Kennedy’s disease
Kennedy’s syndrome also known as Kennedy’s disease, X-linked spinal and bulbar muscular atrophy, X-linked spinobulbar muscular atrophy or Spinal and Bulbar Muscular Atrophy (SBMA), is a rare X-linked recessive inherited progressive motor neuron disorder that primarily affects specialized nerve cells that control muscle movement or lower motor neurons 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15. These nerve cells or lower motor neurons originate in your spinal cord and the part of your brain that is connected to your spinal cord called the brainstem. Kennedy’s syndrome is characterized by the degeneration of lower motor neurons, leading to symptoms like progressive muscle weakness, wasting of muscles (muscle atrophy), and muscle twitches (fasciculations) that usually begins in adulthood and worsens slowly over time. Muscle wasting in your arms and legs results in cramping; leg muscle weakness can also lead to difficulty walking and a tendency to fall. Certain muscles in your face and throat (bulbar muscles) are also affected, which causes progressive problems with swallowing (dysphagia) and speech (dysarthria) 16. Affected patients are at risk of choking on food and aspiration pneumonia because of weakness of the bulbar muscles 17. Additionally, muscle twitches (fasciculations) are common. Some males with the disorder experience unusual breast development (gynecomastia), impotence (inability to achieve an erection), testicular atrophy (shrunken testicles) and may be unable to father a child (infertile) and metabolic changes, suggesting a multisystem involvement in the disease 14. Kennedy’s disease is slowly progressive. Individuals tend to remain ambulatory until late in the disease, although some may be wheelchair-bound during later stages.
Neurological signs suggestive of Kennedy’s disease 15:
- Muscle twitches (fasciculations)
- Cramps
- Muscle atrophy
- Predominant motor deficit in the lower and proximal limbs,
- Decrease or abolition of osteo-tendinous reflexes
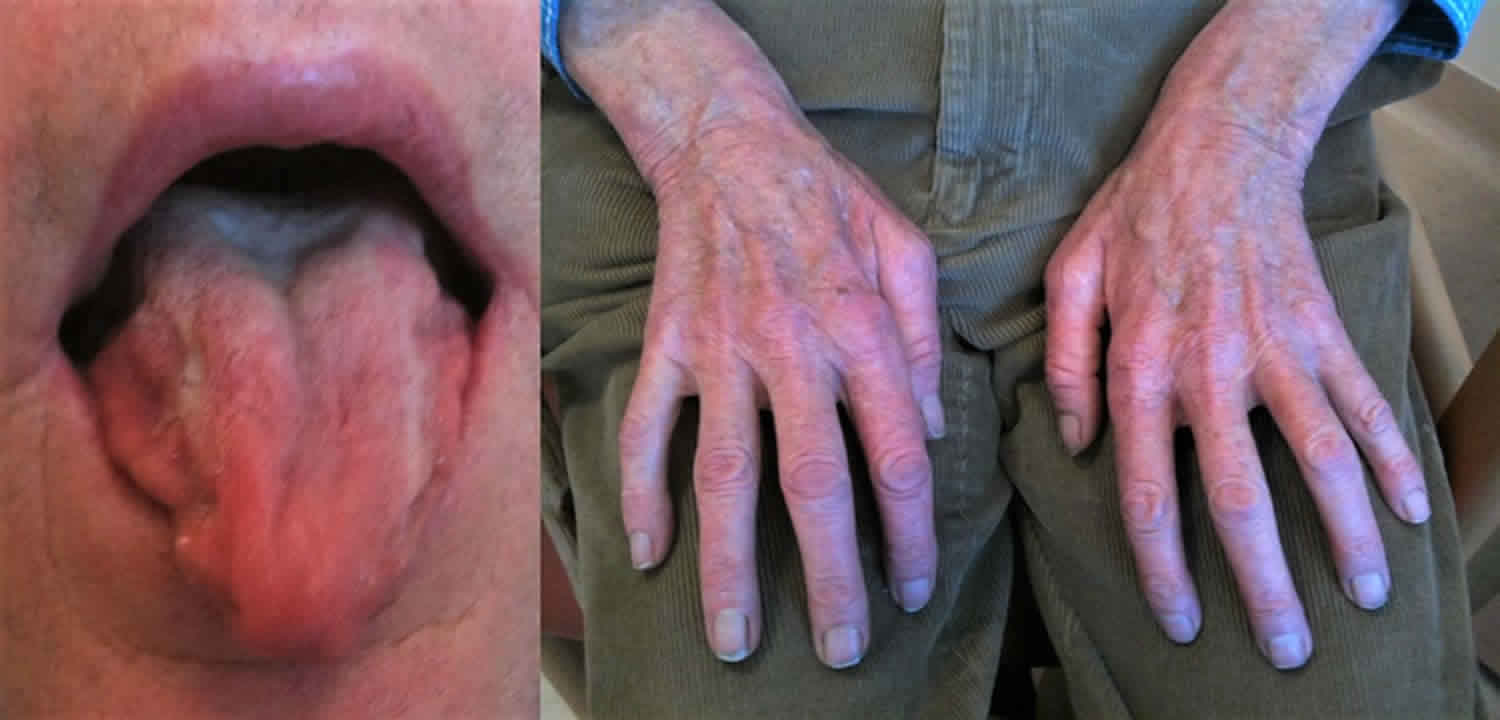
- Atrophy of the tongue and fasciculations that may extend to the lips, chin, or perioral area (often with a myokymic appearance).
- Slurred speech (dysarthria), with potentially a discreet nasal voice
- Difficulty swallowing (dysphagia)
- Sensory involvement most often subclinical and distal (numbness and tingling in your lower limbs)
- Absence of spasticity, clonus, exaggeration and / or diffusion of reflexes, or Babinski’s sign
- Postural tremor of the upper limbs
Kennedy’s disease is caused by CAG repeat expansion in the Androgen Receptor (AR) gene on the X chromosome, which regulates the effects of male sex hormones 18. The AR gene mutation that causes Kennedy’s syndrome or Kennedy’s disease is the abnormal expansion of the CAG trinucleotide repeat. In most people, the number of CAG trinucleotide repeat in the AR gene ranges from fewer than 10 to about 36. In people with Kennedy’s syndrome or Kennedy’s disease, the CAG trinucleotide segment is repeated from 38 times to more than 60 times, and it may be two or three times its usual length 19. Although the extended CAG region changes the structure of the androgen receptor (AR), it is unclear how the altered protein disrupts nerve cells in your brain and spinal cord. Researchers believe that a fragment of the androgen receptor protein containing the CAG segment accumulates within these nerve cells and interferes with normal cell functions. The nerve cells gradually die, leading to the muscle weakness and wasting (atrophy) seen in Kennedy’s disease. People with a higher number of CAG repeats tend to develop signs and symptoms of Kennedy’s disease at an earlier age.
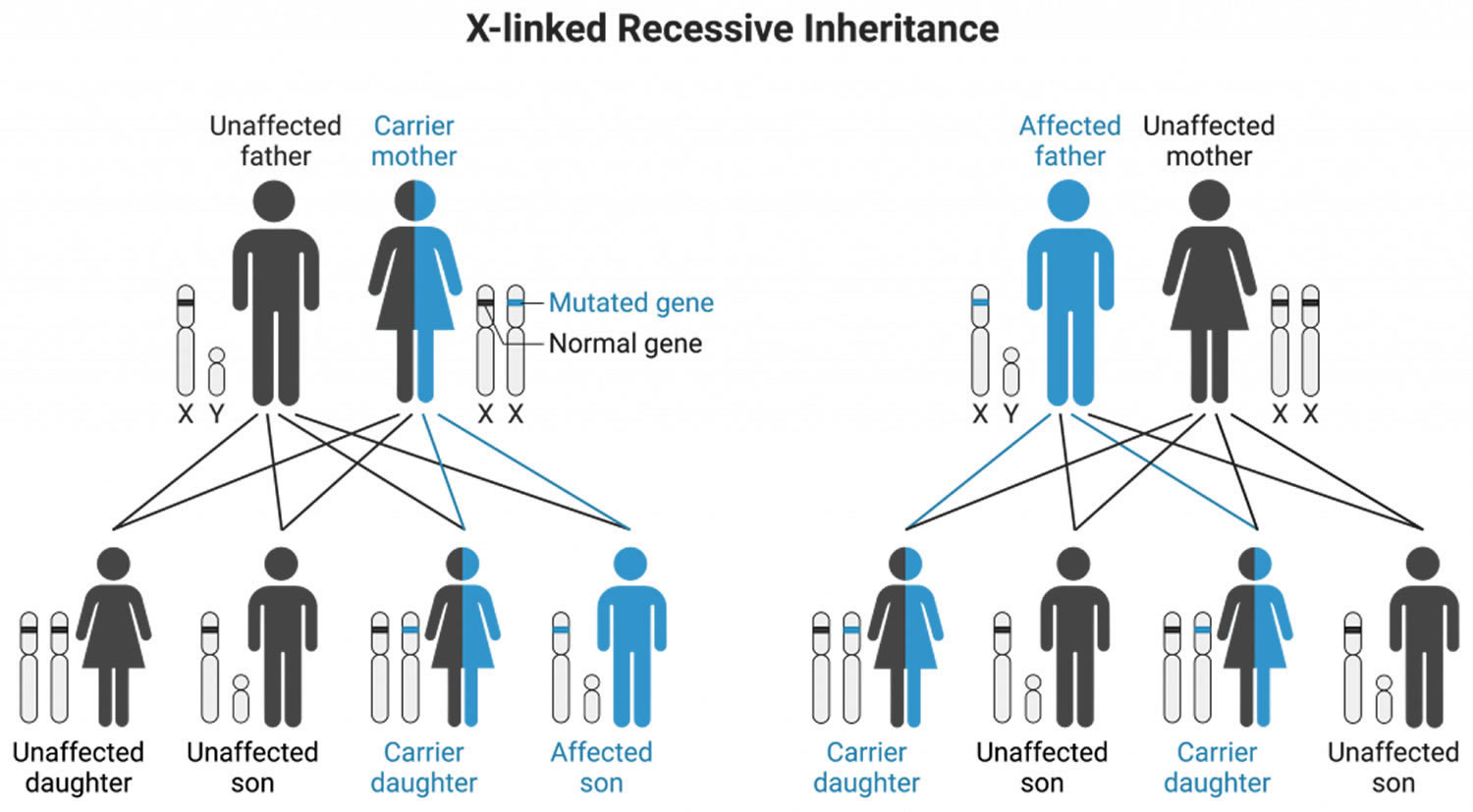
Kennedy’s disease is an X-linked recessive disorder, meaning it is primarily passed down through the X chromosome and mainly affects males. Kennedy’s disease is an X-linked recessive disease, which means the patient’s mother carries the defective AR gene on one of her X chromosomes. Daughters of patients with Kennedy’s disease are also carriers and have a 1 in 2 chance of having a son affected with the disease. Parents with concerns about their children may wish to talk to a genetic counselor.
Kennedy’s disease is a rare disorder that affects fewer than 1 to 2.5 in 100,000 males and is very rare in females who are protected by their low levels of circulating testosterone, accounting for the sex-limited inheritance pattern in this disorder 20, 21. In most cases, females who inherit the AR gene are carriers, while males who inherit the AR gene develop the disease. Kennedy disease has been diagnosed in the USA, Europe, Asia, South America, and Australia. It is estimated that 1 in 40,000 individuals worldwide have Kennedy’s Disease 4. The Japanese and Finnish population appears to have a very high prevalence of Kennedy Disease because of a founder effect 22, 23, 24.
Kennedy disease is typically an adult-onset disease where symptoms occur mainly between the ages of 30 and 50, although it has been diagnosed in men from their teens to their 70s 25, 26, 27. Early symptoms include tremor of the outstretched hands, muscle cramps with exertion, and muscle twitches (fasciculations). Eventually, individuals develop limb weakness which usually begins in the pelvic or shoulder regions. Weakness of the facial and tongue muscles may occur later in the course of the disease and often leads to dysphagia (difficulty in swallowing), dysarthria (slurring of speech), and recurrent aspiration pneumonia. Some individuals develop gynecomastia (excessive enlargement of male breasts) and low sperm count (oligospermia) or complete absence of sperm (azoospermia) or infertility. Still others develop non-insulin-dependent diabetes mellitus (type 2 diabetes).
Female carriers develop some symptoms of Kennedy’s disease such as distal muscle weakness and wasting, cramping and/or muscle twitching (fasciculations) 2, but men are more severely affected by Kennedy’s disease with symptoms including difficulty speaking (dysarthria), difficulty swallowing (dysphagia), enlarged breasts (gynecomastia), cramping, muscle twitching (fasciculations) and tremor 28, 2.
Kennedy disease diagnosis is often made based on symptoms, family history, and genetic testing using a sample of whole blood to identify the mutation in the AR gene. The genetic testing is rapid and accurate and could also be used to detect pre-symptomatic individuals (i.e., those who have the mutated AR gene but do not show any signs of the disease), or carriers, and for prenatal diagnosis. Testing is done only after appropriate genetic counseling has been undertaken.
There is no cure for Kennedy’s disease, but treatment focuses on managing symptoms and improving quality of life. This may include physical therapy, speech therapy, and medications to manage specific symptoms.
Figure 1. Motor neurons
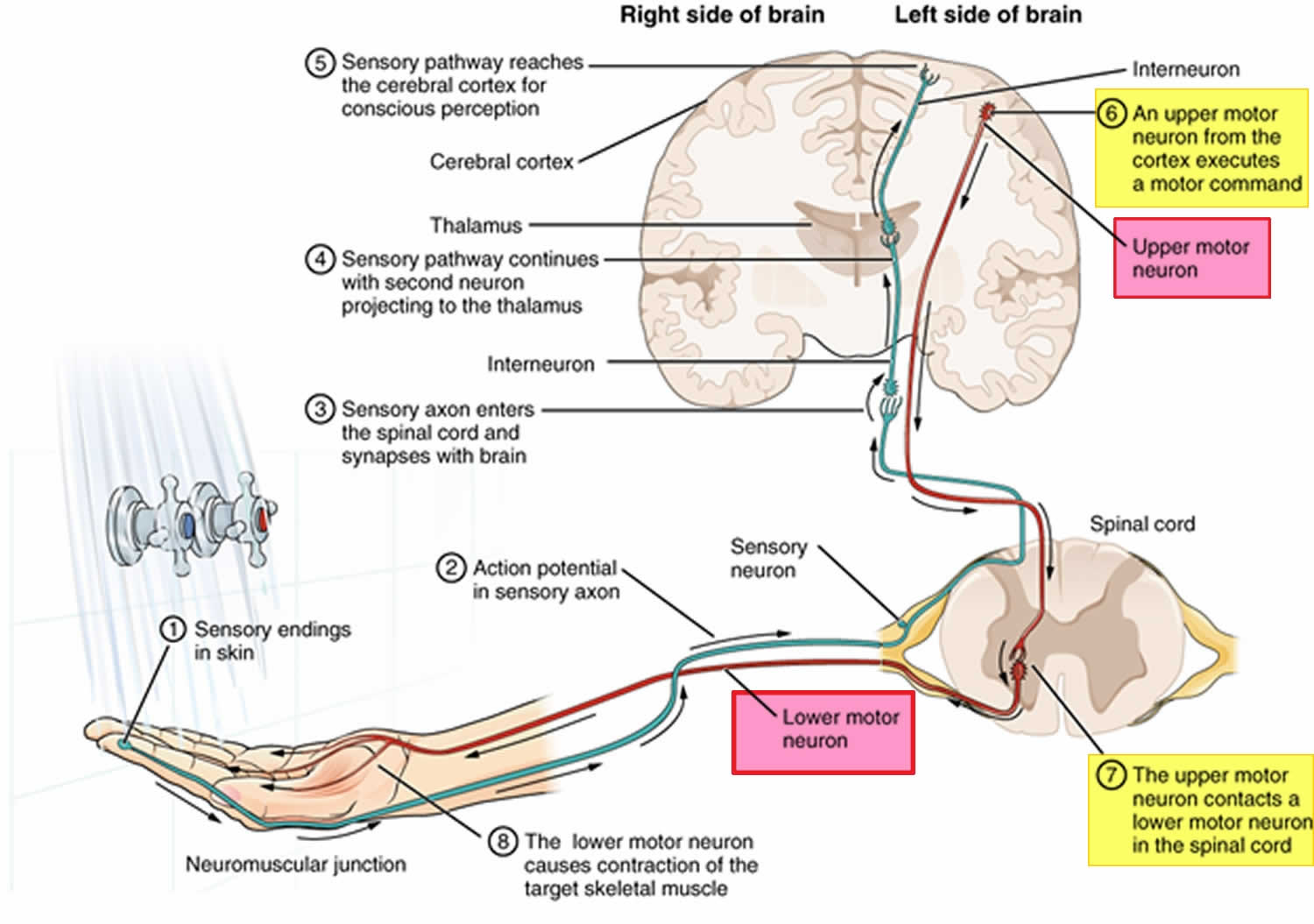
Figure 2. Kennedy’s syndrome
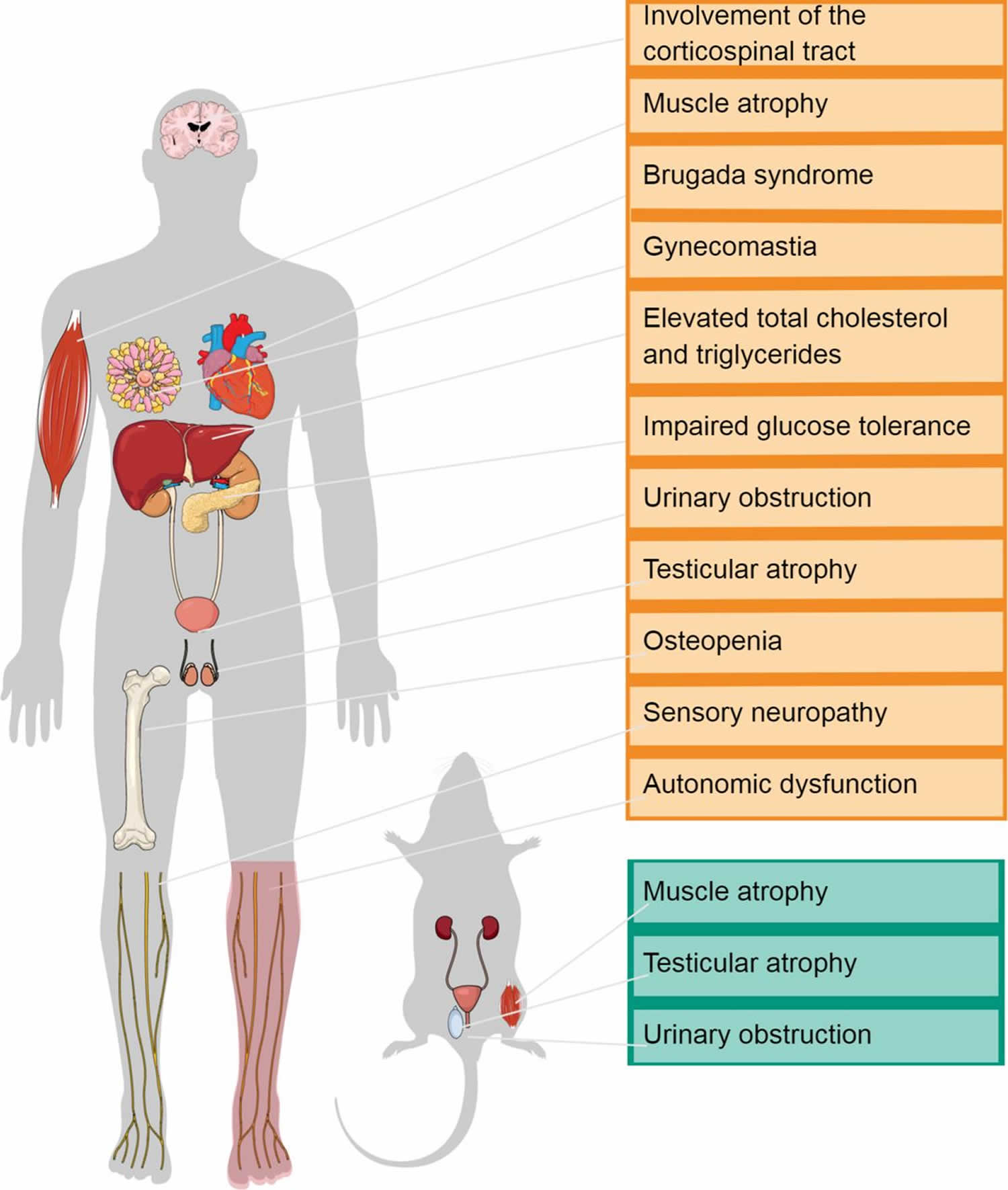
Figure 3. Kennedy’s disease
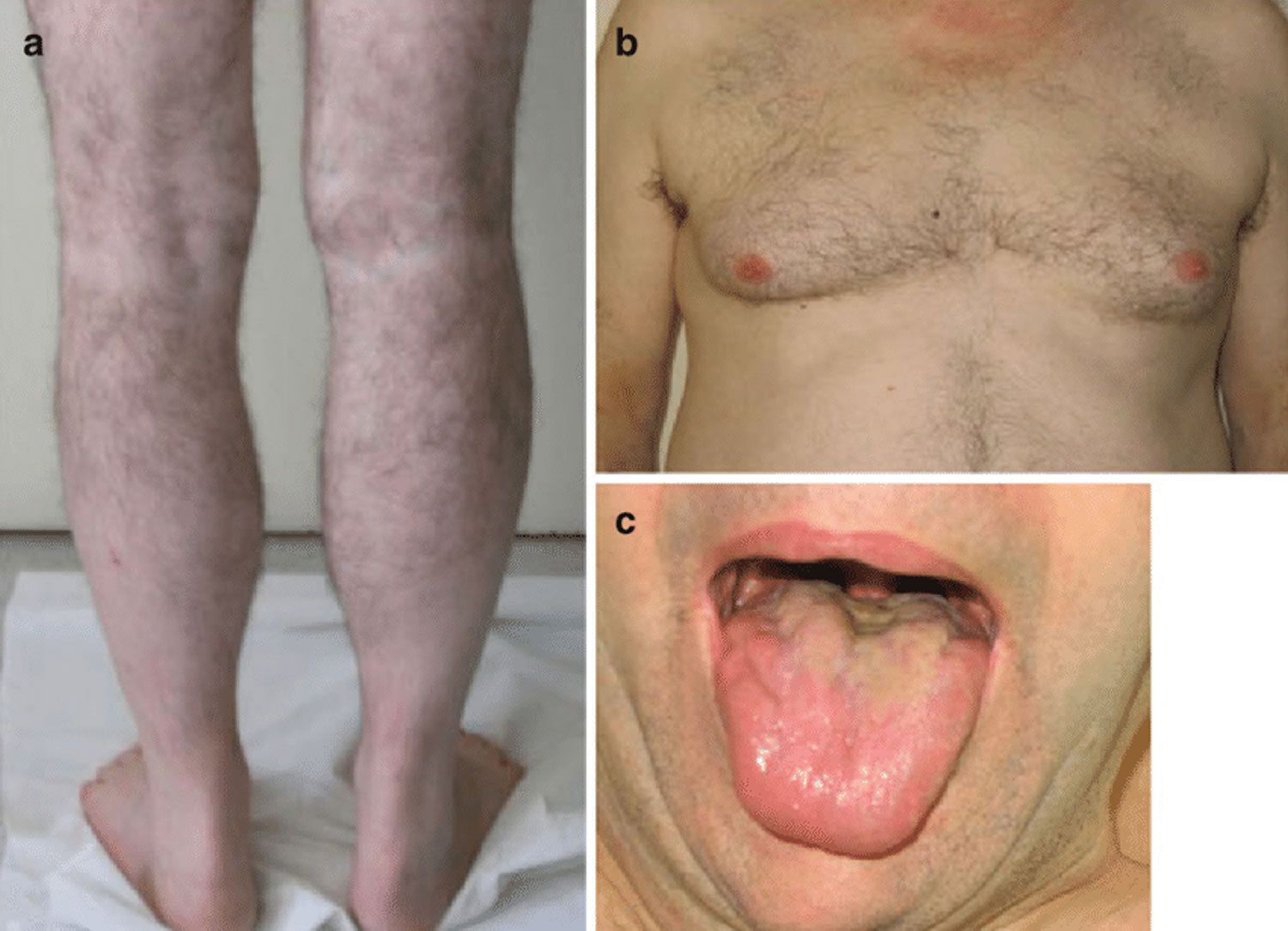
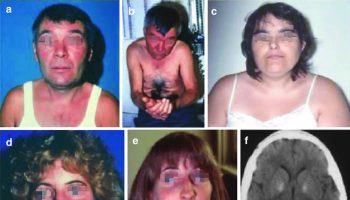
Footnotes: Kennedy’s disease patient with slight calf hypertrophy (a), gynecomastia (b), and tongue atrophy (c)
[Source 29 ]What can I do about tightening in my throat (laryngospasm)?
Sometimes you may experience tightening in your throat known as a laryngospasm or “dry drowning” 30, 31, 15. It is the result of a spasm in the adductor muscles – the muscles closing or bringing the vocal folds or vocal cords together, which can make it difficult to breathe and swallow. Laryngospasm happens when your voice box or the area of the windpipe below the voice box detect the entry of water or another substance. The vocal folds spasm and the airway shuts down. All of us have experienced it when a bug flies down your throat while you were starting to inhale, or you inhale a glass of water. The vocal cords very immediately and very effectively close. That closure is a benefit to protect the airway, but it makes “breathing in”, very difficult. The throat muscles weaken (atrophy) in a person with Kennedy’s Disease. Because of this, it becomes more difficult to swallow or clear the throat. Laryngospasms is a common occurrence reported in up to half of the patients with Kennedy’s disease and usually last for around 60 seconds, but can be longer or shorter. Some people experience more than one laryngospasm at a time. Although this can be distressing it will pass. It can help to try to remain calm and relaxed during a laryngospasm. Some people report that ‘straw breathing’ or ‘pursed-lip breathing’ during a laryngospasm is also helpful.
‘Straw breathing’ or ‘pursed-lip breathing’ is when you make the shape of your mouth as if you are sucking through a straw and slowly taking breaths in this way. It is thought that this slows down your breathing and allows your vocal folds to relax. Breathing in quickly can make the vocal folds move closer together, making it more difficult to breathe.
Some people have also found drinking small sips of water and/or the sniff-blow technique – taking two consecutive sniffs through the nose followed by a slow exhale through the mouth with pursed lips – helpful.
If you do experience laryngospasm, discuss this with your neurologist, as medications are available to control it. In some cases, Lorazepam medication placed under the tongue is prescribed to help manage laryngospasm.
In a study of 49 Kennedy’s disease patients, laryngospasm (47%) and heartburn (60%) were common. In patients with both laryngospasm and heartburn, anti-reflux therapy reduced laryngospasm frequency 31. There are many approaches to treating gastroesophageal reflux disease (GERD). These include behavioral changes, dietary changes, medications, and even surgery for particularly refractory cases. Lifestyle modifications for gastroesophageal reflux disease (GERD) include avoiding eating before lying down, sleeping with the back elevated, and not smoking. Avoiding foods that worsen gastroesophageal reflux disease (GERD) can help. This includes fatty foods, alcohol, caffeinated beverages, peppermint, and acidic liquids. Medications for gastroesophageal reflux disease (GERD) range from over-the-counter antacids to H2 receptor blockers and proton pump inhibitors. Fundoplication is a procedure to tighten the junction between the esophagus and stomach. This has traditionally been done surgically, but there are now endoscopic techniques that are less invasive.
Frequent choking often occurs as the person ages. Chewing foods longer, not trying to talk while eating, and not gulping liquids will all help. It is also found that daily throat exercises have helped minimize choking.
What can I do about swallowing difficulties?
In Kennedy’s disease, chewing and difficulty in swallowing (dysphagia) is associated with weakness of muscles in your mouth, tongue, and throat, and lead to coughing or a bout of choking, which can result in aspiration of solids, liquids or saliva. This can lead to an increased risk of inhaling small pieces of food or saliva into your lungs, which can cause chest infections and pneumonia. When foreign material enters the tracheobronchial tree, it can result in inflammation, increased mucus production, recurrent cough, chest congestion and fluid accumulation. Symptoms may develop gradually with increased difficulty breathing when active or lying flat, a feeling of suffocating or drowning, wheezing or gasping for breath, and a productive cough with frothy or foamy sputum.
Swallowing difficulties can also lead to weight loss, dehydration and lack of energy.
Ask your doctor for a referral to a speech and language therapist and a dietitian who will be able to assess your ability to chew and swallow safely. They will also be able to give you information about the types of diet, supplements or aids that might help you maintain a healthy diet.
Swallowing management approaches include:
- Changes in eating habits (posture, location, avoiding distractions);
- Training in safer and more effective swallowing techniques;
- Adjusting food consistency or texture by avoiding difficult to chew foods, food textures (avoidance of dry crumbly foods), and changing food consistency with thickeners.
Your nutritional status should be monitored and easy to swallow food supplements should be added to facilitate adequate protein, energy and vitamin intake.
Enteral nutrition through gastrostomy may be indicated in rare cases 32.
What can I do about ineffective cough?
An ineffective cough is coughing that fails to adequately clear your airways of mucus or other foreign substances. Coughing can help clear your breathing passages of foreign materials, fluids, irritants, and microbes. However, an effective cough has three phases: [1] deep inhalation; [2] forced exhalation against a closed glottis (opening in the vocal folds); and [3] a violent release of air when the glottis is forced open. The rapid release of air during an effective cough can expel particles and move secretions upward. Coughing also assists in clearing secretions from the lower airway, preventing the pooling of secretions in the air sacs (alveoli) which may reduce oxygen (O2) and carbon dioxide (CO2) exchange.
Muscle weakness from Kennedy’s disease can make it difficult to inhale a large breath, to tightly close the vocal folds preventing air leakage, and then to forcefully exhale. A weak cough due to Kennedy’s disease can make it more difficult to expel foreign materials leading to inflammation, increased mucus production and infections. In some cases, a weakened cough can lead to formation of mucus plugs that reduce airflow in larger airways and collapsed air sacs (alveoli) in smaller airways. If enough alveoli are blocked, a person’s oxygen levels will be negatively impacted over time.
A mechanical insufflator-exsufflator device or “cough assist” machine (Philips Cough Assist™ or Baxter Synclara) can assist your coughing by delivering a brief positive inspiratory pressure to your airway through a facemask or mouthpiece followed by a rapid switch to negative pressure 15. This simulates a natural cough, increasing inspiratory and expiratory volumes and moving tracheal and bronchial secretions up towards your mouth. Some insufflator-exsufflator devices or “cough assist” machines vibrate or oscillate the respiratory system to loosen secretions. Mechanical insufflator-exsufflator device has been shown to increase Peak Cough Flow in patients with neuromuscular disorders 33, 34. Several men with Kennedy’s disease have reported these devices to be very helpful and describe them as a “vacuum cleaner for your lungs” and “pulls the mucus up to your throat where you can spit it out”. Non-invasive mechanical ventilation is seldom required, and is best initiated after a joint neurological-respiratory consultation based on overnight pulse oximetry readings and early morning arterial blood gas analyses.
Oxygen therapy should only be considered in a palliative management setting as without pressure support it may lead to reduced respiratory drive.
What can I do about problems with my speech?
Kennedy’s disease may affect the muscles in your tongue and lips, making it difficult to speak. It’s rare for someone with Kennedy’s disease to lose their voice completely, but your speech may become unclear and slurred. Ask your doctor for a referral to a speech and language therapist for assessment of your needs.
Your speech and language therapist can advise:
- ways to manage your speech
- equipment that can help
- recording your voice to bank it for future use on communication aids.
Kennedy’s syndrome cause
Kennedy’s syndrome or Kennedy’s disease is caused by mutation in the androgen receptor (AR) gene on the X chromosome 35, 36, 18. The AR gene provides instructions for making a protein called an androgen receptor. Androgens are male sex hormones such as testosterone that are important for normal male sexual development before birth and during puberty and for the maintenance of male characteristics. Androgens have other important roles too, like keeping your bones strong, helping red blood cell production and sexual desire and function. Both males and females have androgens, but males naturally have more. Androgen receptors (AR) allow your body to respond appropriately to these male sex hormones.
The androgen receptors (ARs) are present in many of your body’s tissues where they attach (bind) to androgens. The resulting androgen-receptor complex then binds to DNA and regulates the activity of certain genes that play a role in male sexual development. By turning the genes on or off as necessary, the androgen receptor complex helps direct the development of male sex characteristics. Androgens and androgen receptors also have other important functions in both males and females, such as regulating hair growth and sex drive.
In one region of the AR gene, a DNA segment known as Cytosine, Adenine, Guanine (CAG) trinucleotide is repeated multiple times. This CAG segment is called a triplet or trinucleotide repeat. In most people, the number of CAG trinucleotide repeat in the AR gene ranges from fewer than 10 to about 36. In people with Kennedy’s syndrome or Kennedy’s disease, the CAG segment is repeated from 38 times to more than 60 times, and it may be two or three times its usual length. Although the extended CAG region changes the structure of the androgen receptor, it is unclear how the altered protein disrupts nerve cells in your brain and spinal cord. Researchers believe that a fragment of the androgen receptor protein containing the CAG segment accumulates within these nerve cells and interferes with normal cell functions. The nerve cells gradually die, leading to the muscle weakness and wasting (atrophy) seen in Kennedy’s disease. People with a higher number of CAG repeats tend to develop signs and symptoms of Kennedy’s disease at an earlier age.
Kennedy’s disease inheritance pattern
Kennedy’s disease is inherited in X-linked recessive pattern. A condition is considered X-linked if the mutated gene that causes the disorder is located on the X chromosome, one of the two sex chromosomes. Males have only one X chromosome (XY) and will develop Kennedy disease if they inherit the X chromosome containing the disease gene. In most cases, males experience more severe symptoms of Kennedy’s disease than females who have two X chromosomes (XX). Affected males with X-linked disorders will always pass the gene to their daughters but will only pass their normal Y chromosome to their sons. Therefore, all the daughters of an affected male will be carriers for Kennedy’s disease, while sons of an affected male will not have the disease. Sons of female carriers have a 50 percent chance of inheriting the disease, while daughters have a 50 percent chance of becoming carriers.
Normal females have two X chromosomes (XX), in which one is an activated chromosome and the other is inactivated. Female carriers with a mutation in one copy of the AR gene in each cell for Kennedy disease typically do not show symptoms because the androgen receptor must bind to its ligand, testosterone, to translocate to the nucleus and perform its functions. As females have low circulating levels of testosterone, Kennedy disease female carriers do not activate their mutant androgen receptors, thus rendering the mutant state of the androgen receptor protein harmless.
A few females with mutations in both copies of the AR gene have had mild features related to Kennedy’s disease, including muscle cramps and occasional tremors. Researchers believe that the milder signs and symptoms in females may be related to lower androgen levels.
A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Figure 4. Kennedy’s disease X-linked inheritance pattern
Genotype-phenotype correlations
Studies of the number of CAG repeats in the AR gene in males with Kennedy’s disease have established a correlation between number of CAG repeats and disease severity 37. In general, CAG repeat number inversely correlates with the age of onset of muscle weakness, difficulty climbing stairs, and wheelchair dependence 38. Therefore, males with Kennedy’s disease whose AR gene have a larger number of CAG repeats tend to have earlier disease onset and more rapid progression 39. For example, early onset (age 8-15 years) and rapid progression have been described in a family in which affected individuals have AR gene of 50-54 CAG repeats 40.
However, these correlations are only generalizations and exceptions have been reported. For example, a male in a family with Kennedy’s disease with AR gene of 37 CAG repeats (the average number of repeats in affected males) has been reported to be asymptomatic at age 46 years 41. The largest AR gene CAG repeat expansion reported in a person with Kennedy’s disease is 68 42.
CAG repeat number may account for approximately 60% of clinical heterogeneity in Kennedy’s syndrome patients, indicating that other factors in addition to CAG repeat number determine age of disease onset and rate of disease progression. Indeed, relatives with Kennedy’s disease with an identical CAG repeat number may have considerably different disease courses. Genetic, environmental and epigenetic factors also likely to play some role in influencing Kennedy’s disease progression 2.
Kennedy’s syndrome signs and symptoms
People with Kennedy’s syndrome begin to develop neurological symptoms between 30 to 50 years of age, although cases from the ages of 15 up to 59 have been described. These early symptoms include:
- Muscle weakness and wasting in arms and legs muscles (proximal > distal)
- Muscle cramps and spasms in arms and legs muscles (proximal > distal)
- Face, mouth and tongue muscle weakness (bulbar muscles) leading to difficulty with swallowing, speech and breathing
- Difficulty with speaking (dysarthria) and difficulty swallowing (dysphagia)
- Muscle twitching (fasciculations)
- Hand tremors and trembling in certain positions
- Enlarged breasts (gynecomastia)
- Numbness
- Reduced fertility or infertility
- Testicular atrophy (shrunken testicles) with low sperm count (oligospermia) or complete absence of sperm (azoospermia)
- Erectile dysfunction.
Kennedy’s syndrome affects the lower motor neurons that are responsible for the movement of many muscles in your legs, arms, mouth and throat. Affected males show signs of muscle twitching (fasciculations), often in the tongue, face and/or hand, followed by muscle weakness and problems with facial muscles. These lower motor neurons, which connect your spinal cord to your muscles, become defective and die, so the muscles cannot contract. The destruction of these nerves is the main reason for numbness, muscle weakness and inability to control muscle contraction. With lack of normal neuromuscular function, a patient may experience hypertrophied calves in which the calf muscles thicken due to muscle cramps. Some patients may have one side of the body more affected than the other side.
Kennedy’s disease also affects nerves that control the bulbar (throat and neck) muscles, which are important for breathing, speaking and swallowing. Androgen insensitivity can also occur, sometimes beginning in adolescence and continuing through adulthood, characterized by enlarged breasts (gynecomastia), decreased masculine appearance and infertility. Patients may experience problems such as low sperm count or infertility and erectile dysfunction (inability to achieve an erection). Still others develop non-insulin-dependent diabetes mellitus (type 2 diabetes).
Although it is known that the age of onset of non-motor manifestations of Kennedy’s syndrome is independent of CAG repeat length, medical literature indicates that there is also a clear negative correlation between the age of onset of motor signs and symptoms and CAG repeat number 28.
Affected Males
Neurologic findings
Neurologic symptoms typically begin between age 30 and 50 years 25, 26. Onset of neurologic symptoms does not usually occur in childhood or adolescence.
Early signs are difficulty with walking and a tendency to fall 6. Many individuals have muscle cramps, while others complain of an action tremor 43. Deep tendon reflexes are decreased.
After one to two decades of symptoms, most affected individuals have difficulty climbing stairs 6. With time, atrophy of the proximal and distal musculature becomes evident. About one third of affected individuals require a wheelchair 20 years after the onset of symptoms 6.
Most individuals eventually show involvement of the bulbar muscles and have difficulty with speech articulation and swallowing 6. Severely affected individuals (many of whom are nonambulatory) are at risk for aspiration pneumonia and ventilatory failure because of weakness of the bulbar and respiratory musculature 44. This complication is the main life-threatening problem in Kennedy’s disease, and likely becomes an issue for only a minority of individuals. Therefore, the majority of individuals with Kennedy’s disease have a normal life expectancy and do not die from direct complications of their motor neuron disease. Fifteen of 223 persons in the Atsuta et al 17 study died at a mean age of 65 years.
Affected males may also have degeneration of the dorsal root ganglia, leading to mild (usually subclinical) abnormalities in sensory function in the distal extremities 43.
Electrodiagnostic studies are consistent with diffuse denervation atrophy, anterior horn cell loss, and sensory neuronopathy 45.
Heart and metabolic abnormalities
Two reports have noted that some individuals with Kennedy’s syndrome develop abnormal heart rhythms and may occasionally show hypertrophic cardiomyopathy-type changes 46, 47. While the pathologic significance of these findings is unclear, there has also been a growing appreciation that most individuals with Kennedy’s syndrome exhibit hyperlipidemia and insulin resistance, and that these metabolic changes often qualify such individuals for a diagnosis of nonalcoholic fatty liver disease (NAFLD) 26, 48, 49. As hyperlipidemia and insulin resistance can predispose to coronary artery disease, there is growing concern that individuals with Kennedy’s syndrome may be at elevated risk for heart attack (myocardial infarction) with aging, especially given a decline in physical activity and exercise as a result of ongoing neuromuscular disease. Hence, it is prudent to follow lipids, cholesterol, and blood sugars annually and refer individuals with Kennedy’s syndrome to a cardiologist or endocrinologist for management of any metabolic or heart abnormalities.
Genito-urinary disorders
The genitourinary tract involvement is among the previously unrecognized non-neuromuscular features of Kennedy’s syndrome. 30-40% of men living with Kennedy’s disease may experience lower urinary tract symptoms, such as urinary urgency or discomfort during urination. Querin et al 50 investigated a cohort of 71 patients with Kennedy’s syndrome using the International Prostate Symptom Score (IPSS), followed by urodynamic and ultrasound investigations, when abnormalities in IPSS were found. Forty per cent of cases were either moderately or severely symptomatic for lower urinary tract symptoms (LUTS). The most prevalent finding was that of a bladder outlet blockage in the absence of any indication for benign prostatic hyperplasia from biochemical and ultrasound investigations. It is not currently known whether lower urinary tract symptoms are due to androgen insensitivity or a toxic AR expansion gain of function. It is nonetheless intriguing that knock-in mice carrying the AR expansion die due to alterations in the neuromuscular bladder axis, which determine acute urinary retention in the absence of a physical obstruction, thus further supporting the direct mechanistic link between the AR expansion and urinary dysfunction 51. A small proportion may need to use a catheter.
Histopathology
Degeneration of anterior horn cells in the spinal cord of affected individuals is observed 25, 52. Changes in muscle include evidence of myopathy 53, 54, in addition to neurogenic muscle atrophy. Immunohistochemistry shows inclusions of mutated androgen receptor (AR) protein 55.
Androgen insensitivity
Symptoms of androgen insensitivity typically begin in adolescence with gynecomastia, which is observed frequently in affected males 56. Variability in disease severity and progression occurs both within and between families 57. This is especially true of the androgen insensitivity signs of testicular atrophy and low sperm count (oligospermia) or complete absence of sperm (azoospermia) with reduced fertility. Males with Kennedy’s disease may not be able to grow a thick beard and may have difficulty conceiving.
The androgen insensitivity can be of greater concern to affected individuals than the motor neuron disease, especially early in the course of the disorder 58.
Female carriers
Neurologic findings
Females who are carriers of full-penetrance alleles of greater than 38 CAG repeats in AR gene are usually asymptomatic. While number of carriers have experienced muscle cramps or occasional tremors, female carriers usually do not have significant motor neuron disease 59.
Females who are symptomatic may have an abnormal electromyography 60.
Androgen insensitivity
Kennedy’s disease is a sex-limited disorder, with females protected by having low levels of circulating androgens leading to lower levels of androgen receptor stimulation. In addition, due to X-chromosome inactivation, females have only a portion of actively transcribed full-penetrance alleles (CAG>37), but it is the low level of circulating androgen that likely accounts for limited to absent symptoms in heterozygous female carriers or in females with biallelic full-penetrance AR alleles.
Kennedy’s syndrome diagnosis
A neurologist first takes a careful record of your medical history and family history and performs a neurological exam. A diagnosis of Kennedy disease or spinal and bulbar muscular atrophy (SBMA) is suspected based on physical signs and symptoms, and sometimes family history.
Kennedy’s disease should be suspected in males with the following clinical features and family history.
Clinical features:
- Adolescent-onset signs of androgen insensitivity (e.g., gynecomastia)
- Post-adolescent onset of:
- Spinal lower motor neuron disease with muscle weakness of the limbs or muscle cramps
- Bulbar lower motor neuron disease with fasciculations of the tongue, lips, or perioral region; dysarthria and difficulty swallowing
- No signs of upper motor neuron disease (e.g., hyperreflexia, spasticity)
Family history consistent with X-linked inheritance
Note: Lack of a family history consistent with X-linked inheritance does not preclude the diagnosis.
You may have the following tests:
- Blood tests. Blood tests check for infections or other possible causes of muscle weakness.
- Serum creatine kinase (CK) testing is not indicated. Increased creatine kinase (CK) levels may be detected in 80 to 94% of Kennedy’s disease patients, often in association with increased transaminases (AST, ALT) 17. CK values range from 31 to 4955 IU / L, with an average serum level of 863 IU / L (normal < 200 IU / L) 17. Rarely, the work-up for an unexplained elevation of creatine kinase (CK) and transaminase (AST, ALT) level may lead to the diagnosis of Kennedy’s disease 61.
- Decreased serum creatinine is thought to strongly correlate with clinical disability 62, and may already be detected in the preclinical phase of the disease 63.
- Brain MRI. Magnetic resonance imaging (MRI) to look at your brain or spine for areas of damage or nerve cells that have broken down. An MRI also can show other causes of symptoms, such as structural problems, multiple sclerosis or spinal cord tumors.
- Existing brain studies have primarily used tractography and morphometry and identified widespread white matter and grey matter abnormalities compared to controls 64, 65, 66. In addition to the existing brain studies, innovative studies also examined the biomarker potential of muscle MRI 67, 68. Despite these efforts, no unifying Kennedy’s disease imaging signature has been established to date, and a recent imaging study failed to capture differences between patients and healthy controls 69.
- Electromyogram (EMG). The test evaluates the electrical activity of your muscles when they contract and when they’re at rest. This test also measures the involvement of lower motor neurons. This can help tell the difference between an upper motor neuron disease and upper motor neuron disease. During an EMG, your doctor inserts a needle electrode through your skin into different muscles.
- Nerve conduction studies. A nerve conduction study can determine if you have nerve damage. A low amount of electrical current measures your nerves’ ability to send impulses to muscles in different areas of your body.
- Lumbar puncture, also known as a spinal tap. A spinal tap can help rule out multiple sclerosis, infections and other conditions. A thin, hollow needle inserted into your spinal canal removes small samples of cerebrospinal fluid. This is the fluid that surrounds your brain and spinal cord. The samples are then analyzed in a lab.
- Nerve and muscle biopsy. Muscle and nerve biopsy is not commonly performed and is seldom necessary to establish the diagnosis. In some instances, nerve and muscle biopsy may be performed in cases of Kennedy’s syndrome when the diagnosis is not clear.
- Muscle biopsy typically reveals both myopathic (nuclear centralisation, myofibrillar disorganisation) and neurogenic (angular fibres or type grouping) changes 70. These abnormalities may occasionally be useful for differentiating Kennedy’s syndrome from diagnosis with amyotrophic lateral sclerosis (ALS) 71.
- Sural-nerve biopsy may show loss of large-diameter myelinated axons 72.
- In a report of 6 cases, muscle biopsy showing marked predominance of type 1 (slow-twitch) muscle fibers with variable and diffuse atrophy was present in two patients in whom biopsy material was available 73.
- Predominance of type 2 (fast-twitch) muscle fibers has been described in other cases.
- In another series, both neurogenic and myopathic changes were reported in muscle biopsies from patients with Kennedy’s syndrome, as well as in female carriers of Kennedy’s syndrome 70.
Kennedy’s disease diagnosis can be confirmed by molecular genetic testing on a blood sample for CAG trinucleotide repeat expansion in the AR gene. Individuals with greater than 36 CAG trinucleotide repeats in the AR gene are diagnosed with the condition. Genetic counseling also may be recommended.
Clinical testing and work-up
Annual examinations to assess muscle strength may be appropriate. It has recently been noted that individuals with Kennedy’s syndrome are at increased risk of developing heart disease and liver disease due to hyperlipidemia (high cholesterol) and insulin resistance. Some patients develop irregular heartbeats (heart arrhythmias) or occasionally hypertrophic cardiomyopathy (a heart condition where the heart muscle becomes abnormally thickened, making it harder for the heart to pump blood), and hyperlipidemia and insulin resistance may predispose to coronary artery disease. Some patients display significant metabolic changes consistent with non-alcoholic fatty liver disease (NAFLD). Hence, Kennedy’s disease patients should receive an ECG and annual lipid, cholesterol and blood sugar testing and referred to a cardiologist or endocrinologist for management of any metabolic or heart abnormalities.
Kennedy’s syndrome differential diagnosis
Symptoms of the following disorders can be similar to those of Kennedy’s disease.
- Adrenoleukodystrophy (ALD) is one of many different leukodystrophies. The adolescent or adult-onset form of the disorder is called adrenomyeloneuropathy (AMN), and symptoms of this form of adrenoleukodystrophy (ALD) may be similar to those of Kennedy disease. Symptoms typically appear between the ages of 21 and 35. They may include progressive leg stiffness, spastic partial paralysis of the lower extremities and ataxia (clumsiness in walking). Decreased function of the sex glands may be present. Adult-onset adrenoleukodystrophy (ALD) progresses slowly; however, it can ultimately result in deterioration of brain function.
- Amyotrophic lateral sclerosis (ALS) also called called Lou Gehrig’s disease is one of a group of disorders known as motor neuron diseases. Amyotrophic lateral sclerosis (ALS) is characterized by the progressive degeneration and eventual death of motor neurons in the brain, brainstem and spinal cord (upper and lower motor neurons) that facilitate communication between the nervous system and voluntary muscles of the body. Ordinarily, motor neurons in the brain (upper motor neurons) send messages to motor neurons in the spinal cord (lower motor neurons) and then to various muscles. ALS affects both the upper and lower motor neurons, so that the transmission of messages is interrupted, and muscles gradually weaken and waste away. As a result, the ability to initiate and control voluntary movement is lost. Ultimately, ALS leads to respiratory failure because affected individuals lose the ability to control muscles in the chest and diaphragm. As many as 10% of Kennedy disease patients may be misdiagnosed with ALS prior to determining that they really have Kennedy disease.
- Spinal muscular atrophy type 3 (SMA type 3) is inherited in an autosomal recessive pattern. Major symptoms may include wasting and weakness in the muscles of the arms and legs, twitching, clumsiness in walking and eventually loss of reflexes. Spinal muscular atrophy type 3 is not apparent at birth but typically appears during the first ten to twenty years of life.
- Myasthenia gravis is a neuromuscular disorder primarily characterized by muscle weakness and muscle fatigue. Although the disorder usually becomes apparent during adulthood, symptom onset may occur at any age. Myasthenia gravis may be restricted to certain muscle groups, particularly those of the eyes (ocular myasthenia gravis) or may become more generalized (generalized myasthenia gravis), involving multiple muscle groups. Most individuals with myasthenia gravis develop weakness and drooping of the eyelids (ptosis); weakness of eye muscles, resulting in double vision (diplopia); and excessive muscle fatigue following activity. Additional features commonly include weakness of facial muscles; impaired articulation of speech (dysarthria); difficulties chewing and swallowing (dysphagia); and weakness of the upper arms and legs (proximal limb weakness). About 10 percent of affected individuals may develop potentially life-threatening complications due to severe involvement of muscles used during breathing (myasthenic crisis). Myasthenia gravis results from an abnormal immune reaction in which the body’s natural immune defenses (i.e., antibodies) inappropriately attack and gradually destroy certain receptors at the neuromuscular junction that receive nerve impulses (antibody-mediated autoimmune response).
- Oculopharyngeal muscular dystrophy (OPMD) is a rare genetic muscle disorder with onset during adulthood, most often between 40 and 60 years of age. Oculopharyngeal muscular dystrophy is characterized by slowly progressive muscle disease (myopathy) affecting the muscles of the upper eyelids and the throat. Affected individuals may develop drooping of the eyelids (ptosis), double vision (diplopia) and/or difficulty swallowing (dysphagia). Eventually, additional muscles may become involved including those of the upper legs and arms (proximal limb weakness). In some patients, muscle weakness of the legs may eventually cause difficulty walking. Oculopharyngeal muscular dystrophy is typically inherited in an autosomal dominant pattern.
- Charcot-Marie-Tooth (CMT) disease is a group of disorders in which the motor and/or sensory peripheral nerves are affected, resulting in muscle weakness and atrophy as well as sensory loss. Symptoms occur first in the distal legs and later in the hands. The nerve cells in individuals with this disorder are not able to send electrical signals properly because of abnormalities in the nerve axon or abnormalities in the insulation (myelin) around the axon. In Charcot-Marie-Tooth specific gene mutations are responsible for the abnormal function of the peripheral nerves. In many forms of Charcot-Marie-Tooth these genes are known and in others, while the condition is known to be inherited, the specific gene has not yet been identified.
- Polymyositis is a systemic connective tissue disorder characterized by inflammatory and degenerative changes in the muscles, leading to symmetric weakness and some degree of muscle atrophy. The areas principally affected are the hip, shoulders, arms, pharynx and neck.
Kennedy’s syndrome treatment
Currently, there is no known treatment or cure for Kennedy disease. Physical therapy, occupational therapy, and speech therapy are commonly used to adapt to the progressing disease and maintain an individual’s skills 74. Braces, walkers, and wheel chairs are used for ambulation. Breast reduction surgery is sometimes used as needed in patients with gynecomastia 75. Testosterone is not an appropriate treatment, as it can make the disease worse 76.
Table 1. Recommended evaluations following initial diagnosis of Kennedy’s syndrome
| System/Concern | Evaluation | Comment |
|---|---|---|
| Neurologic | Complete neurologic exam | Assess:
|
| Musculoskeletal/ activities of daily living | Orthopedics / physical medicine & rehab / physical therapy (PT) eval | To include assessment of:
|
| Occupational therapy (OT) | Assess:
| |
| Dysarthria | For those with dysarthria: speech-language evaluation | Referral for speech therapy as needed |
| Dysphagia | For those with frequent choking or severe dysphagia, assess nutritional status & aspiration risk. | Consider involving a gastroenterology/nutrition/feeding team, incl formal swallowing eval. |
| Respiratory function | By pulmonologist | Assess respiratory function & need for respiratory support. |
| Endocrine |
| Consider mastectomy if gynecomastia presents in teenage years or young adult years causing psychosocial stress to self-image / gender identity. |
| Genetic counseling | By genetics professionals 1 | To obtain a pedigree & inform affected persons & their families re nature, mode of inheritance (MOI), & implications of Kennedy disease to facilitate medical & personal decision making |
Footnotes: 1 Clinical geneticist, certified genetic counselor, certified genetic nurse, genetics advanced practice provider
[Source 6 ]Table 2. Kennedy’s syndrome treatment options
| Manifestation/Concern | Treatment | Considerations/Other |
|---|---|---|
| Upper motor neuron and lower motor neuron involvement Activities of daily living (ADL) | Physical medicine & rehabilitation / PT & OT | Ankle-foot braces, walkers, wheelchairs, hospital beds, toileting equipment, lifts to improve functionality |
| Dysarthria | Speech-language therapy | Use of augmentative communication devices |
| Dysphagia | Feeding evaluation & therapy |
|
| Gynecomastia | Breast reduction surgery as needed | |
| Cardiac & other manifestations | Standard treatment per cardiologist &/or endocrinologist | Monitor blood sugar, lipids, & cholesterol for insulin resistance & nonalcoholic fatty liver disease. |
| Family/caregiver support & resources | Psychosocial support & education via caregiver & patient support groups | To ↓ stress & burden on caregivers |
Clinical follow-up
Appropriate measures include:
- Strength testing (annually or as needed)
- Pulmonary function tests (annually in advanced cases)
- Dysarthria: Speech-language therapy (annually or as needed)
- Dysphagia Feeding evaluation (annually or as needed)
- Heart complications (annually)
- Cholesterol & triglycerides
- Liver function testing in those with high cholesterol &/or triglycerides
- Any additional assessment of cardiac health per cardiologist
- Family/caregiver support & resources (annually or as needed)
Agents and circumstances to avoid
Individuals with a tendency to fall should avoid slippery or rough walking surfaces.
Therapies under investigation
High-dose testosterone. At least one clinical trial of high-dose oral testosterone has been undertaken; no significant benefit was reported for the androgen treatment group 76. Based on research in Drosophila and mouse models of Kennedy’s disease, many investigators believe that androgen treatment may be harmful. Nevertheless, some rationale supports the use of testosterone in Kennedy disease 77.
Anti-androgen therapy. There is no consensus or clear evidence as to whether anti-androgen therapy is an effective form of treatment for the neurologic complications.
- Anti-androgen therapy shows promise based on studies in Drosophila and mouse models as well as knowledge of the molecular basis of Kennedy’s disease. For these reasons, a Japanese group 78 performed a clinical trial of leuprorelin in individuals with Kennedy’s disease who were followed over 48 weeks: significant improvement was observed in cricopharyngeal opening duration, but in no other outcome measures. In particular, there was no effect on the primary outcome measure (the ALS Functional Rating Scale or ALSFRS) in the period of randomization. Although the trial was continued as an open label extension, and encouraging results were reported, the conclusion was that this clinical trial did not establish efficacy for anti-androgen therapy in Kennedy’s disease 79.
- A larger subsequent study with swallow function as the primary outcome measure also did not show an overall benefit, except in post hoc analysis of subjects in whom disease duration was less than ten years 80.
- Another anti-androgen therapy approach was attempted 81: individuals with Kennedy’s disease were randomized to placebo or dutasteride, a drug that blocks the conversion of testosterone to dihydrotestosterone (DHT). The rationale was that DHT may mediate many of the toxic effects, and this drug would permit affected individuals to retain the anabolic effects of testosterone, thereby diminishing the side effects of anti-androgen therapy. However, this study did not show a significant effect of dutasteride on the progression of muscle weakness in Kennedy’s disease.
Hence, the utility of anti-androgen therapy as a treatment for Kennedy’s disease remains unclear. Furthermore, it is possible that anti-androgen therapies, even if effective, would need to be administered prior to disease onset or early on in the neurodegenerative process. More importantly, the side effects of anti-androgen therapies would probably far outweigh the therapeutic benefit for most individuals, and likely should be reserved for people with Kennedy’s disease who are wheelchair bound or exhibit pronounced bulbar weakness.
Creatine supplementation. Studies of amyotrophic lateral sclerosis (ALS) suggest that creatine supplementation may temporarily enhance muscle strength and exercise performance in this motor neuron disease 82, prompting speculation that it may offer a similar benefit to individuals with Kennedy’s disease; this remains to be tested.
Experimental therapies in animal models
- Other interventions that have been shown to have benefit in mouse models of Kennedy’s disease include the HSP-90 inhibitors 17-AAG and 17-DMAG, the synthetic curcumin derivative ASC-J9, and insulin-like growth factor 1 83.
- More recently, one group directly examined the role of muscle expression of mutated AR in Kennedy’s disease disease pathogenesis by developing a BAC transgenic mouse model featuring a floxed first exon to permit cell-type specific excision of a human AR transgene 84. They engineered the human AR transgene to carry 121 CAG repeats (BAC fxAR121), and found that BAC fxAR121 mice develop a gender-restricted, progressive neuromuscular phenotype, characterized by weight loss, motor deficits, muscle atrophy, myopathy, and shortened life span. By terminating expression of mutated AR in the skeletal muscles of BAC fxAR121 male mice, this study revealed a crucial role for muscle expression of mutated AR in Kennedy’s disease disease pathogenesis. Hence, this work predicts that muscle-directed therapies hold great promise as definitive treatments for Kennedy’s disease motor neuron degeneration.
- Another study sought to ameliorate toxicity in mouse models of Kennedy’s disease by suppressing polyQ-AR expression using antisense oligonucleotides (ASOs) 85. This investigation developed compounds to specifically target AR expression in the periphery, and using two mouse models, found that peripheral gene suppression of mutated AR rescues deficits in muscle weight, fiber size, and grip strength; reverses changes in muscle gene expression; and extends the life span of mutated males. Interestingly, delivery of an anti-AR ASO to the CNS also elicited a modest improvement in these disease read-outs in a Kennedy’s disease mouse model, but was much less effective than peripheral delivery. Hence, this report, together with the genetic rescue study of Kennedy’s disease 84, strongly suggests that peripheral administration of therapies directed to muscle should be explored in humans with Kennedy’s disease. Preparations are underway to initiate a clinical trial of anti-AR ASO therapy via peripheral delivery in males with Kennedy’s disease.
Kennedy’s syndrome prognosis
Generally, Kennedy’s syndrome follows a gradual progression, starting with postural tremors of the upper limb in the early thirties 86, 87. Compared with other motor neuron diseases, such as amyotrophic lateral sclerosis (ALS), disease progression is relatively slow, with muscle strength declining by 2% per year 87. In addition to the onset of difficulty speaking (dysarthria) and difficulty swallowing (dysphagia) in the forties, lower limb motor deficits also develop, eventually requiring the use of walking aids by the fifth decade 86. Finally, patients become wheelchair-bound by, or during, their sixth decade 86.
The life span of individuals with Kennedy’s disease is usually normal. Patients with early-stage involvement of bulbar (throat and neck) muscles often die from recurrent aspiration pneumonia in their fifties 88.
Kennedy’s syndrome life expectancy
Kennedy’s disease life expectancy is normal, though a small percentage of patients (about 10%) succumb to the disease in their 60’s or 70’s due to swallowing complications such as aspiration pneumonia and asphyxiation resulting from the weak muscles in the neck and throat, causing difficulty with speech, swallowing, and breathing 88.
- Kennedy WR, Alter M, Sung JH. Progressive proximal spinal and bulbar muscular atrophy of late onset. A sex-linked recessive trait. Neurology. 1968 Jul;18(7):671-80. doi: 10.1212/wnl.18.7.671[↩]
- Pradat P.-F., Bernard E., Corcia P., Couratier P., Jublanc C., et al. The French national protocol for Kennedy’s disease (SBMA): consensus diagnostic and management recommendations. Orphanet J. Rare Dis. 2020 Dec;15(90):1–21. doi: 10.1186/s13023-020-01366-z[↩][↩][↩][↩]
- Müller KI, Nilssen Ø, Nebuchenykh M, Løseth S, Jonsrud C, Hoem G, Van Ghelue M, Arntzen KA. Kennedy disease in two sisters with biallelic CAG expansions of the androgen receptor gene. Neuromuscul Disord. 2022 Jan;32(1):75-79. doi: 10.1016/j.nmd.2021.11.007[↩]
- What is Kennedy’s Disease. https://kennedysdisease.org/what-is-kd/overview.html[↩][↩]
- Finsterer J, Soraru G. Onset Manifestations of Spinal and Bulbar Muscular Atrophy (Kennedy’s Disease). J Mol Neurosci. 2016 Mar;58(3):321-9. doi: 10.1007/s12031-015-0663-x[↩]
- La Spada A. Spinal and Bulbar Muscular Atrophy. 1999 Feb 26 [Updated 2022 Dec 15]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1333[↩][↩][↩][↩][↩][↩][↩]
- Bellai DJ, Rae MG. A systematic review of the association between the age of onset of spinal bulbar muscular atrophy (Kennedy’s disease) and the length of CAG repeats in the androgen receptor gene. eNeurologicalSci. 2024 Jan 28;34:100495. doi: 10.1016/j.ensci.2024.100495[↩]
- Grunseich C., Fischbeck K.H. Spinal and bulbar muscular atrophy. Neurol. Clin. 2015 Nov;33(4):847–854. doi: 10.1016/j.ncl.2015.07.002[↩]
- SPINAL AND BULBAR MUSCULAR ATROPHY, X-LINKED 1; SMAX1. https://www.omim.org/entry/313200[↩]
- Finsterer J, Scorza FA. Central nervous system abnormalities in spinal and bulbar muscular atrophy (Kennedy’s disease). Clin Neurol Neurosurg. 2019 Sep;184:105426. doi: 10.1016/j.clineuro.2019.105426[↩]
- Deng X, Tan EK. Spinal bulbar muscular atrophy-Kennedy’s disease. QJM. 2024 Apr 12;117(4):289. doi: 10.1093/qjmed/hcad268[↩]
- Grunseich C, Fischbeck KH. Molecular pathogenesis of spinal bulbar muscular atrophy (Kennedy’s disease) and avenues for treatment. Curr Opin Neurol. 2020 Oct;33(5):629-634. doi: 10.1097/WCO.0000000000000856[↩]
- Teive HAG, Coutinho L, Cardoso FEC, Tsuji S. Spinal and bulbar muscular atrophy: Kennedy’s disease and its first description by Hiroshi Kawahara in 1897. Rev Neurol (Paris). 2023 Oct 26:S0035-3787(23)01071-8. doi: 10.1016/j.neurol.2023.07.016[↩]
- Manzano R, Sorarú G, Grunseich C, et al. Beyond motor neurons: expanding the clinical spectrum in Kennedy’s disease. Journal of Neurology, Neurosurgery & Psychiatry 2018;89:808-812. https://jnnp.bmj.com/content/89/8/808[↩][↩][↩]
- Pradat, PF., Bernard, E., Corcia, P. et al. The French national protocol for Kennedy’s disease (SBMA): consensus diagnostic and management recommendations. Orphanet J Rare Dis 15, 90 (2020). https://doi.org/10.1186/s13023-020-01366-z[↩][↩][↩][↩]
- Sumner CJ, Fischbeck KH. Jaw drop in Kennedy’s disease. Neurology 2002;59:1471–2. doi:10.1212/01.WNL.0000033325.01878.13[↩]
- Atsuta N, Watanabe H, Ito M, Banno H, Suzuki K, Katsuno M, Tanaka F, Tamakoshi A, Sobue G. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006 Jun;129(Pt 6):1446-55. doi: 10.1093/brain/awl096[↩][↩][↩][↩]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991 Jul 4;352(6330):77-9. doi: 10.1038/352077a0[↩][↩]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002 Aug 29;35(5):843-54. doi: 10.1016/s0896-6273(02)00834-6[↩]
- Lefter S, Hardiman O, Ryan AM. A population-based epidemiologic study of adult neuromuscular disease in the Republic of Ireland. Neurology. 2017 Jan 17;88(3):304-313. doi: 10.1212/WNL.0000000000003504[↩]
- Müller KI, Ghelue MV, Lund I, Jonsrud C, Arntzen KA. The prevalence of hereditary neuromuscular disorders in Northern Norway. Brain Behav. 2021 Jan;11(1):e01948. doi: 10.1002/brb3.1948[↩]
- Lund A., Udd B., Juvonen V., Andersen P.M., Cederquist K., Davis M., et al. Multiple founder effects in spinal and bulbar muscular atrophy (SBMA, Kennedy disease) around the world. Eur. J. Hum. Genet. 2001 Jun;9(6):431–436. doi: 10.1038/sj.ejhg.5200656[↩]
- Udd B, Juvonen V, Hakamies L, Nieminen A, Wallgren-Pettersson C, Cederquist K, Savontaus ML. High prevalence of Kennedy’s disease in Western Finland — is the syndrome underdiagnosed? Acta Neurol Scand. 1998 Aug;98(2):128-33. doi: 10.1111/j.1600-0404.1998.tb01732.x[↩]
- Tanaka F, Doyu M, Ito Y, Matsumoto M, Mitsuma T, Abe K, Aoki M, Itoyama Y, Fischbeck KH, Sobue G. Founder effect in spinal and bulbar muscular atrophy (SBMA). Hum Mol Genet. 1996 Sep;5(9):1253-7. doi: 10.1093/hmg/5.9.1253[↩]
- Breza M, Koutsis G. Kennedy’s disease (spinal and bulbar muscular atrophy): a clinically oriented review of a rare disease. J Neurol. 2019 Mar;266(3):565-573. doi: 10.1007/s00415-018-8968-7[↩][↩][↩]
- Rhodes LE, Freeman BK, Auh S, Kokkinis AD, La Pean A, Chen C, Lehky TJ, Shrader JA, Levy EW, Harris-Love M, Di Prospero NA, Fischbeck KH. Clinical features of spinal and bulbar muscular atrophy. Brain. 2009 Dec;132(Pt 12):3242-51. doi: 10.1093/brain/awp258[↩][↩][↩]
- Kennedy’s Disease Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Kennedys-Disease-Information-Page[↩]
- Finsterer J., Soraru G. Onset manifestations of spinal and bulbar muscular atrophy (Kennedy’s disease) J. Mol. Neurosci. 2016 Mar;58(3):321–329. doi: 10.1007/s12031-015-0663-x[↩][↩]
- Angelini, Corrado. (2014). Spinal Bulbar Muscular Atrophy, Kennedy Disease. 10.1007/978-3-319-07500-6_69. DOI:10.1007/978-3-319-07500-6_69[↩]
- Sperfeld AD, Karitzky J, Brummer D, Schreiber H, Haussler J, Ludolph AC, Hanemann CO. X-linked bulbospinal neuronopathy: Kennedy disease. Arch Neurol. 2002;59:1921–1926. doi: 10.1001/archneur.59.12.1921[↩]
- Sperfeld AD, Hanemann CO, Ludolph AC, Kassubek J. Laryngospasm: an underdiagnosed symptom of X-linked spinobulbar muscular atrophy. Neurology. 2005 Feb 22;64(4):753-4. doi: 10.1212/01.WNL.0000151978.74467.E7[↩][↩]
- Pedroso JL, Vale TC, Barsottini OG, Oliveira ASB, Espay AJ. Perioral and tongue fasciculations in Kennedy’s disease. Neurol Sci. 2018 Apr;39(4):777-779. doi: 10.1007/s10072-017-3170-8[↩]
- Chatwin M, Ross E, Hart N, Nickol AH, Polkey MI, Simonds AK. Cough augmentation with mechanical insufflation/exsufflation in patients with neuromuscular weakness. Eur Respir J. 2003 Mar;21(3):502-8. doi: 10.1183/09031936.03.00048102[↩]
- Chatwin M, Wakeman RH. Mechanical Insufflation-Exsufflation: Considerations for Improving Clinical Practice. J Clin Med. 2023 Mar 31;12(7):2626. doi: 10.3390/jcm12072626[↩]
- AR gene. https://medlineplus.gov/genetics/gene/ar/[↩]
- Spinal and bulbar muscular atrophy. https://medlineplus.gov/genetics/condition/spinal-and-bulbar-muscular-atrophy[↩]
- Neuromuscular Disease Center Motor Syndromes, Hereditary (SMA, ALS + …) 2023. https://neuromuscular.wustl.edu/synmot.html#bsma[↩]
- La Spada AR, Roling DB, Harding AE, Warner CL, Spiegel R, Hausmanowa-Petrusewicz I, Yee WC, Fischbeck KH. Meiotic stability and genotype-phenotype correlation of the trinucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat Genet. 1992;2:301–4.[↩]
- Igarashi S, Tanno Y, Onodera O, Yamazaki M, Sato S, Ishikawa A, Miyatani N, Nagashima M, Ishikawa Y, Sahashi K, et al. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology. 1992;42:2300–2.[↩]
- Echaniz-Laguna A, Rousso E, Anheim M, Cossée M, Tranchant C. A family with early-onset and rapidly progressive X-linked spinal and bulbar muscular atrophy. Neurology. 2005;64:1458–60.[↩]
- Kuhlenbäumer G, Kress W, Ringelstein EB, Stögbauer F. Thirty-seven CAG repeats in the androgen receptor gene in two healthy individuals. J Neurol. 2001;248:23–6.[↩]
- Grunseich C, Kats IR, Bott LC, Rinaldi C, Kokkinis A, Fox D, Chen KL, Schindler AB, Mankodi AK, Shrader JA, Schwartz DP, Lehky TJ, Liu CY, Fischbeck KH. Early onset and novel features in a spinal and bulbar muscular atrophy patient with a 68 CAG repeat. Neuromuscul Disord. 2014a;24:978–81.[↩]
- Grunseich C, Rinaldi C, Fischbeck KH. Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis. 2014 Jan;20(1):6-9. doi: 10.1111/odi.12121[↩][↩]
- Hashizume A, Katsuno M, Suzuki K, Hirakawa A, Hijikata Y, Yamada S, Inagaki T, Banno H, Sobue G. Long-term treatment with leuprorelin for spinal and bulbar muscular atrophy: natural history-controlled study. J Neurol Neurosurg Psychiatry. 2017 Dec;88(12):1026-1032. doi: 10.1136/jnnp-2017-316015[↩]
- Jokela ME, Udd B. Diagnostic Clinical, Electrodiagnostic and Muscle Pathology Features of Spinal and Bulbar Muscular Atrophy. J Mol Neurosci. 2016 Mar;58(3):330-4. doi: 10.1007/s12031-015-0684-5[↩]
- Araki A, Katsuno M, Suzuki K, Banno H, et al. Brugada syndrome in spinal and bulbar muscular atrophy. Neurology. 2014 May 20;82(20):1813-21. doi: 10.1212/WNL.0000000000000434[↩]
- Steinmetz K, Rudic B, Borggrefe M, Müller K, Siebert R, Rottbauer W, Ludolph A, Buckert D, Rosenbohm A. J wave syndromes in patients with spinal and bulbar muscular atrophy. J Neurol. 2022 Jul;269(7):3690-3699. doi: 10.1007/s00415-022-10992-5[↩]
- Guber RD, Takyar V, Kokkinis A, Fox DA, Alao H, Kats I, Bakar D, Remaley AT, Hewitt SM, Kleiner DE, Liu CY, Hadigan C, Fischbeck KH, Rotman Y, Grunseich C. Nonalcoholic fatty liver disease in spinal and bulbar muscular atrophy. Neurology. 2017 Dec 12;89(24):2481-2490. doi: 10.1212/WNL.0000000000004748[↩]
- Francini-Pesenti F, Querin G, Martini C, Mareso S, Sacerdoti D. Prevalence of metabolic syndrome and non-alcoholic fatty liver disease in a cohort of italian patients with spinal-bulbar muscular atrophy. Acta Myol. 2018 Sep 1;37(3):204-209. https://pmc.ncbi.nlm.nih.gov/articles/PMC6390113[↩]
- Querin G, Bertolin C, Da Re E, et al. Non-neural phenotype of spinal and bulbar muscular atrophy: results from a large cohort of Italian patients. J Neurol Neurosurg Psychiatry 2016;87:810–6. doi:10.1136/jnnp-2015-311305[↩]
- Yu Z, Dadgar N, Albertelli M, Gruis K, Jordan C, Robins DM, Lieberman AP. Androgen-dependent pathology demonstrates myopathic contribution to the Kennedy disease phenotype in a mouse knock-in model. J Clin Invest. 2006 Oct;116(10):2663-72. doi: 10.1172/JCI28773[↩]
- Ogata A, Matsuura T, Tashiro K, Moriwaka F, Demura T, Koyanagi T, Nagashima K. Expression of androgen receptor in X-linked spinal and bulbar muscular atrophy and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 1994;57:1274–5.[↩]
- Sorarù G, D’Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L, Gellera C, Moretto G, Pegoraro E, Angelini C. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. 2008;264:100–5.[↩]
- Katsuno M, Banno H, Suzuki K, Adachi H, Tanaka F, Sobue G. Molecular pathophysiology and disease-modifying therapies for spinal and bulbar muscular atrophy. Arch Neurol. 2012 Apr;69(4):436-40. doi: 10.1001/archneurol.2011.2308[↩]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005 Mar;128(Pt 3):659-70. doi: 10.1093/brain/awh381[↩]
- Sinnreich M, Sorenson EJ, Klein CJ. Neurologic course, endocrine dysfunction and triplet repeat size in spinal bulbar muscular atrophy. Can J Neurol Sci. 2004;31:378–82.[↩]
- Lee JH, Shin JH, Park KP, Kim IJ, Kim CM, Lim JG, Choi YC, Kim DS. Phenotypic variability in Kennedy’s disease: implication of the early diagnostic features. Acta Neurol Scand. 2005;112:57–63.[↩]
- Warner CL, Griffin JE, Wilson JD, Jacobs LD, Murray KR, Fischbeck KH, Dickoff D, Griggs RC. X-linked spinomuscular atrophy: a kindred with associated abnormal androgen receptor binding. Neurology. 1992;42:2181–4.[↩]
- Mariotti C, Castellotti B, Pareyson D, Testa D, Eoli M, Antozzi C, Silani V, Marconi R, Tezzon F, Siciliano G, Marchini C, Gellera C, Donato SD. Phenotypic manifestations associated with CAG-repeat expansion in the androgen receptor gene in male patients and heterozygous females: a clinical and molecular study of 30 families. Neuromuscul Disord. 2000;10:391–7.[↩]
- Sobue G, Doyu M, Kachi T, Yasuda T, Mukai E, Kumagai T, Mitsuma T. Subclinical phenotypic expressions in heterozygous females of X-linked recessive bulbospinal neuronopathy. J Neurol Sci. 1993;117:74–8.[↩]
- Sorenson EJ, Klein CJ. Elevated creatine kinase and transaminases in asymptomatic SBMA. Amyotroph Lateral Scler. 2007 Feb;8(1):62-4. doi: 10.1080/17482960600765040[↩]
- Lombardi V, Querin G, Ziff OJ, Zampedri L, et al. Muscle and not neuronal biomarkers correlate with severity in spinal and bulbar muscular atrophy. Neurology. 2019 Mar 12;92(11):e1205-e1211. doi: 10.1212/WNL.0000000000007097. Erratum in: Neurology. 2020 May 12;94(19):852. doi: 10.1212/WNL.0000000000009392[↩]
- Hijikata Y, Hashizume A, Yamada S, Inagaki T, Ito D, Hirakawa A, Suzuki K, Atsuta N, Tsuboi T, Hattori M, Hori A, Banno H, Sobue G, Katsuno M. Biomarker-based analysis of preclinical progression in spinal and bulbar muscular atrophy. Neurology. 2018 Apr 24;90(17):e1501-e1509. doi: 10.1212/WNL.0000000000005360[↩]
- Kassubek J, Juengling FD, Sperfeld AD. Widespread white matter changes in Kennedy disease: a voxel based morphometry study. J Neurol Neurosurg Psychiatry. 2007 Nov;78(11):1209-12. doi: 10.1136/jnnp.2006.112532[↩]
- Unrath A, Müller HP, Riecker A, Ludolph AC, Sperfeld AD, Kassubek J. Whole brain-based analysis of regional white matter tract alterations in rare motor neuron diseases by diffusion tensor imaging. Hum Brain Mapp. 2010 Nov;31(11):1727-40. doi: 10.1002/hbm.20971[↩]
- Pieper CC, Konrad C, Sommer J, Teismann I, Schiffbauer H. Structural changes of central white matter tracts in Kennedy’s disease – a diffusion tensor imaging and voxel-based morphometry study. Acta Neurol Scand. 2013 May;127(5):323-8. doi: 10.1111/ane.12018[↩]
- Dahlqvist JR, Oestergaard ST, Poulsen NS, Thomsen C, Vissing J. Refining the spinobulbar muscular atrophy phenotype by quantitative MRI and clinical assessments. Neurology. 2019 Feb 5;92(6):e548-e559. doi: 10.1212/WNL.0000000000006887[↩]
- Dahlqvist JR, Fornander F, de Stricker Borch J, Oestergaard ST, Poulsen NS, Vissing J. Disease progression and outcome measures in spinobulbar muscular atrophy. Ann Neurol. 2018 Nov;84(5):754-765. doi: 10.1002/ana.25345[↩]
- Spinelli EG, Agosta F, Ferraro PM, Querin G, Riva N, Bertolin C, Martinelli I, Lunetta C, Fontana A, Sorarù G, Filippi M. Brain MRI shows white matter sparing in Kennedy’s disease and slow-progressing lower motor neuron disease. Hum Brain Mapp. 2019 Jul;40(10):3102-3112. doi: 10.1002/hbm.24583[↩]
- Sorarù G, D’Ascenzo C, Polo A, Palmieri A, Baggio L, Vergani L, Gellera C, Moretto G, Pegoraro E, Angelini C. Spinal and bulbar muscular atrophy: skeletal muscle pathology in male patients and heterozygous females. J Neurol Sci. 2008 Jan 15;264(1-2):100-5. doi: 10.1016/j.jns.2007.08.012[↩][↩]
- Jokela M, Huovinen S, Raheem O, Lindfors M, Palmio J, Penttilä S, Udd B. Distinct Muscle Biopsy Findings in Genetically Defined Adult-Onset Motor Neuron Disorders. PLoS One. 2016 Mar 21;11(3):e0151376. doi: 10.1371/journal.pone.0151376[↩]
- Li M, Sobue G, Doyu M, Mukai E, Hashizume Y, Mitsuma T. Primary sensory neurons in X-linked recessive bulbospinal neuropathy: histopathology and androgen receptor gene expression. Muscle Nerve. 1995 Mar;18(3):301-8. doi: 10.1002/mus.880180306[↩]
- Barkhaus PE, Kennedy WR, Stern LZ, Harrington RB. Hereditary proximal spinal and bulbar motor neuron disease of late onset. A report of six cases. Arch Neurol. 1982 Feb;39(2):112-6. doi: 10.1001/archneur.1982.00510140046012[↩]
- Yunusova Y, Plowman EK, Green JR, Barnett C, Bede P. Clinical Measures of Bulbar Dysfunction in ALS. Front Neurol. 2019 Feb 19;10:106. doi: 10.3389/fneur.2019.00106[↩]
- Sperfeld AD, Karitzky J, Brummer D, Schreiber H, Häussler J, Ludolph AC, Hanemann CO. X-linked bulbospinal neuronopathy: Kennedy disease. Arch Neurol. 2002 Dec;59(12):1921-6. doi: 10.1001/archneur.59.12.1921[↩]
- Goldenberg JN, Bradley WG. Testosterone therapy and the pathogenesis of Kennedy’s disease (X-linked bulbospinal muscular atrophy). J Neurol Sci. 1996 Feb;135(2):158-61. doi: 10.1016/0022-510x(95)00285-a[↩][↩]
- Greenland KJ, Zajac JD. Kennedy’s disease: pathogenesis and clinical approaches. Intern Med J. 2004 May;34(5):279-86. doi: 10.1111/j.1444-0903.2004.00588.x[↩]
- Banno H, Katsuno M, Suzuki K, Takeuchi Y, Kawashima M, Suga N, Takamori M, Ito M, Nakamura T, Matsuo K, Yamada S, Oki Y, Adachi H, Minamiyama M, Waza M, Atsuta N, Watanabe H, Fujimoto Y, Nakashima T, Tanaka F, Doyu M, Sobue G. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:140–50.[↩]
- Fischbeck KH, Bryan WW. Anti-androgen treatment for spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:119–20.[↩]
- Katsuno M, Banno H, Suzuki K, Takeuchi Y, Kawashima M, Yabe I, Sasaki H, Aoki M, Morita M, Nakano I, Kanai K, Ito S, Ishikawa K, Mizusawa H, Yamamoto T, Tsuji S, Hasegawa K, Shimohata T, Nishizawa M, Miyajima H, Kanda F, Watanabe Y, Nakashima K, Tsujino A, Yamashita T, Uchino M, Fujimoto Y, Tanaka F, Sobue G., Japan SBMA Interventional Trial for TAP-144-SR (JASMITT) study group. Efficacy and safety of leuprorelin in patients with spinal and bulbar muscular atrophy (JASMITT study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9:875–84.[↩]
- Fernández-Rhodes LE, Kokkinis AD, White MJ, Watts CA, Auh S, Jeffries NO, Shrader JA, Lehky TJ, Li L, Ryder JE, Levy EW, Solomon BI, Harris-Love MO, La Pean A, Schindler AB, Chen C, Di Prospero NA, Fischbeck KH. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011;10:140–7.[↩]
- Mazzini L, Balzarini C, Colombo R, Mora G, Pastore I, De Ambrogio R, Caligari M. Effects of creatine supplementation on exercise performance and muscular strength in amyotrophic lateral sclerosis: preliminary results. J Neurol Sci. 2001;191:139–44.[↩]
- Fischbeck KH. Developing treatment for spinal and bulbar muscular atrophy. Prog Neurobiol. 2012;99:257–61.[↩]
- Cortes CJ, Ling SC, Guo LT, Hung G, Tsunemi T, Ly L, Tokunaga S, Lopez E, Sopher BL, Bennett CF, Shelton GD, Cleveland DW, La Spada AR. Muscle expression of mutant androgen receptor accounts for systemic and motor neuron disease phenotypes in spinal and bulbar muscular atrophy. Neuron. 2014;82:295–307.[↩][↩]
- Lieberman AP, Yu Z, Murray S, Peralta R, Low A, Guo S, Yu XX, Cortes CJ, Bennett CF, Monia BP, La Spada AR, Hung G. Peripheral androgen receptor gene suppression rescues disease in mouse models of spinal and bulbar muscular atrophy. Cell Rep. 2014;7:774–84.[↩]
- Atsuta N. Natural history of spinal and bulbar muscular atrophy (SBMA): a study of 223 Japanese patients. Brain. 2006 Apr 13;129(6):1446–1455. doi: 10.1093/brain/awl096[↩][↩][↩]
- Fernández-Rhodes LE, Kokkinis AD, White MJ, et al. Efficacy and safety of dutasteride in patients with spinal and bulbar muscular atrophy: a randomised placebo-controlled trial. Lancet Neurol. 2011 Feb;10(2):140-7. doi: 10.1016/S1474-4422(10)70321-5[↩][↩]
- Spada A.R.L., Roling D.B., Harding A.E., Warner C.L., Spiegel R., Hausmanowa-Petrusewicz I., et al. Meiotic stability and genotype- phenotype correlation of the binucleotide repeat in X-linked spinal and bulbar muscular atrophy. Nat. Genet. 1992;2:301–304. doi: 10.1038/ng1292-301[↩][↩]
{kind=link}