231-3[↩]
- Thomas S, Wright KJ, Le Corre S, Micalizzi A, Romani M, Abhyankar A, Saada J, Perrault I, Amiel J, Litzler J, Filhol E, Elkhartoufi N, Kwong M, Casanova JL, Boddaert N, Baehr W, Lyonnet S, Munnich A, Burglen L, Chassaing N, Encha-Ravazi F, Vekemans M, Gleeson JG, Valente EM, Jackson PK, Drummond IA, Saunier S, Attié-Bitach T. A homozygous PDE6D mutation in Joubert syndrome impairs targeting of farnesylated INPP5E protein to the primary cilium. Hum Mutat. 2014 Jan;35(1):137-46. doi: 10.1002/humu.22470[↩]
- Vilboux T, Malicdan MC, Roney JC, Cullinane AR, Stephen J, Yildirimli D, Bryant J, Fischer R, Vemulapalli M, Mullikin JC; NISC Comparative Sequencing Program; Steinbach PJ, Gahl WA, Gunay-Aygun M. CELSR2, encoding a planar cell polarity protein, is a putative gene in Joubert syndrome with cortical heterotopia, microophthalmia, and growth hormone deficiency. Am J Med Genet A. 2017 Mar;173(3):661-666. doi: 10.1002/ajmg.a.38005[↩]
- Vilboux T, Doherty DA, Glass IA, Parisi MA, Phelps IG, Cullinane AR, Zein W, Brooks BP, Heller T, Soldatos A, Oden NL, Yildirimli D, Vemulapalli M, Mullikin JC, Nisc Comparative Sequencing Program, Malicdan MCV, Gahl WA, Gunay-Aygun M. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med. 2017 Aug;19(8):875-882. doi: 10.1038/gim.2016.204[↩]
- Kumada S, et al. Renal disease in Arima syndrome is nephronophthisis as in other Joubert-related Cerebello-oculo-renal syndromes. Am J Med Genet A. 2004;131:71–6. doi: 10.1002/ajmg.a.30294[↩]
- Hildebrandt F. Juvenile nephronophthisis. In: Barratt TMAE, Harmon WE, editors. Pediatric Nephrology. Williams & Wilkins; Baltimore: 1999. pp. 453–458.[↩]
- Dempsey JC, Phelps IG, Bachmann-Gagescu R, Glass IA, Tully HM, Doherty D. Mortality in Joubert syndrome. Am J Med Genet A. 2017 May;173(5):1237-1242. doi: 10.1002/ajmg.a.38158[↩][↩][↩]
- Fleming LR, Doherty DA, Parisi MA, Glass IA, Bryant J, Fischer R, Turkbey B, Choyke P, Daryanani K, Vemulapalli M, Mullikin JC, Malicdan MC, Vilboux T, Sayer JA, Gahl WA, Gunay-Aygun M. Prospective Evaluation of Kidney Disease in Joubert Syndrome. Clin J Am Soc Nephrol. 2017 Dec 7;12(12):1962-1973. doi: 10.2215/CJN.05660517[↩]
- Salomon R, Saunier S, Niaudet P. Nephronophthisis. Pediatr Nephrol. 2009 Dec;24(12):2333-44. doi: 10.1007/s00467-008-0840-z[↩]
- Nephronophthisis. https://medlineplus.gov/genetics/condition/nephronophthisis[↩]
- Parisi MA, et al. AHI1 mutations cause both retinal dystrophy and renal cystic disease in Joubert syndrome. J Med Genet. 2006;43:334–9. doi: 10.1136/jmg.2005.036608[↩]
- Utsch B, et al. Identification of the first AHI1 gene mutations in nephronophthisis-associated Joubert syndrome. Pediatr Nephrol. 2006;21:32–5. doi: 10.1007/s00467-005-2054-y[↩]
- Saraiva JM, Baraitser M. Joubert syndrome: a review. Am J Med Genet. 1992;43:726–731. doi: 10.1002/ajmg.1320430415[↩][↩]
- Gleeson JG, et al. Molar tooth sign of the midbrain-hindbrain junction: occurrence in multiple distinct syndromes. Am J Med Genet. 2004;125A:125–34. doi: 10.1002/ajmg.a.20437. discussion 117[↩]
- Verloes A, Lambotte C. Further delineation of a syndrome of cerebellar vermis hypo/aplasia, oligophrenia, congenital ataxia, coloboma, and hepatic fibrosis. Am J Med Genet. 1989;32:227–232. doi: 10.1002/ajmg.1320320217[↩]
- Strongin A, Heller T, Doherty D, Glass IA, Parisi MA, Bryant J, Choyke P, Turkbey B, Daryanani K, Yildirimli D, Vemulapalli M, Mullikin JC, Malicdan MC, Vilboux T, Gahl WA, Gunay-Aygun M; NISC Comparative Sequencing Program. Characteristics of Liver Disease in 100 Individuals With Joubert Syndrome Prospectively Evaluated at a Single Center. J Pediatr Gastroenterol Nutr. 2018 Mar;66(3):428-435. doi: 10.1097/MPG.0000000000001816[↩][↩]
- Doherty D, Parisi MA, Finn LS, et al. Mutations in 3 genes (MKS3, CC2D2A and RPGRIP1L) cause COACH syndrome (Joubert syndrome with congenital hepatic fibrosis). J Med Genet. 2010 Jan;47(1):8-21. doi: 10.1136/jmg.2009.067249[↩]
- Lehman AM, Eydoux P, Doherty D, Glass IA, Chitayat D, Chung BY, Langlois S, Yong SL, Lowry RB, Hildebrandt F, Trnka P. Co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy. Am J Med Genet A. 2010 Jun;152A(6):1411-9. doi: 10.1002/ajmg.a.33416[↩]
- Halbritter J, Bizet AA, Schmidts M, et al. Defects in the IFT-B component IFT172 cause Jeune and Mainzer-Saldino syndromes in humans. Am J Hum Genet. 2013 Nov 7;93(5):915-25. doi: 10.1016/j.ajhg.2013.09.012[↩]
- Tuz K, Bachmann-Gagescu R, O’Day DR, et al. Mutations in CSPP1 cause primary cilia abnormalities and Joubert syndrome with or without Jeune asphyxiating thoracic dystrophy. Am J Hum Genet. 2014 Jan 2;94(1):62-72. doi: 10.1016/j.ajhg.2013.11.019. Erratum in: Am J Hum Genet. 2014 Feb 6;94(2):310.[↩]
- Shaheen R, Schmidts M, Faqeih E, Hashem A, Lausch E, Holder I, Superti-Furga A; UK10K Consortium; Mitchison HM, Almoisheer A, Alamro R, Alshiddi T, Alzahrani F, Beales PL, Alkuraya FS. A founder CEP120 mutation in Jeune asphyxiating thoracic dystrophy expands the role of centriolar proteins in skeletal ciliopathies. Hum Mol Genet. 2015 Mar 1;24(5):1410-9. doi: 10.1093/hmg/ddu555[↩]
- Ju-Wang JD, Dempsey JC, Zhang C, Doherty D, Witmans M, Tablizo MA, Chen ML. Sleep and breathing in children with Joubert syndrome and a review of other rare congenital hindbrain malformations. Ther Adv Respir Dis. 2025 Jan-Dec;19:17534666241308405. doi: 10.1177/17534666241308405[↩]
- Hodgkins PR, Harris CM, Shawkat FS, Thompson DA, Chong K, Timms C, Russell-Eggitt I, Taylor DS, Kriss A. Joubert syndrome: long-term follow-up. Dev Med Child Neurol. 2004 Oct;46(10):694-9. doi: 10.1017/s0012162204001161[↩]
- Braddock BA, Farmer JE, Deidrick KM, Iverson JM, Maria BL. Oromotor and communication findings in joubert syndrome: further evidence of multisystem apraxia. J Child Neurol. 2006 Feb;21(2):160-3. doi: 10.1177/08830738060210020501[↩]
- Hannah WB, DeBrosse S, Kinghorn B, Strausbaugh S, Aitken ML, Rosenfeld M, Wolf WE, Knowles MR, Zariwala MA. The expanding phenotype of OFD1-related disorders: Hemizygous loss-of-function variants in three patients with primary ciliary dyskinesia. Mol Genet Genomic Med. 2019 Sep;7(9):e911. doi: 10.1002/mgg3.911[↩]
- Braddock SR, Henley KM, Maria BL. The face of Joubert syndrome: a study of dysmorphology and anthropometry. Am J Med Genet A. 2007;143A:3235–42. doi: 10.1002/ajmg.a.32099[↩]
- Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999 Sep;14(9):583-90; discussion 590-1. doi: 10.1177/088307389901400906[↩]
- Akcan N, Bas F, Poyrazoglu S, Bundak R. Joubert syndrome with multiple pituitary hormone deficiency. BMJ Case Rep. 2019 Apr 23;12(4):e229016. doi: 10.1136/bcr-2018-229016[↩]
- Lorda-Sanchez I, Ayuso C, Ibañez A. Situs inversus and hirschsprung disease: two uncommon manifestations in Bardet-Biedl syndrome. Am J Med Genet. 2000 Jan 3;90(1):80-1. doi: 10.1002/(sici)1096-8628(20000103)90:1<80::aid-ajmg14>3.0.co;2-e[↩]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300). Eur J Hum Genet. 2007;15(5):511–521. doi:10.1038/sj.ejhg.5201648[↩]
- Poretti A, Huisman TA, Scheer I, Boltshauser E. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. AJNR Am J Neuroradiol. 2011 Sep;32(8):1459-63. doi: 10.3174/ajnr.A2517[↩]
- Parikh FR, Athalye AS, Naik NJ, Naik DJ, Sanap RR, Madon PF. Preimplantation Genetic Testing: Its Evolution, Where Are We Today? J Hum Reprod Sci. 2018 Oct-Dec;11(4):306-314. doi: 10.4103/jhrs.JHRS_132_18[↩]
- Joubert Syndrome & Related disorders Foundation. https://jsrdf.org[↩]
- Vodopich DJ, Gordon GJ. Anesthetic management in Joubert syndrome. Paediatr Anaesth. 2004;14:871–3.[↩]
- Sriganesh K, Vinay B, Jena S, Sudhir V, Saini J, Umamaheswara Rao GS. Anesthetic management of patients with Joubert syndrome: a retrospective analysis of a single-institutional case series. Paediatr Anaesth. 2014;24:1180–4.[↩]
- Maria BL, Bozorgmanesh A, Kimmel KN, Theriaque D, Quisling RG. Quantitative assessment of brainstem development in Joubert syndrome and Dandy-Walker syndrome. J Child Neurol. 2001;16:751–758. doi: 10.1177/088307380101601008[↩]
- Genel F, Atlihan F, Ozdemir D, Targan S. Development of hydrocephalus in a patient with Joubert syndrome. J Postgrad Med. 2004 Apr-Jun;50(2):153.[↩]
- Haug K, Khan S, Fuchs S, König R. OFD II, OFD VI, and Joubert syndrome manifestations in 2 sibs. Am J Med Genet. 2000 Mar 13;91(2):135-7. doi: 10.1002/(sici)1096-8628(20000313)91:2<135::aid-ajmg11>3.0.co;2-1[↩][↩]
- Zamponi N, Rossi B, Messori A, Polonara G, Regnicolo L, Cardinali C. Joubert syndrome with associated corpus callosum agenesis. Eur J Paediatr Neurol. 2002;6(1):63-6. doi: 10.1053/ejpn.2001.0542[↩]
- Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A, Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG. Mutations in the AHI1 gene encoding jouberin cause Joubert syndrome with cortical polymicrogyria. Am J Hum Genet. 2004;75:979–987. doi: 10.1086/425985[↩][↩]
- Giordano L, Vignoli A, Pinelli L, Brancati F, Accorsi P, Faravelli F, Gasparotti R, Granata T, Giaccone G, Inverardi F, Frassoni C, Dallapiccola B, Valente EM, Spreafico R. Joubert syndrome with bilateral polymicrogyria: clinical and neuropathological findings in two brothers. Am J Med Genet A. 2009;149A:1511–1515. doi: 10.1002/ajmg.a.32936[↩]
- Wang P, Chang FM, Chang CH, Yu CH, Jung YC, Huang CC. Prenatal diagnosis of Joubert syndrome complicated with encephalocele using two-dimensional and three-dimensional ultrasound. Ultrasound Obstet Gynecol. 1999;14:360–362. doi: 10.1046/j.1469-0705.1999.14050360.x[↩]
- Shian WJ, Chi CS, Mak SC, Chen CH. Joubert syndrome in Chinese infants and children: a report of four cases. Zhonghua Yi Xue Za Zhi (Taipei). 1993 Nov;52(5):342-5.[↩]
- Poretti A, Snow J, Summers AC, Tekes A, Huisman TAGM, Aygun N, Carson KA, Doherty D, Parisi MA, Toro C, Yildirimli D, Vemulapalli M, Mullikin JC; NISC Comparative Sequencing Program; Cullinane AR, Vilboux T, Gahl WA, Gunay-Aygun M. Joubert syndrome: neuroimaging findings in 110 patients in correlation with cognitive function and genetic cause. J Med Genet. 2017 Aug;54(8):521-529. doi: 10.1136/jmedgenet-2016-104425[↩]
- Bachmann-Gagescu R, Dempsey JC, Phelps IG, O’Roak BJ, Knutzen DM, Rue TC, Ishak GE, Isabella CR, Gorden N, Adkins J, Boyle EA, de Lacy N, O’Day D, Alswaid A, Ramadevi A R, Lingappa L, Lourenço C, Martorell L, Garcia-Cazorla À, Ozyürek H, Haliloğlu G, Tuysuz B, Topçu M; University of Washington Center for Mendelian Genomics; Chance P, Parisi MA, Glass IA, Shendure J, Doherty D. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet. 2015 Aug;52(8):514-22. doi: 10.1136/jmedgenet-2015-103087[↩]
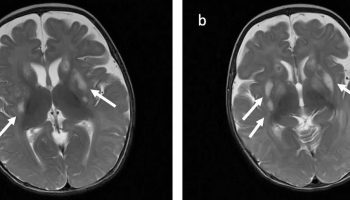

{kind=link}