What is Long QT syndrome
Long QT syndrome (LQTS) is a heart rhythm disorder due to problems with the heart’s electrical system that can cause sudden, fast uncontrollable, and irregular heartbeats (arrhythmia), which may lead to sudden death 1, 2, 3, 4, 5, 6, 7, 8, 9. However, not everyone who has long QT syndrome has dangerous heart rhythms. When they do occur, though, they can be fatal. Long QT syndrome (LQTS) affects the heart’s electrical process called repolarization, which is the recharging of the heart after each heartbeat. When your heart contracts, it sends out an electrical signal. The signal is produced by the flow of ions (potassium, sodium, and calcium) within your heart’s cells. The ions flow in and out of the heart’s cells through ion channels. Long QT syndrome occurs as the result of a defect in the ion channels, causing a delay in the time it takes for the electrical system to recharge (repolarize) after each heartbeat 1. The prolonged QT interval can results from delayed inactivation of inward sodium (INa) or calcium (ICa) currents or a loss of function in outward potassium (slow delayed rectifier potassium channel [IKs], rapid delayed rectifier potassium channel [IKr], or inward rectifier potassium current [IK1]) currents, leading to an extended action potential duration in heart muscle cells 10, 11. This prolonged repolarization (prolonged Q-T interval) can increase your risk for a type of ventricular arrhythmia called Torsades de Pointes, a polymorphic ventricular tachycardia. When Torsade de Pointes occurs, your heart cannot pump enough oxygen-rich blood to the rest of your body, especially your brain. Torsade de Pointes can also lead to ventricular fibrillation, a dangerous form of arrhythmia that causes rapid, uncoordinated contractions in the muscle fibers of the ventricles. With ventricular fibrillation, the heart cannot pump oxygen-rich blood to the rest of the body, which can lead to sudden cardiac death 12.
If you have long QT syndrome, your heart takes longer than usual to recharge between beats. In long QT syndrome, the lower chambers of the heart (ventricles) take too long to contract and release. The gap of time needed to complete a cycle can be measured and compared to normal averages. When seen on an electrocardiogram (EKG or ECG), which records the electrical activity of your heart, this delay is called a prolonged (long) QT interval. Long QT syndrome is characterized by QT prolongation and T-wave abnormalities on the ECG that are associated with fast chaotic heartbeats, typically the ventricular tachycardia Torsades de Pointes 13. The Q-T interval is the section on the electrocardiogram (ECG) – that represents the time it takes for the electrical system to fire an impulse through the ventricles and then recharge. It is translated to the time it takes for the heart muscle to contract and then recover (see Figures 1 and 2).
There are 2 types of Long QT syndrome 14, 15, 16, 1:
- Congenital long QT syndrome. You’re born with this type of long QT syndrome. It’s estimated to affect about 1 in 2,000 people 17, 18. It’s caused by changes in DNA that are passed down through families. That means it is inherited. Congenital long QT syndrome has been shown to be caused by mutations in one of at least 15 different ion-channel genes 19: the KCNQ1 gene causing long QT syndrome type 1 (LQTS1); KCNH2 causing long QT syndrome type 2 (LQTS2); SCN5A causing long QT syndrome type 3 (LQTS3); ANK2 causing LQTS4; KCNE1 causing LQTS5; KCNE2 causing LQT6; KCNJ2 causing LQTS7; CACNA1c causing LQTS8; CAV3 causing LQTS9; SCN4B causing LQTS10; AKAB9 causing LQTS11; SNTA1 causing LQTS12; KCNJ5 causing LQTS13; CALM1 causing LQTS14; and CALM2 causing LQTS15. Mutations in KCNQ1, KCNH2, and SCN5A correlate to Long QT types 1-3 and account for the majority (80-90%) of genetically identifiable cases 20, 21.
- There are 2 types of congenital long QT syndrome:
- Romano-Ward syndrome. This more common type happens in people who get only a single gene change from one parent. Receiving a changed gene from one parent is known as an autosomal dominant inheritance pattern.
- Jervell and Lange-Nielsen syndrome. This rare form of long QT syndrome usually happens very early in life and is severe. Children with this type of long QT syndrome also are deaf. In Jervell and Lange-Nielsen syndrome, children get the gene change from both parents. This is called an autosomal recessive inheritance pattern.
- Congenital Long QT syndrome is autosomal in genetic transmission but shows a greater frequency of expression and a greater lengthening of the QT interval in women than in men. You should avoid competitive sports if you have congenital long QT syndrome 22. If you inherited Long QT syndrome from a parent, you should talk to a heart rhythm specialist before your start exercising. In congenital long QT syndrome, the mortality rate for untreated patients is 50% in 10 years, which can be reduced to 3-4% with therapeutic intervention. A systematic review of babies born with long QT syndrome from 83 studies comprising 265 newborns with postnatal confirmation of long QT syndrome found that a longer fetal QTc was more predictive of death than any other antenatal factor, and the mortality risk was significantly raised when the fetal QTc was longer than 600 ms 23. Other factors that were highly predictive of death included the combination of ventricular tachycardia/Torsade de Pointes or functional 2:1 heart block and lack of a family history of long QT syndrome 23. However, fetal heart rate and heart z-score did not predict death 23.
- There are 2 types of congenital long QT syndrome:
- Acquired long QT syndrome. Acquired long QT syndrome is caused by another health condition or medicine 24, 25. It usually can be reversed when the specific cause is found and treated.
- If a medicine causes acquired long QT syndrome, the disorder may be called drug-induced long QT syndrome. More than 100 medicines can cause prolonged QT intervals in otherwise healthy people. Medicines that can cause long QT syndrome include 26:
- Some antibiotics, such as erythromycin (Eryc, Erythrocin, others), azithromycin (Zithromax) and others.
- Some antifungal medicines used to treat yeast infections.
- Water pills, also called diuretics, that cause the body to remove too much potassium or other minerals.
- Heart rhythm medicines called anti-arrhythmics, which can make the QT interval longer.
- Some medicines used to treat mental health conditions such as anxiety and depression.
- Some medicines used to treat upset stomach.
- Always tell your doctor about all the medicines you take, including those you buy without a prescription.
- Health conditions that can cause acquired long QT syndrome include 26:
- Body temperature below 95 degrees Fahrenheit (35 degrees Celsius), a condition called hypothermia.
- Low calcium, also called hypocalcemia.
- Low magnesium, also called hypomagnesemia.
- Low potassium, also called hypokalemia.
- A tumor of the adrenal gland that usually is not cancer, called pheochromocytoma.
- Stroke or brain bleed.
- Underactive thyroid, also called hypothyroidism.
- If a medicine causes acquired long QT syndrome, the disorder may be called drug-induced long QT syndrome. More than 100 medicines can cause prolonged QT intervals in otherwise healthy people. Medicines that can cause long QT syndrome include 26:
Normally your heart circulates blood throughout your body during each heartbeat. Your heart’s chambers contract and relax to pump blood. These actions are controlled by electrical impulses that travel through your heart and cause it to beat. After each heartbeat, your heart’s electrical system recharges itself in preparation for the next heartbeat.
In long QT syndrome, your heart muscle takes longer than normal to recharge between beats. This electrical disturbance, which often can be seen on an electrocardiogram (ECG), is called a prolonged QT interval.
The electrical activity of the heart is produced by the flow of ions (electrically charged particles of sodium, calcium, potassium, and chloride) in and out of the cells of the heart. Tiny ion channels control this flow. In most cases, long QT syndrome delays the flow of potassium ions out of heart muscle cells. However, for a small number of people, the sodium channels are affected and too many sodium ions are allowed into the cells. This causes a delay in the electrical impulse known as ‘prolonged repolarization’. This shows up on an ECG as a lengthened QT interval, which is part of the heartbeat cycle.
The electrical activity that occurs between the Q and T waves is called the QT interval. This interval shows electrical activity in the heart’s lower chambers, the ventricles. The timing of the heart’s electrical activity is complex, and your body carefully controls it. Normally the QT interval is about a third of each heartbeat cycle. However, in people who have long QT syndrome, the QT interval lasts longer than normal.
A long QT interval can upset the careful timing of the heartbeat and trigger dangerous heart rhythms.
Long QT syndrome is a rare disorder. Experts think that about 1 in 7,000 people in the United States has long QT syndrome. But no one knows for sure, because long QT syndrome often goes undiagnosed 27.
Long QT syndrome causes about 3,000 to 4,000 sudden deaths in children and young adults each year in the United States 28. Unexplained sudden deaths in children are rare. When they do occur, long QT syndrome often is the cause.
Inherited long QT syndrome usually is first detected during childhood or young adulthood. Half of all people who have long QT syndrome have their first abnormal heart rhythm by the time they’re 12 years old, and 90 percent by the time they’re 40 years old. The condition rarely is diagnosed after age 40.
In boys who have long QT syndrome, the QT interval (which can be seen on an EKG test) often returns toward normal after puberty. If this happens, the risk of long QT syndrome symptoms and complications goes down.
Long QT syndrome is more common in women than men. Women who have long QT syndrome are more likely to faint or die suddenly from the disorder during menstruation and shortly after giving birth.
Children who are born deaf also are at increased risk for long QT syndrome. This is because the same genetic problem that affects hearing also affects the function of ion channels in the heart.
The following techniques are commonly used to diagnose long QT syndrome:
- A standard electrocardiogram (EKG or ECG) is the best test for diagnosing LQTS. The EKG machine records your heart’s electrical activity in waveforms, which can show a prolonged Q-T interval.
- An exercise ECG, also known as a stress test, can show an abnormal Q-T interval that may otherwise be normal during a resting EKG.
- Holter monitoring gets a continuous reading of your heart rate and rhythm over a 24-hour period (or longer). Doctors can then look at the recording to see if it shows a prolonged Q-T interval.
Some people with long QT syndrome may not have a prolonged Q-T interval all the time, so the disorder is sometimes overlooked during a routine physical exam. This is why it is important to know your family’s medical history. In any family where there are repeated episodes of fainting or a history of sudden death, long QT syndrome may be the cause.
As Long QT syndrome is sometimes inherited, you may also be referred for genetic testing to test for any faulty genes that are linked with the condition. Your immediate family members (such as your parents, siblings and your children) may also be invited for an assessment.
Treatment for long QT syndrome includes lifestyle changes and medicines to prevent dangerous heartbeats or surgery. Beta blocker medication is the primary treatment for long QT syndrome. Beta blockers do not cure long QT syndrome, but they have been shown to reduce or prevent the symptoms. Beta blockers, such as Propranolol or Nadolol, help control irregular heartbeats and slow your heart rate to make the prolonged QT interval less likely.
Sodium channel blockers can be useful as additional drug therapy for patients with a QTc interval >500 ms (normal QTc interval is 440 ms).
If you’re at risk of a life-threatening arrhythmia and you need more than just medicine to manage this risk, you may need a pacemaker or an implantable cardioverter defibrillator (ICD) fitted.Pacemaker or an implantable cardioverter defibrillator (ICD) are fitted to help control the rhythm and rate of your heart or help correct life-threatening heart rhythms.Implantable cardioverter-defibrillators (ICDs) and/or left cardiac sympathetic denervation (LCSD) surgery are for those with beta-blocker-resistant symptoms, inability to take beta blockers, and/or history of cardiac arrest.
Others with long QT syndrome may need to be prescribed potassium supplements from their doctor. They might also suggest you increase your uptake of potassium-rich foods like bananas, vegetables and pulses. If you’re taking any new medicine or supplements, always check with your pharmacist that what you’re taking is okay with long QT syndrome.
If you are active in competitive sports, talk to your heart doctor about how this may affect your long QT syndrome. Often, once treatment in started, patients with long QT syndrome can participate in recreational sports or other activities in moderation. If you have episodes of fainting while you exercise, you may want to think about exercising with a friend or a family member who can call for help if you need it.
Do You Need to be Screened for Long QT Syndrome?
Long QT Syndrome is a medical condition that can be passed on from generation to generation. It is important for you to be screened for this condition if you have a first-degree relative with long QT Syndrome. First-degree relatives are your parents, siblings and children. All first-line relatives (brothers, sisters, parents and children) should have electrocardiogram (EKG) testing. Any other family members who have a history of seizures or fainting should also undergo testing.
The first step is to tell your doctor that you have a family history of Long QT Syndrome. He or she may want to do diagnostic tests to check your heart. If these tests are positive, you should be seen by a cardiologist who is familiar with Long QT Syndrome.
Can I live a normal life with long QT syndrome?
There are very few things you need to change if you have long QT syndrome. However, you should be aware that:
- Competitive sports are prohibited in patients with congenital long QT syndrome 22.
- Prolonged (longer than a day) or severe episodes of vomiting or diarrhea can affect your sodium and potassium levels. You should discuss this situation with your doctor who may wish to prescribe oral rehydration supplements. These supplements can help to replenish sodium and potassium levels but should be used under medical supervision.
- Over-the-counter medicines and supplements should also first be discussed with your doctor. Some of these may induce symptoms or react with medicines you may be taking to help reduce your risk of abnormal heart rhythms.
- You should always inform medical staff that you have long QT syndrome when you speak to them.
What is the QT interval?
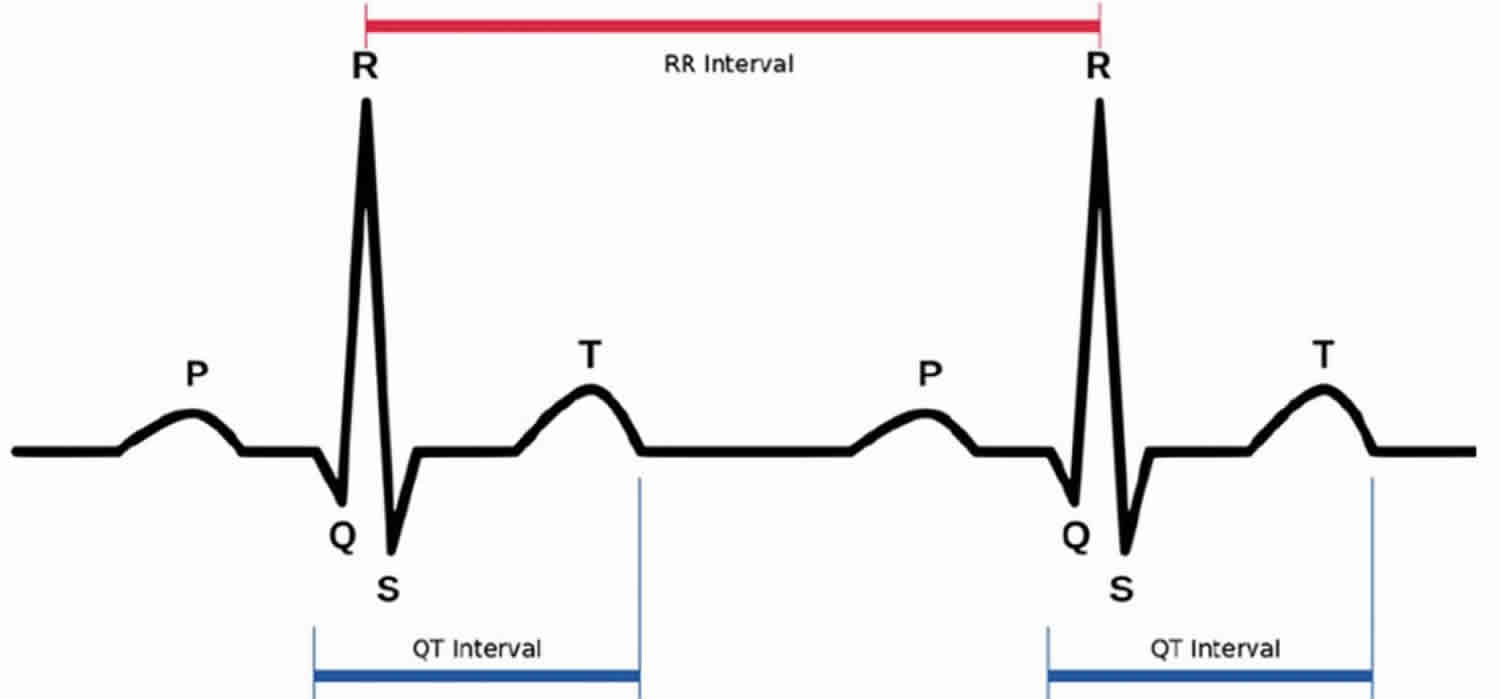
An ECG measures electrical impulses as five distinct waves (Figure 1). Doctors label these five waves using the letters P, Q, R, S and T. The waves labeled Q through T show electrical activity in your heart’s lower chambers (ventricles). The QT interval is a time interval on the ECG.
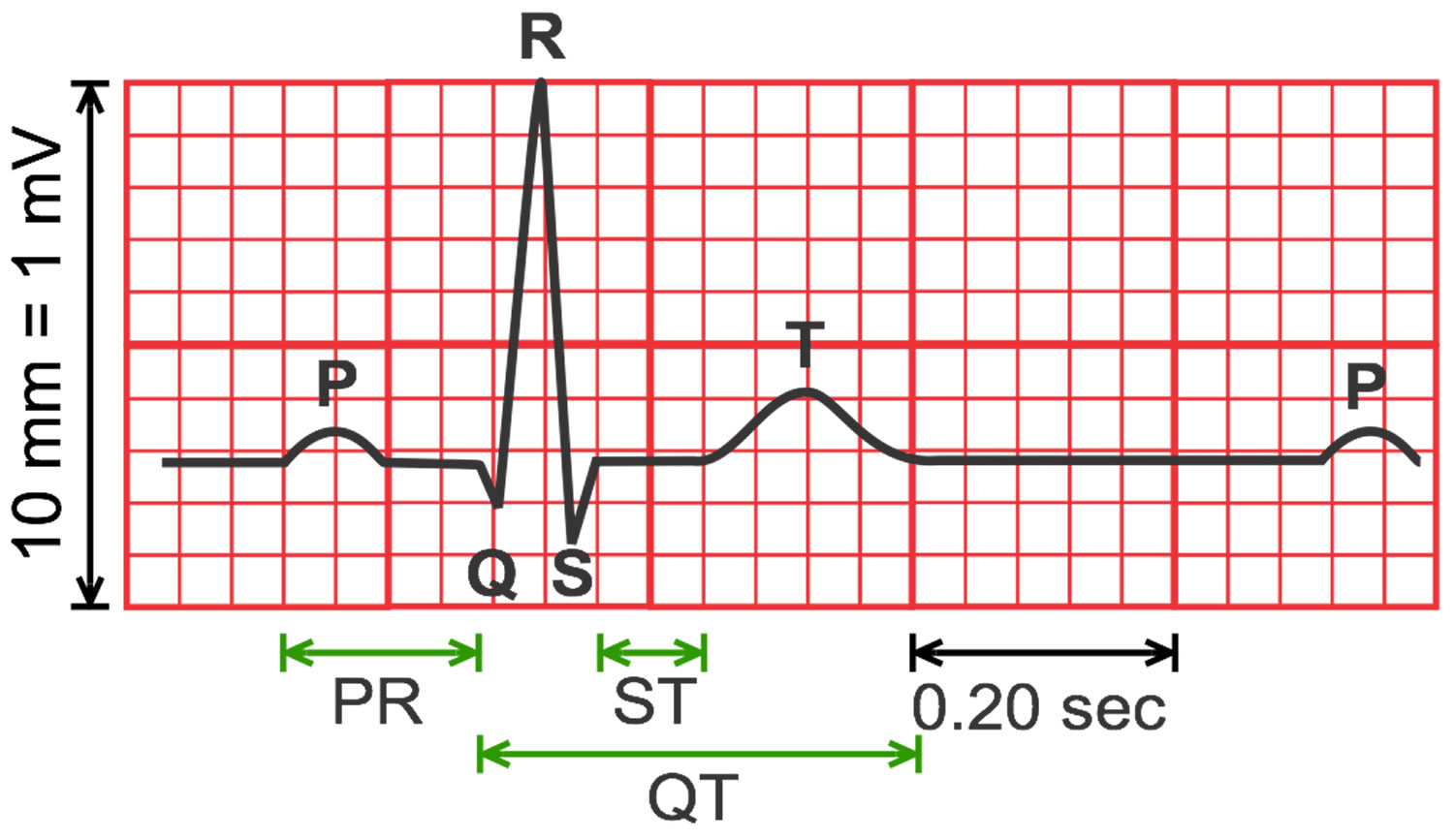
A typical ECG tracing is shown in Figure 1 below. The different waves that comprise the ECG represent the sequence of depolarization and repolarization of the atria and ventricles. The ECG is recorded at a speed of 25 mm/second (5 large squares/second or 5 dark red boxes), and the voltages are calibrated so that 1 mV = 10 mm (2 large squares) in the vertical axis. Therefore, each large dark red 5 mm square represents 0.2 sec (20 msec [millisecond]) and each small 1 mm square represents 0.04 sec (40 msec [millisecond]) in time and 0.10 mV in voltage. Because the recording speed is standardized, you can calculate the heart rate from the intervals between different waves. One method is to divide 1500 by the number of small squares between two R waves. For example, the rate between beats 1 and 2 in Figure 4 below is 1500/22, which equals 68 beats/min. Alternatively, you can divide 300 by the number of large squares, which is 300/4.4 (68 beats/min). Another method, which gives a rough approximation, is the “count off” method. Count the number of large squares between R waves with the following rates: 300 – 150 – 100 – 75 – 60. For example, if there are three large boxes between R waves, then the rate is 100 beats/min. You must extrapolate, however, between boxes. Atrial rate can be determined like the ventricular rate, but using the P waves. Remember, if the heart is in sinus rhythm and there is a one-to-one correspondence between P waves and QRS completes, then the atrial rate will be the same as the ventricular rate. The rate is normal if the interval lies between 5 and 3 large squares (60 – 100 beats/min). Intervals less than 3 large squares or greater than 5 large squares represent tachycardia or bradycardia, respectively.
The space between the start of the Q wave and the end of the T wave (QT interval) corresponds to the time it takes for your heart to contract and then refill with blood before beginning the next contraction. The QT interval represents the time from the electrical stimulation (depolarization) of the heart’s pumping chambers (ventricles), to the end of the recharging of the electrical system (repolarization) and therefore roughly estimates the duration of an average ventricular action potential 29. The interval between the letters Q and T defines the action of the ventricles. The QT interval is measured in milliseconds (msec) and closely approximates the time from the beginning of the heart ventricles’ contraction until the end of relaxation.
The QT interval interval can range from 0.20 to 0.40 seconds (200 to 400 msec), depending upon your heart rate. At high heart rates, ventricular action potentials shorten in duration, which decreases the QT interval 29. Long QT syndrome means that time period is too long, even if by fractions of a second. Because prolonged QT intervals can be diagnostic for susceptibility to certain types of heart arrhythmias, it is important to determine if a QT interval is excessively long. The QT interval is often expressed as a corrected QT (QTc) by taking the QT interval and dividing it by the square root of the R-R interval (interval between ventricular depolarizations) using Bazett formula: QTc = QT / √ RR 7. Corrected QT (QTc) allows an assessment of the QT interval that is independent of the heart rate. Normal corrected QTc intervals are 0.44 seconds or less (<440 msec). QTc is prolonged if it is greater than 440 ms in men or greater than 460 ms in women 30. A QTc greater than 500 is associated with an increased risk of Torsade de Pointes. While several equations exist to help correct heart rate variation, the Bazett formula (QTC = QT / √ RR) is the most commonly used. Though the Bazett formula seems relatively accurate in heart rates between 60 to 100 beats/min, it tends to overcorrect with higher heart rates and undercorrect with lower heart rates.
An occasional prolonged QT interval can be precipitated by everyday circumstances, including:
- When startled by a noise
- Physical activity or exercise
- Intense emotion (such as fright, anger or pain)
In these instances, the heartbeat usually regains its normal contraction rhythm quickly.
Long QT syndrome results from abnormalities in the heart’s electrical recharging system. However, the heart’s structure is normal. Abnormalities in your heart’s electrical system might be inherited. Or, they may be acquired due to an underlying medical condition or a medication.
Figure 1. Normal ECG pattern
Footnotes:
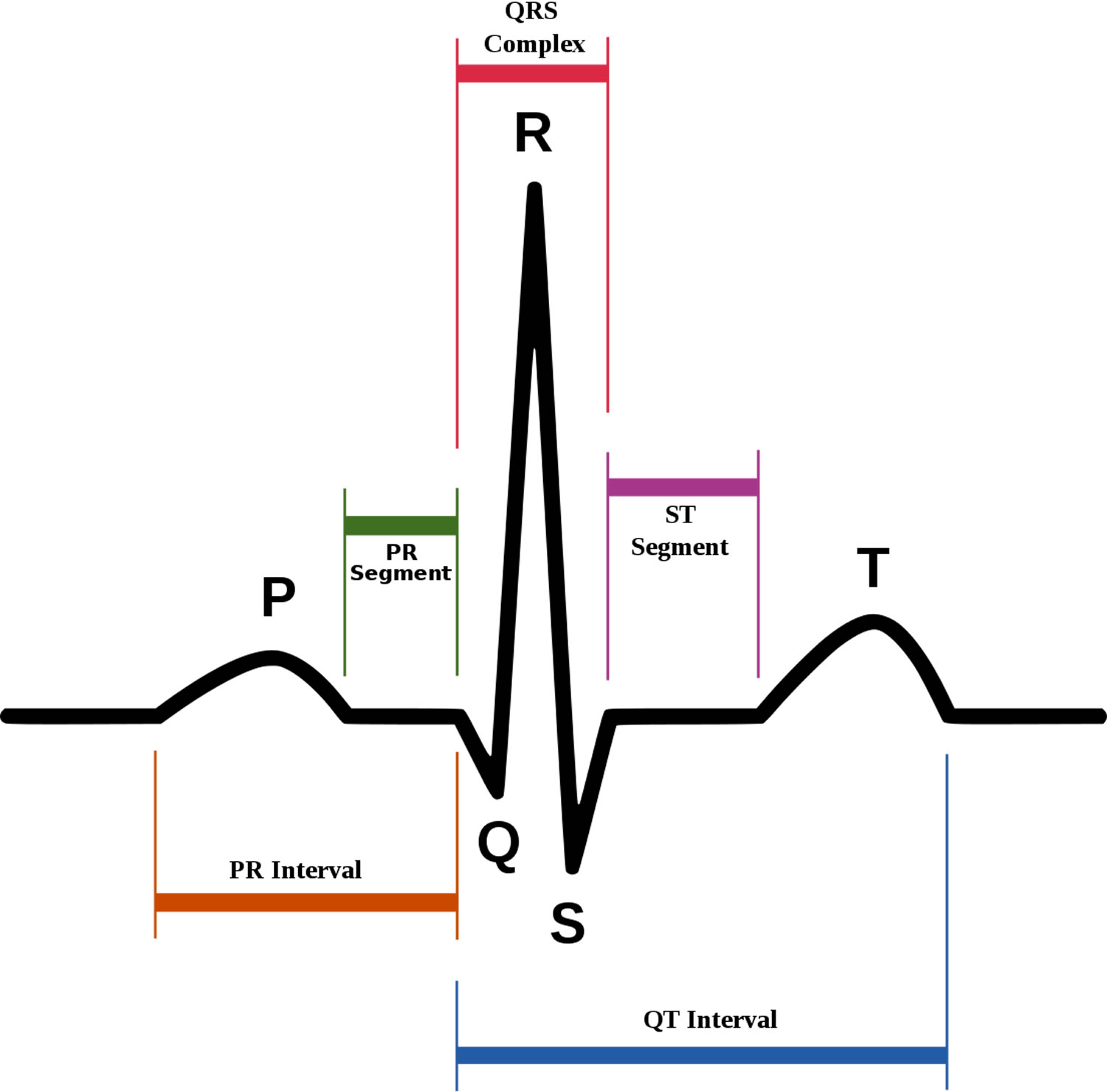
- P wave represents the wave of depolarization that spreads from the SA node throughout the atria and is usually 0.08 to 0.10 seconds (80-100 ms) in duration. There is no distinctly visible wave representing atrial repolarization in the ECG because it occurs during ventricular depolarization. Because the wave of atrial repolarization is relatively small in amplitude (i.e., has low voltage), it is masked by the much larger ventricular-generated QRS complex.
- QRS complex represents ventricular depolarization. Ventricular rate can be calculated by determining the time interval between QRS complexes. The duration of the QRS complex is normally 0.06 to 0.10 seconds.
- ST segment is the isoelectric period following the QRS and ending at the beginning of the T wave. This represents the period at which both ventricles are completely depolarized. This segment roughly corresponds to the plateau phase of the ventricular action potentials. The ST segment is crucial in the diagnosis of ventricular ischemia or hypoxia because under those conditions, the ST segment can become either depressed or elevated.
- T wave represents ventricular repolarization. The T wave exhibits a positive deflection. This is because the last cells to depolarize in the ventricles are the first to repolarize. This occurs because the last cells to depolarize are in the subepicardial region of the ventricles, and these cells have shorter action potentials than found in the subendocardial regions of the ventricular wall. Therefore, although the depolarization of the subepicardial cells occurs after subendocardial depolarization, the subepicardial cells undergo phase 3 repolarization before the subendocardial cells. Therefore, repolarization waves generally are oriented opposite of depolarization waves and repolarization waves moving away from a positive recording electrode produce a positive voltage.
- QT interval represents the time for both ventricular depolarization and repolarization to occur, and therefore roughly estimates the duration of an average ventricular action potential. This interval can range from 0.20 to 0.40 seconds, depending upon heart rate. At high heart rates, ventricular action potentials shorten in duration, which decreases the QT interval.
Figure 2. Long QT syndrome
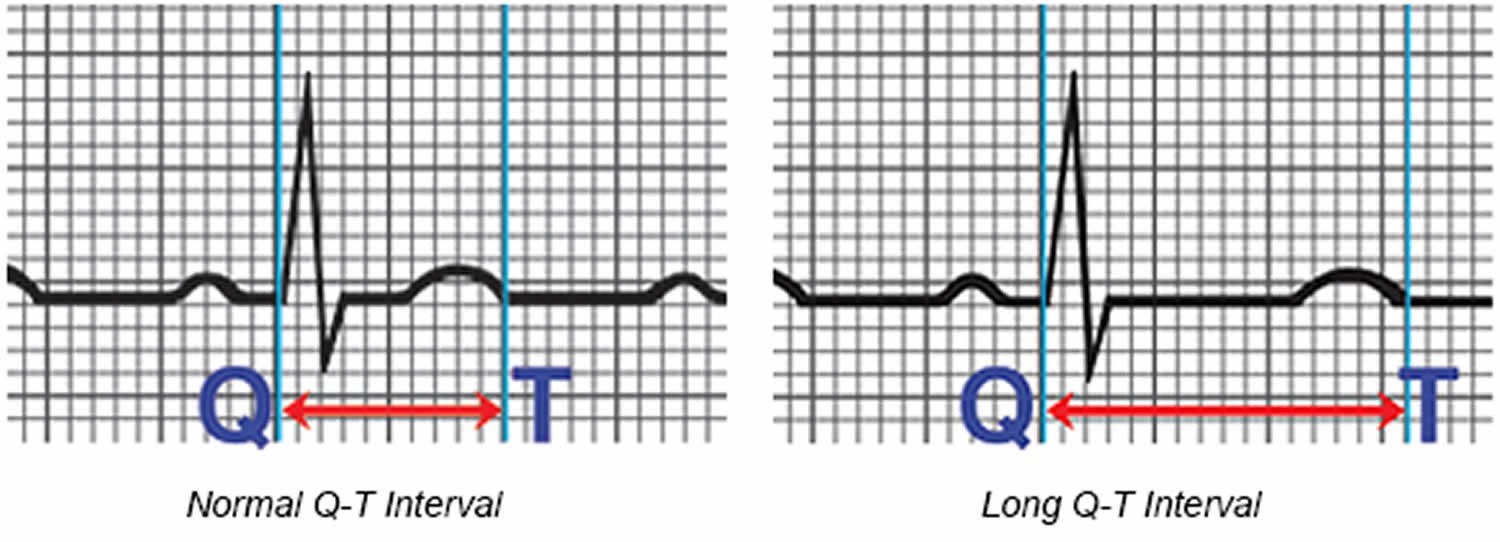
Figure 3. The heart’s electrical system
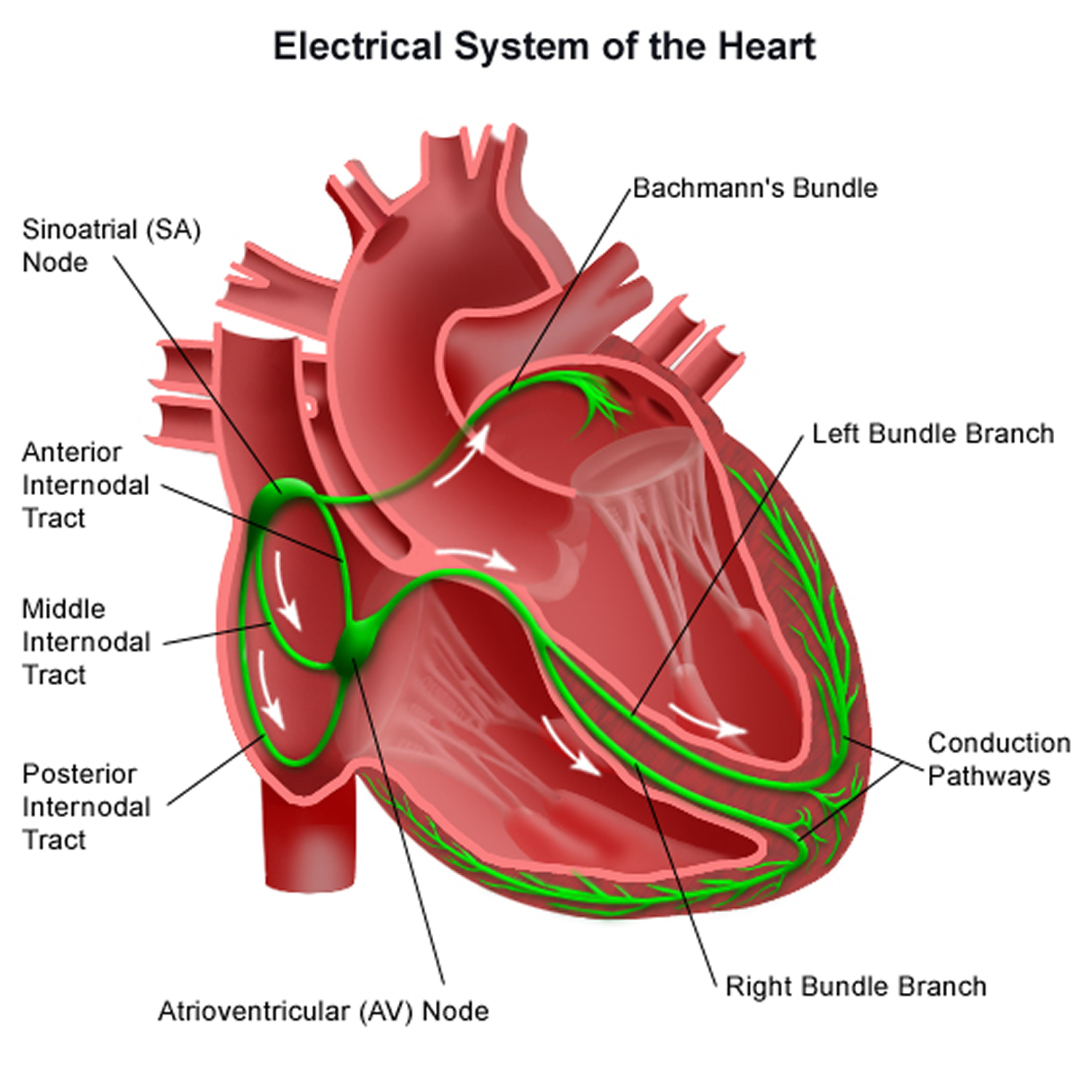
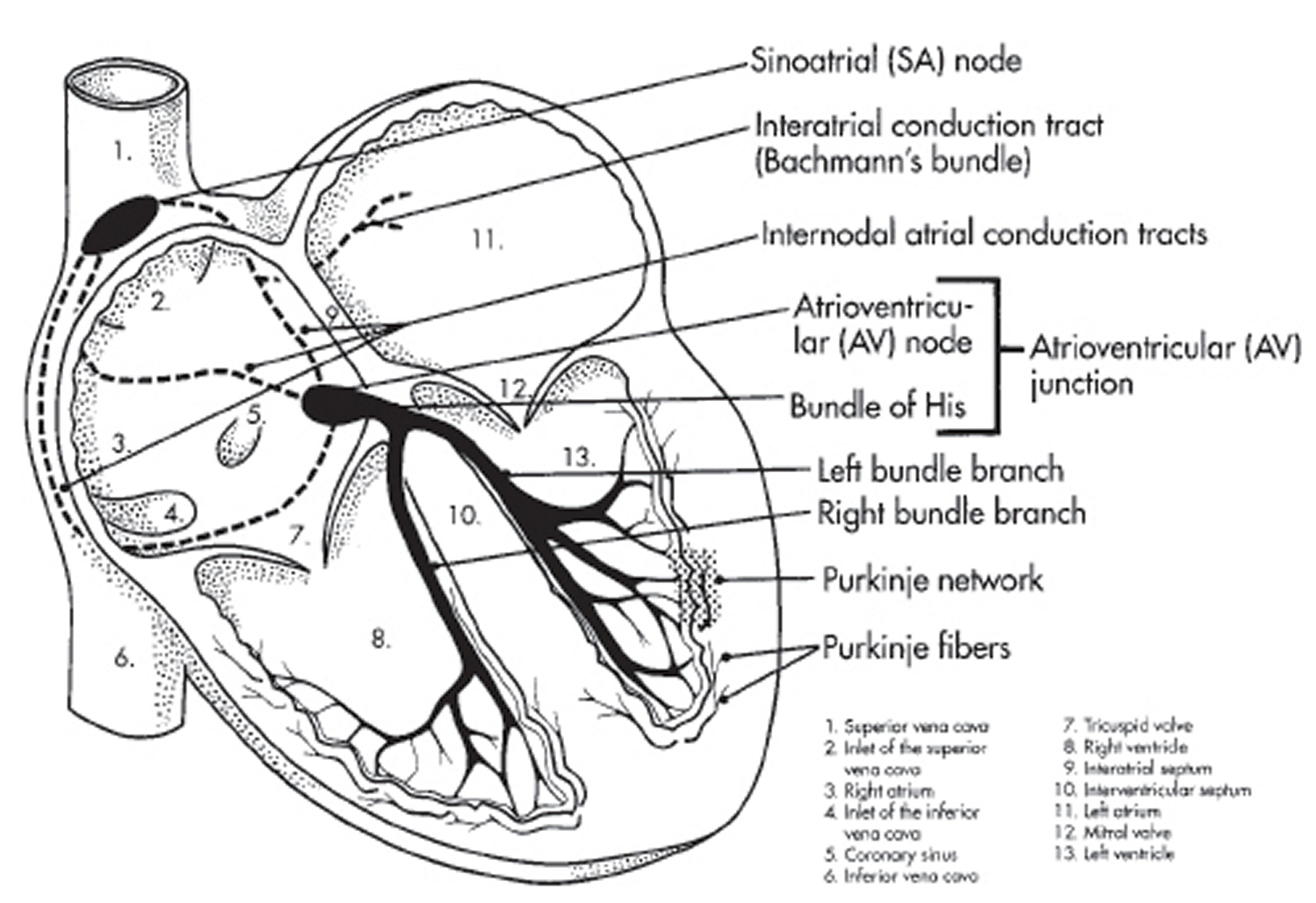 Figure 4. Normal ECG
Figure 4. Normal ECG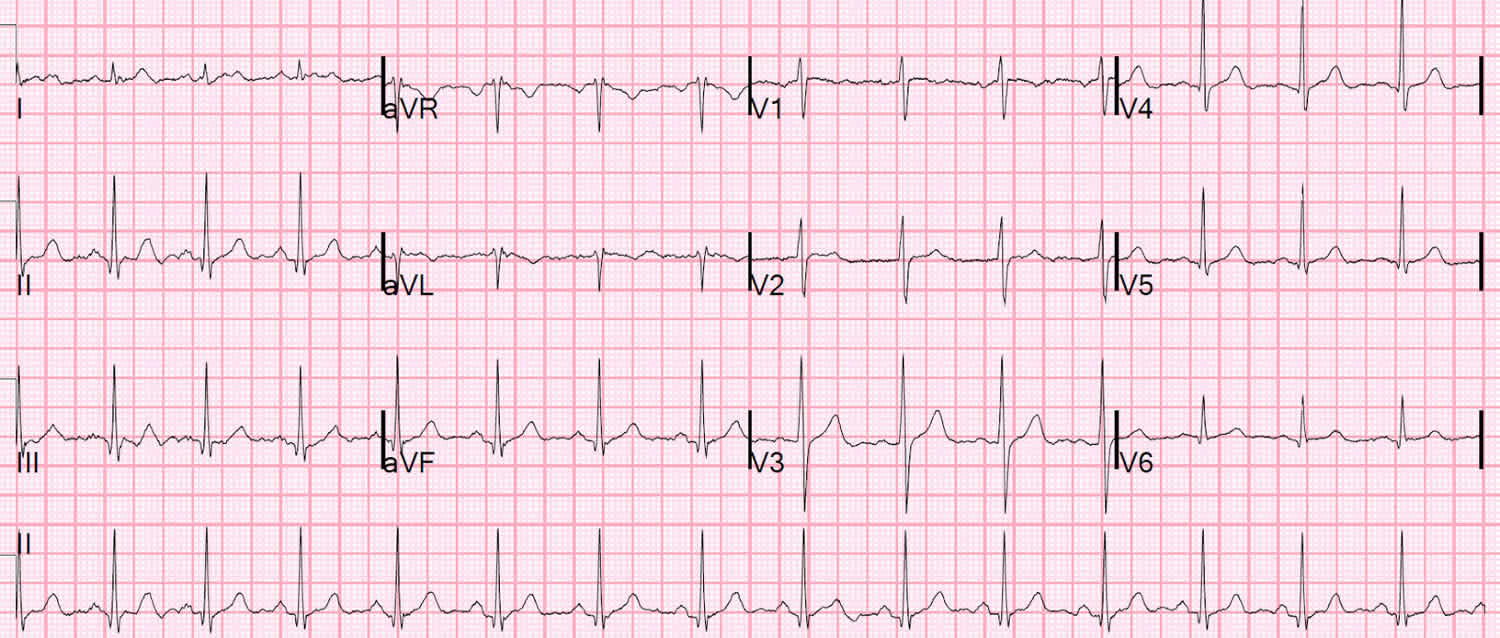
Long QT syndrome causes
Long Q-T syndrome can be acquired or congenital 1:
- Acquired long QT syndrome is caused by another health condition or medicine 24, 25. Sensitivity to these medications may be related to genetic causes. It usually can be reversed when the specific cause is found and treated.
- Congenital long QT syndrome is usually inherited caused by changes in DNA that are passed down through families. Congenital long QT syndrome is estimated to affect about 1 in 2,000 people 17, 18. Congenital long QT syndrome is caused by an abnormality in the gene code for the ion channels. The abnormality of the ion channels slows the recovery phase of the heartbeat. Congenital long QT syndrome has been shown to be caused by mutations in one of at least 15 different ion-channel genes: the KCNQ1 gene causing long QT syndrome type 1 (LQTS1); KCNH2 causing long QT syndrome type 2 (LQTS2); SCN5A causing long QT syndrome type 3 (LQTS3); ANK2 causing LQTS4; KCNE1 causing LQTS5; KCNE2 causing LQT6; KCNJ2 causing LQTS7; CACNA1c causing LQTS8; CAV3 causing LQTS9; SCN4B causing LQTS10; AKAB9 causing LQTS11; SNTA1 causing LQTS12; KCNJ5 causing LQTS13; CALM1 causing LQTS14; and CALM2 causing LQTS15 19. Mutations in KCNQ1, KCNH2, and SCN5A correlate to Long QT types 1-3 and account for the majority (80-90%) of genetically identifiable cases 20, 21. Forms of inherited long QT syndrome include:
- Recent Classifications – Multiple ion channel abnormalities have been discovered. The most common ones include LQTS1, LQTS2, LQTS3, LQTS4, LQTS5; these are classified by the type of channel which causes the long QT syndrome. The type of long QT syndrome classification is related to the risk of future cardiac events, those with LQTS3 having the highest risk of life-threatening arrhythmias.
- Jervell and Lange-Nielsen Syndrome (autosomal recessive inheritance pattern) – Both parents are carriers of the abnormal gene, but they may not manifest long QT syndrome. Each child has a 25-percent chance of inheriting long QT syndrome. This syndrome is associated with deafness at birth and is extremely rare, as there is a small chance that both parents would carry the long QT syndrome gene.
- Romano-Ward Syndrome (autosomal dominant inheritance pattern) – In 1963 and 1964, Romano 31 and Ward 32 independently described families with a prolonged QT interval and syncope without deafness, which became known as Romano-Ward syndrome. Romano-Ward syndrome is an autosomal dominant form of long QT syndrome and is more common than Jervell and Lange-Nielsen Syndrome. Studies in the 1970s and 1980s further characterized the clinical features and familial nature of LQTS, leading to the recognition of its genetic heterogeneity 33. One parent has long QT syndrome and the other parent usually does not. Each child has a 50-percent chance of inheriting the abnormal gene. In this syndrome, hearing is normal; however the likelihood that children in this family would have long QT syndrome is greater. The gene may be present in all the couple’s children, some of them or none at all.
- Timothy Syndrome. Timothy syndrome is a rare, autosomal dominant disorder caused by mutations in the CACNA1C gene that affects other parts of your body as well as your heart including prolonged QT interval (QT prolongation), heart arrhythmias, structural heart defects, syndactyly (webbed fingers or toes), and neurological problems like autism spectrum disorders 34. Mutations in the CACNA1C gene that cause Timothy syndrome change the structure of CaV1.2 channels that transports positively charged calcium atoms (calcium ions) into cardiac cells (cardiomyocytes) and nerve cells (neurons) in the brain. Calcium ions are important for many cellular functions, including regulating the electrical activity of cells, cell-to-cell communication, the tensing of muscle fibers (muscle contraction), and the regulation of certain genes, particularly those involved in the development of the brain and bones before birth. The CACNA1C gene changes lead to altered CaV1.2 channels that stay open much longer than usual, which allows calcium ions to continue flowing into cells abnormally. The resulting overload of calcium ions within heart muscle cells changes the way the heart beats and can cause abnormal heart muscle contractions and arrhythmia. It is thought that the altered channels and flow of calcium ions also impair regulation of certain genes, resulting in the facial, dental, and neurological abnormalities in Timothy syndrome. Other variants in the CACNA1C gene can cause isolated features of Timothy syndrome without the other associated health problems of the condition. For example, some people with CACNA1C gene variants may have only long QT syndrome or only neurodevelopmental disorders.
Those at risk for long QT syndrome include:
- Children who are deaf at birth
- Children and young adults who have unexplained sudden death or syncope in family members
- Blood relatives of family members with long QT syndrome
- Those with long QT syndrome taking medications that can further prolong the QT intervals.
Congenital long QT syndrome
At least 17 genes associated with long QT syndrome have been found so far, and hundreds of mutations within these genes have been identified. Mutations in three of these genes account for about 75 percent of long QT syndrome cases, while mutations in the other minor genes contribute a small percent of long QT syndrome cases.
About 20 to 25 percent of people who definitely have congenital long QT syndrome have a negative genetic test result. On the other hand, among families with genetically established long QT syndrome, between 10 percent and 37 percent of the relatives with a positive long QT syndrome genetic test have a normal QT interval.
Doctors have described two forms of inherited long QT syndrome:
- Romano-Ward syndrome. This more common form occurs in people who inherit only a single genetic variant from one parent.
- Jervell and Lange-Nielsen syndrome. This rare form usually occurs earlier and is more severe. In this syndrome, children inherit genetic variants from both parents. They have long QT syndrome and also are born deaf.
Additionally, scientists have been investigating a possible link between sudden infant death syndrome (SIDS) and long QT syndrome and have discovered that approximately five to 10 percent of babies affected by SIDS (sudden infant death syndrome) had a genetic defect or mutation for long QT syndrome.
Acquired long QT syndrome
Acquired long QT syndrome can be caused by certain medications, electrolyte abnormalities such as low body potassium (hypokalemia), low blood calcium (hypocalcemia), low blood magnesium (hypomagnesemia) or medical conditions 24, 25. More than 100 medications — many of them common — can lengthen the QT interval in otherwise healthy people and cause a form of acquired long QT syndrome known as drug-induced long QT syndrome.
Medications
Various medications across multiple therapeutic classes have been linked to QT prolongation and an increased risk of Torsade de Pointes.
Medications that can lengthen the QT interval and upset heart rhythm include:
- Certain antibiotics
- Certain antidepressant and antipsychotic medications
- Some antihistamines
- Diuretics
- Medications used to maintain normal heart rhythms (antiarrhythmic medications)
- Cholesterol-lowering medicines and some diabetes medicines
- Some anti-nausea medications
People who develop drug-induced long QT syndrome might also have some subtle genetic defects in their hearts. These defects make them more likely to have disruptions in their heart rhythm from certain medications.
Some people who have medication-induced long QT syndrome also may have an inherited form of the disorder. They may not have symptoms unless they take medicines that lengthen the QT interval or lower potassium levels in the blood. When long QT syndrome doesn’t cause symptoms, it’s called silent long QT syndrome.
Electrolyte imbalances
Severe diarrhea or vomiting that causes a major loss of potassium or sodium ions from the bloodstream may cause acquired long QT syndrome. The long QT syndrome lasts until these ion levels return to normal.
- Hypokalemia, defined as serum potassium below 3.5 mEq/L, can lead to prolonged repolarization and increased risk of Torsade de Pointes 35. Low extracellular potassium reduces the conductance of the inward rectifier potassium current [IK1], essential for maintaining resting membrane potential and terminal repolarization of cardiac myocytes. It also enhances Na+/Ca2+ exchanger activity, increasing intracellular calcium levels and prolonging action potential duration 36. The eating disorders anorexia nervosa and bulimia and some thyroid disorders may cause a drop in potassium ion levels in the blood, causing long QT syndrome.
- Hypomagnesemia, defined as serum magnesium below 1.7 mg/dL, is another important risk factor for acquired long QT syndrome 37. Magnesium is a critical cofactor for various ion channels and transporters involved in cardiac repolarization. Low magnesium levels can impair the function of inward rectifier potassium current [IK1], slow delayed rectifier potassium channel [IKs], and Na+/K+-ATPase, leading to prolonged action potential duration and increased arrhythmia risk 38.
- Hypocalcemia has also been associated with QT prolongation, although the mechanisms are less well understood 39. Calcium plays a crucial role in cardiac electrophysiology, and low extracellular calcium levels can alter the function of various ion channels and transporters involved in repolarization.
Identifying and correcting electrolyte imbalances is essential in managing patients with acquired long QT syndrome or those at risk. Monitoring serum electrolyte levels, particularly in patients treated with QT-prolonging medications or those with predisposing conditions, can help prevent life-threatening arrhythmias 40.
Other medical conditions
Several medical conditions can contribute to the development of acquired long QT syndrome by directly affecting heart repolarization or indirectly influencing the risk of QT prolongation through altered pharmacokinetics or pharmacodynamics of QT-prolonging medications.
- Heart conditions
- Bradycardia: Slow heart rates can prolong the QT interval and increase Torsade de Pointes risk 41, 42
- Heart failure: Associated with an increased risk of QT prolongation and Torsade de Pointes because of altered ion channel expression, neurohumoral activation, and electrolyte disturbances 43
- Myocardial ischemia and infarction: Can lead to QT prolongation by altering ion channel function and increasing repolarization dispersion 44
- Endocrine disorders
- Hypothyroidism (underactive thyroid): Can prolong the QT interval by altering cardiac ion channel expression and function 45
- Pheochromocytoma: Catecholamine excess can lead to QT prolongation and increased ventricular arrhythmia risk 46
- Neurological conditions
- Subarachnoid hemorrhage (SAH): Increased risk of QT prolongation and Torsade de Pointes because of autonomic dysfunction, electrolyte disturbances, and QT-prolonging medications 47
- Stroke: Acute stroke, particularly involving the insular cortex, can lead to QT prolongation and increased ventricular arrhythmia risk 48
- Liver disease: Hepatic dysfunction can alter QT-prolonging drug metabolism, leading to increased plasma levels and higher acquired long QT syndrome risk 49
- Kidney disease: Chronic kidney disease and end-stage renal disease are associated with increased QT prolongation and Torsade de Pointes risk 50
- Eating disorders: Anorexia nervosa and bulimia nervosa can lead to QT prolongation and increased sudden cardiac death risk 51
- Autoimmune disorders: Systemic lupus erythematosus (SLE) and other autoimmune disorders have been associated with QT prolongation 52, 53, 54
Recognizing and managing these underlying medical conditions is crucial for minimizing acquired long QT syndrome risk. Patients with these conditions should be closely monitored for QT prolongation, especially when treated with QT-prolonging medications, and appropriate interventions should be implemented to correct modifiable risk factors.
Risk factors for long QT syndrome
People who may have a higher risk of inherited or acquired long QT syndrome may include:
- Children, teenagers and young adults with unexplained fainting, unexplained near drownings or other accidents, unexplained seizures, or a history of cardiac arrest
- Family members of children, teenagers and young adults with unexplained fainting, unexplained near drownings or other accidents, unexplained seizures, or a history of cardiac arrest
- First-degree relatives of people with known long QT syndrome
- People taking medications known to cause prolonged QT intervals
- People with low body potassium (hypokalemia), low blood calcium (hypocalcemia), low blood magnesium (hypomagnesemia) levels — such as those with the eating disorder anorexia nervosa.
Inherited long QT syndrome often goes undiagnosed or is misdiagnosed as a seizure disorder, such as epilepsy. However, long QT syndrome might be responsible for some otherwise unexplained deaths in children and young adults. For example, an unexplained drowning of a young person might be the first clue to inherited long QT syndrome in a family.
- You’re at risk of having long QT syndrome if anyone in your family has ever had it. Unexplained fainting or seizures, drowning or near drowning, and unexplained sudden death are all possible signs of long QT syndrome.
- You’re also at risk for long QT syndrome if you take medicines that make the QT interval longer. Your doctor can tell you whether your prescription or over-the-counter medicines might do this.
- You also may develop long QT syndrome if you have excessive vomiting or diarrhea or other conditions that cause low blood levels of potassium or sodium. These conditions include the eating disorders anorexia nervosa and bulimia, as well as some thyroid disorders.
Long QT syndrome prevention
If you have inherited long QT syndrome, be careful about which medications you take. Some medications — including certain appetite suppressants, decongestants and common antibiotics — might trigger dangerous heart rhythms. Ask your doctor what you can and can’t take safely. Illegal drugs, such as cocaine and amphetamines, pose a serious risk for people with long QT syndrome.
In addition, seek medical treatment right away for illnesses that could result in low blood-potassium levels, especially if you have a lot of vomiting and diarrhea. Such illnesses could trigger an episode of long QT syndrome. Your doctor might advise you not to take some drugs, such as diuretics, that lower blood-potassium levels.
Some people — especially older adults with long QT syndrome who haven’t had signs or symptoms of the condition in decades — may not need any treatment other than preventive measures.
If Long QT syndrome runs in your family, your doctor can test your family members to see if they have it. With proper treatment, you can manage and prevent the dangerous heartbeats that can lead to Long QT syndrome complications.
Regular health checkups and good communication with your doctor also may help prevent causes of some types of acquired long QT syndrome. It’s especially important not to take medicines that can affect the heart rhythm and cause a prolonged QT interval.
Long QT syndrome drugs to avoid
There are many medications that can prolong the QT interval. Those with long QT syndrome may be more prone to the effects of these medications. If you have long QT syndrome, you should:
- NOT take over-the-counter medications (except for plain aspirin or acetaminophen) without first talking to your health care provider.
- Tell all your health care providers you have long QT syndrome, as there are many drugs you cannot take.
- Talk to your doctor before taking any medications prescribed for other medical conditions. The following types of medications may affect you if you have long QT syndrome:
- Antihistamines
- Antidepressants, mental illness medications
- Heart medications
- Antibiotics, antifungals, antivirals
- Intestinal medications
- Anticonvulsants
- Diuretics
- Antihypertensives
- Migraine medications
- Cholesterol lowering medications
Table 1. Long QT syndrome drugs to avoid
| Generic Name | Brand Names (Partial List) | Drug Class | Therapeutic Use |
|---|---|---|---|
| Abarelix (Only on Non US Market) | Plenaxis | GnRH Antagonist | Cancer (prostate) |
| Aclarubicin (Only on Non US Market) | Aclacin, Aclacinomycine, Aclacinon, Aclaplastin, Jaclacin | Anti-cancer | Cancer |
| Albuterol (salbutamol) | Proventil, Ventolin, Ventolin-HFA, Accuneb, Combivent, Vospire-ER, ProAir HFA, Duoneb | Bronchodilator | Asthma |
| Alfuzosin | Uroxatral | Alpha-1 adrenergic blocker | Benign prostatic hyperplasia |
| Amantadine | Symmetrel, Symadine | Antiviral | Viral infection (Influenza), Parkinson’s disease |
| Amiodarone | Cordarone, Pacerone, Nexterone | Antiarrhythmic | Arrhythmia |
| Amisulpride (Only on Non US Market) | Solian, Supitac, Soltus, Amitrex, Amazeo | Antipsychotic, atypical | Schizophrenia |
| Amitriptyline | Elavil (Discontinued 6/13), Tryptomer, Tryptizol, Laroxyl, Saroten, Sarotex Lentizol, Endep | Antidepressant, Tricyclic | Depression |
| Amphetamine (Amfetamine) | Adderal-XR, Dexedrine, Dextroamp | CNS stimulant | ADHD |
| Amphotericin B | Fungilin, Fungizone, Abelcet, AmBisome, Fungisome, Amphocil, Amphotec | Antifungal | Fungal infection |
| Amsacrine (acridinyl anisidide) (Only on Non US Market) | Amsidine | Antineoplastic Agent | Acute Lymphoblastic Leukemia |
| Anagrelide | Agrylin, Xagrid | Phosphodiesterase 3 inhibitor | Thrombocythemia |
| Apalutamide | Erleada | Nonsteroidal antiandrogen | Cancer (prostate) |
| Apomorphine | Apokyn, Ixense, Spontane, Uprima | Dopamine agonist | Parkinson’s disease |
| Arformoterol | Brovana | Bronchodilator | Asthma, COPD |
| Aripiprazole | Abilify, Aripiprex | Antipsychotic, atypical | Schizophrenia, depression (adjunct) |
| Arsenic trioxide | Trisenox | Anti-cancer | Cancer (leukemia) |
| Artenimol+piperaquine (Only on Non US Market) | Eurartesim | Antimalarial | Malaria |
| Asenapine | Saphris, Sycrest | Antipsychotic, atypical | Schizophrenia |
| Astemizole (Removed from Market) | Hismanal | Antihistamine | Allergic rhinitis |
| Atazanavir | Reyataz, Evotaz | Antiviral | Viral infection (HIV/AIDS) |
| Atomoxetine | Strattera | CNS stimulant | ADHD |
| Azithromycin | Zithromax, Zmax | Antibiotic | Bacterial infection |
| Bedaquiline | Sirturo | Antibiotic | Tuberculosis, Multi-drug resistant |
| Bendamustine | Treanda, Treakisym, Ribomustin, Levact | Anti-cancer | Cancer (Leukemia, lymphoma) |
| Bendroflumethiazide or bendrofluazide | Aprinox, Corzide | Diuretic, thiazide | Hypertension, diuresis |
| Benperidol (Only on Non US Market) | Anquil, Glianimon | Antipsychotic | Schizophrenia |
| Bepridil (Removed from Market) | Vascor | Antianginal | Angina Pectoris (heart pain) |
| Betrixaban | Bevyxxa | Anticoagulant | Anticoagulant |
| Bortezomib | Velcade, Bortecad | Proteasome inhibitor | Cancer (multiple myeloma,lymphoma) |
| Bosutinib | Bosulif | Anti-cancer | Cancer (leukemia) |
| Buprenorphine | Butrans, Belbuca, Bunavail, Buprenex, Suboxone, Zubsolv | Opiate | Narcotic addiction and pain |
| Cabozantinib | Cometriq | Anti-cancer | Cancer (renal cell) |
| Capecitabine | Xeloda | Anti-cancer | Cancer (GI, Breast) |
| Ceritinib | Zykadia | Anti-cancer | Cancer (Lung) |
| Chloral hydrate | Aquachloral, Novo-Chlorhydrate, Somnos, Noctec, Somnote | Sedative | Sedation, insomnia |
| Chloroquine | Aralen | Antimalarial | Malaria |
| Chlorpromazine | Thorazine, Largactil, Megaphen | Antipsychotic / Antiemetic | Schizophrenia, nausea, many others |
| Cilostazol | Pletal | Phosphodiesterase 3 inhibitor | Intermittent claudication |
| Cimetidine | Tagamet and others | Antacid | Gastric hyperacidity, GERD |
| Ciprofloxacin | Cipro, Cipro-XR, Neofloxin | Antibiotic | Bacterial infection |
| Cisapride (Removed from Market) | Propulsid | GI stimulant | Increase GI motility |
| Citalopram | Celexa, Cipramil | Antidepressant, SSRI | Depression |
| Clarithromycin | Biaxin, Prevpac | Antibiotic | Bacterial infection |
| Clofazimine (Only on Non US Market) | Lamprene | Antibiotic | Leprosy |
| Clomipramine | Anafranil | Antidepressant, Tricyclic | Depression |
| Clotiapine (Only on Non US Market) | Entumine | Antipsychotic, atypical | Psychosis |
| Clozapine | Clozaril, Fazaclo, Versacloz | Antipsychotic, atypical | Schizophrenia |
| Cocaine | Cocaine | Local anesthetic | Anesthesia (topical) |
| Crizotinib | Xalkori | Anti-cancer | Cancer (Non-small cell lung cancer, metastatic) |
| Cyamemazine (cyamepromazine) (Only on Non US Market) | Tercian | Antipsychotic | Schizophrenia, sedation |
| Dabrafenib | Tafinlar | Anti-cancer | Cancer (melanoma) |
| Dasatinib | Sprycel | Anti-cancer | Cancer (leukemia) |
| Degarelix | Firmagon, Ferring | Gonadotropin Releasing Hormone Agonist/antagonist | Cancer (prostate) |
| Delamanid (Only on Non US Market) | Deltyba | Antibiotic | Tuberculosis, Multi-drug resistant |
| Desipramine | Pertofrane, Norpramine | Antidepressant, Tricyclic | Depression |
| Deutetrabenazine | Austedo | Vesicular monamine transporter 2 inhibitor | Chorea (Huntington’s disease) |
| Dexmedetomidine | Precedex, Dexdor, Dexdomitor | Sedative | Sedation |
| Dexmethylphenidate | Focalin, Focalin-XR, Attenade | CNS stimulant | ADHD |
| Dextroamphetamine (Dexamfetamine) | Dexedrine, dexamphetamine, dexamfetamine, (S)-(+)-amphetamine, Dextrostat, Dexedrine, Metamina, Attentin, Zenzedi, Procentra, Amfexa | CNS stimulant | ADHD, obesity |
| Diphenhydramine | Benadryl, Nytol, Unisom, Sominex, Dimedrol, Daedalon | Antihistamine | Allergic rhinitis, insomnia |
| Disopyramide | Norpace | Antiarrhythmic | Arrhythmia |
| Dobutamine | Dobutrex | Inotrope | Heart failure, shock (low blood pressure) |
| Dofetilide | Tikosyn | Antiarrhythmic | Arrhythmia |
| Dolasetron | Anzemet | Antiemetic | Nausea, vomiting |
| Domperidone (Only on Non US Market) | Motilium, Motillium, Motinorm Costi, Nomit | Antiemetic | Nausea, vomiting |
| Donepezil | Aricept | Cholinesterase inhibitor | Dementia (Alzheimer’s Disease) |
| Dopamine | Intropine | Inotrope | Heart failure, shock (low blood pressure) |
| Doxepin | Sinequan, Silenor, Aponal, Adapine, Doxal, Deptran, Sinquan | Antidepressant, Tricyclic | Depression |
| Dronedarone | Multaq | Antiarrhythmic | Arrhythmia |
| Droperidol | Inapsine, Droleptan, Dridol, Xomolix | Antipsychotic / Antiemetic | Anesthesia (adjunct), nausea |
| Droxidopa | Northera | Adrenergic pro-drug | Hypotension (Neurogenic orthostatic) |
| Efavirenz | Sustiva and others | Antiviral | HIV/AIDS |
| Eliglustat | Cerdelga | Glucosylceramide synthase inhibitor | Gaucher’s disease |
| Eperisone (Only on Non US Market) | Myonal, Epry | Antispasmodic | Spasticity |
| Ephedrine | Rynatuss, Broncholate | Bronchodilator, decongestant | Allergic reaction, allergic rhinitis, asthma |
| Epinephrine (adrenaline) | Primatene, Bronkaid | Catecholamine, vasoconstrictor | Allergic reaction, anaphylaxis, cardiac arrest |
| Epirubicin | Ellence, Pharmorubicin, Epirubicin Ebewe | Anti-cancer | Cancer |
| Eribulin mesylate | Halaven | Anti-cancer | Cancer (breast, metastatic) |
| Erythromycin | E.E.S., Robimycin, EMycin, Erymax, Ery-Tab, Eryc Ranbaxy, Erypar, Eryped, Erythrocin Stearate Filmtab, Erythrocot, E-Base, Erythroped, Ilosone, MY-E, Pediamycin, Abboticin, Abboticin-ES, Erycin, PCE Dispertab, Stiemycine, Acnasol, Tiloryth | Antibiotic | Bacterial infection, increase GI motility |
| Escitalopram | Cipralex, Lexapro, Nexito, Anxiset-E (India), Exodus (Brazil), Esto (Israel), Seroplex, Elicea, Lexamil, Lexam, Entact (Greece), Losita (Bangladesh), Reposil (Chile), Animaxen (Colombia), Esitalo (Australia), Lexamil (South Africa) | Antidepressant, SSRI | Depression (major), anxiety disorders |
| Esomeprazole | Nexium, Nexum and others | Proton Pump Inhibitor | Gastric hyperacidity, GERD |
| Ezogabine (Retigabine) | Potiga, Trobalt | Anticonvulsant | Seizures, Partial |
| Famotidine | Pepcid, Fluxid, Quamatel | H2-receptor antagonist | Gastric hyperacidity, GERD |
| Felbamate | Felbatol | Anticonvulsant | Seizures |
| Fenfluramine (Removed from Market) | Pondimin, Ponderax, Adafax | Appetite suppressant | Obesity |
| Fingolimod | Gilenya | Sphingosine phospate receptor modulator | Multiple Sclerosis |
| Flecainide | Tambocor, Almarytm, Apocard, Ecrinal, Flécaine | Antiarrhythmic | Arrhythmia |
| Fluconazole | Diflucan, Trican | Antifungal | Fungal infection |
| Fluorouracil (5-FU) | Adrucil, Carac, Efudex, Efudix, others | Anti-cancer | Cancer |
| Fluoxetine | Prozac, Sarafem, Fontex | Antidepressant, SSRI | Depression |
| Flupentixol (Only on Non US Market) | Depixol, Fluanxol | Antipsychotic | Schizophrenia |
| Fluvoxamine | Faverin, Fevarin, Floxyfral, Dumyrox and Luvox | Selective Serotonin Reuptake Inhibitor | Depression, Obsessive Compulsive Disorder |
| Formoterol | Foradil, Foradile, Oxeze, Oxis, Atock, Atimos, Atimos Modulite, Perforomist, Dulera, Symbicort, Vannair, Quikhale FB | Bronchodilator | Asthma |
| Furosemide (frusemide) | Lasix, Fusid, Frumex | Diuretic | Hypertension, diuresis |
| Galantamine | Reminyl, Nivalin, Razadyne-ER, Lycoremine | Cholinesterase inhibitor | Dementia (Alzheimer’s Disease) |
| Garenoxacin (Only on Non US Market) | Geninax | Antibiotic | Bacterial infection |
| Gatifloxacin (Removed from Market) | Tequin | Antibiotic | Bacterial infection |
| Gemifloxacin | Factive | Antibiotic | Bacterial infection |
| Granisetron | Kytril, Sancuso, Granisol | Antiemetic | Nausea, vomiting |
| Grepafloxacin (Removed from Market) | Raxar | Antibiotic | Bacterial infection |
| Halofantrine (Only on Non US Market) | Halfan | Antimalarial | Malaria |
| Haloperidol | Haldol (US & UK), Aloperidin, Bioperidolo, Brotopon, Dozic, Duraperidol (Germany), Einalon S, Eukystol, Halosten, Keselan, Linton, Peluces, Serenace, Serenase, Sigaperidol | Antipsychotic | Schizophrenia, agitation |
| Hydrochlorothiazide | Apo-Hydro, Aquazide H, BP Zide, Dichlotride, Hydrodiuril, HydroSaluric, Hydrochlorot, Microzide, Esidrex, Oretic | Diuretic | Hypertension, diuresis |
| Hydrocodone – ER | Hysingla™ ER, Zohydro ER | Analgesic | Pain, severe |
| Hydroxychloroquine | Plaquenil, Quineprox | Antimalarial, Anti-inflammatory | Malaria, SLE, rheumatoid arthritis |
| Hydroxyzine | Atarax, Vistaril, Aterax, Alamon, Durrax, Equipose, Masmoran, Orgatrax, Paxistil Quiess, Tran-Q, Tranquizine | Antihistamine | Allergic reaction, anxiety disorders |
| Ibogaine (Only on Non US Market) | None | Psychedelic | Narcotic addiction, unproven |
| Ibutilide | Corvert | Antiarrhythmic | Arrhythmia |
| Iloperidone | Fanapt, Fanapta, Zomaril | Antipsychotic, atypical | Schizophrenia |
| Imipramine (melipramine) | Tofranil | Antidepressant, Tricyclic | Depression |
| Indacaterol | Arcapta Neohaler (US), Onbrez Breezhaler (Canada), Utibron (combo w/ glycopyrrolate) | Bronchodilator | Asthma, COPD |
| Indapamide | Lozol, Natrilix, Insig | Diuretic | Hypertension, diuresis |
| Inotuzumab ozogamicin | Besponsa | Anti-cancer | Cancer (acute lymphocytic leukemia} |
| Isoproterenol | Medihaler-Iso, Isuprel | Bronchodilator | Allergic reaction |
| Isradipine | Dynacirc | Antihypertensive | Hypertension |
| Itraconazole | Sporanox, Onmel | Antifungal | Fungal infection |
| Ivabradine | Procoralan, Coralan, Corlentor, Coraxan, Ivabid, Bradia | Antianginal | Angina Pectoris (heart pain) |
| Ketanserin (Only on Non US Market) | Sufrexal | Antihypertensive | Hypertension |
| Ketoconazole | Nizoral, Sebizole, Ketomed, Keton | Antifungal | Fungal infection |
| Lacidipine (Only on Non US Market) | Lacipil, Motens | Calcium channel blocker | Hypertension |
| Lansoprazole | Prevacid | Proton Pump Inhibitor | Gastric hyperacidity, GERD |
| Lapatinib | Tykerb, Tyverb | Anti-cancer | Cancer (breast, metastatic) |
| Lenvatinib | Lenvima | Anti-cancer | Cancer (Thyroid) |
| Leuprolide | Lupron, Eligard, Viadur, Carcinil, Enanton, Leuplin, Lucrin, Procren, Prostap and others | Gonadotropin receptor agonist/antogist | Cancer (prostate) |
| Levalbuterol (levsalbutamol) | Xopenex, Levolin, Axazest | Bronchodilator | Asthma |
| Levofloxacin | Levaquin, Tavanic | Antibiotic | Bacterial infection |
| Levomepromazine (methotrimeprazine) (Only on Non US Market) | Nosinan, Nozinan, Levoprome | Antipsychotic | Schizophrenia |
| Levomethadyl acetate (Removed from Market) | Orlaam | Opiate | Narcotic dependence |
| Levosulpiride (Only on Non US Market) | Lesuride, Levazeo, Enliva (with rabeprazole) | Antipsychotic | Schizophrenia |
| Lisdexamfetamine | Vyvanse | CNS stimulant | ADHD |
| Lithium | Eskalith, Lithobid | Antimanic | Bipolar disorder |
| Loperamide | Imodium and many other OTC and Rx brands | Opiate | Diarrhea |
| Lopinavir and ritonavir | Kaletra, Aluvia | Antiviral | HIV/AIDS |
| Maprotiline | Ludiomil and others | Anti-depressant, Tetracyclic | Depression |
| Melperone (Only on Non US Market) | Bunil, Buronil, Eunerpan | Antipsychotic, atypical | Schizophrenia |
| Memantine | Namenda XR and many others | NMDA receptor antagonist | Alzheimer’s disease |
| Mesoridazine (Removed from Market) | Serentil | Antipsychotic | Schizophrenia |
| Metaproterenol (orciprenaline) | Metaprel, Alupent | Bronchodilator | Asthma |
| Methadone | Dolophine, Symoron, Amidone, Methadose, Physeptone, Heptadon | Opiate | Narcotic dependence, pain |
| Methamphetamine (Metamfetamine) | Desoxyn, Pervitin, Anadrex, Methedrine, Syndrox | CNS stimulant | Obesity, ADHD |
| Methylphenidate | Ritalin, Concerta, Focalin, Daytrana, Methylin, Metadate CD | CNS stimulant | ADHD |
| Metoclopramide | Reglan, Afipran, Maxolon, Cerucal, Clopamon, Clopra, Maxeran, Maxolon, Metozolv, Plasil, Pramin, Primperan, Perinorm | Antiemetic | Nausea, vomiting |
| Metolazone | Zytanix, Zaroxolyn, and Mykrox | Diuretic | Hypertension, diuresis |
| Metronidazole | Flagyl and many others | Antibiotic | Trichomoniasis, amebiasis, bacterial infection |
| Midodrine | ProAmatine, Amatine, Gutron | Vasoconstrictor | Hypotension |
| Midostaurin | Rydapt | Anti-cancer | Cancer (Acute myeloid leukemia) |
| Mifepristone | Korlym, Mifeprex | Progesterone antagonist | Pregnancy termination |
| Mirabegron | Myrbetriq | Beta3 adrenergic antagonist | Bladder spasm |
| Mirtazapine | Remeron | Antidepressant, Tetracyclic | Depression |
| Moexipril/HCTZ | Uniretic, Univasc | Antihypertensive | Hypertension, diuresis |
| Moxifloxacin | Avelox, Avalox, Avelon | Antibiotic | Bacterial infection |
| Necitumumab | Portrazza | Anti-cancer | Cancer (Lung) |
| Nelfinavir | Viracept | Antiviral | Viral infection (HIV/AIDS) |
| Nicardipine | Cardene | Antihypertensive | Hypertension |
| Nilotinib | Tasigna | Anti-cancer | Cancer (leukemia) |
| Norepinephrine | Levophed | Vasconstrictor, Inotrope | Heart failure, shock (low blood pressure) |
| Norfloxacin (Removed from Market) | Noroxin, Ambigram | Antibiotic | Bacterial infection |
| Nortriptyline | Pamelor, Sensoval, Aventyl, Norpress, Allegron, Noritren, Nortrilen | Antidepressant, Tricyclic | Depression |
| Nusinersen | Spinraza | Antisense oligonucleotide | Spinal Muscular Atrophy |
| Ofloxacin | Floxin | Antibiotic | Bacterial infection |
| Olanzapine | Zyprexa, Zydis, Relprevv | Antipsychotic, atypical | Schizophrenia, bipolar disorder |
| Olodaterol | Striverdi Respimat | Bronchodilator | Asthma |
| Omeprazole | Losec, Prilosec, Zegerid | Proton Pump Inhibitor | Gastric hyperacidity, GERD |
| Ondansetron | Zofran, Anset, Ondemet, Zuplenz, Emetron, Ondavell, Emeset, Ondisolv, Setronax | Antiemetic | Nausea, vomiting |
| Osimertinib | Tagrisso | Anti-cancer | Cancer (EGFR pos. NSC Lung cancer) |
| Oxaliplatin | Eloxatin | Anti-cancer | Cancer |
| Oxytocin | Pitocin, Syntocinon | Oxytocic | Labor stimulation |
| Paliperidone | Invega, Xepilon | Antipsychotic, atypical | Schizophrenia |
| Palonosetron | Aloxi | Antiemetic | Nausea, vomiting |
| Panobinostat | Farydak | Histone deacetylase inhibitor | Cancer, Multiple myeloma |
| Pantoprazole | Protonix and others | Proton Pump Inhibitor | Gastric hyperacidity, GERD |
| Papaverine HCl (Intra-coronary) | none | Vasodilator, Coronary | Diagnostic adjunct |
| Paroxetine | Paxil, Aropax, Pexeva, Seroxat, Sereupin, Seroxat | Antidepressant, SSRI | Depression |
| Pasireotide | Signifor | Somatostatin analog | Cushings Disease |
| Pazopanib | Votrient | Anti-cancer | Cancer (renal cell, sarcoma) |
| Pentamidine | Pentam | Antifungal | Fungal infection (Pneumocystis pneumonia) |
| Perflutren lipid microspheres | Definity, Optison | Imaging contrast agent | Diagnostic adjunct |
| Perphenazine | Trilafon, Etrafon/Triavil, Decentan | Antipsychotic | Schizophrenia |
| Phentermine | Adipex P, Adiphene (India), Anoxine-AM, Ionamin, Duromine, Metermine, Miraprontv, Obephen, Obermine, Obestin-30, Phentremine, Phentrol, Phenterex, Phentromin, Pro-Fast SA, Redusa, Panbesy, Obenix, Oby-Trim, Teramine, Zantryl, Sinpet, Supremin, Suprenza, Umine, Weltmine | Appetite suppressant | Obesity |
| Phenylephrine | Neosynephrine | Vasoconstrictor | Shock (low blood pressure), allergic rhinitis, asthma |
| Phenylpropanolamine (Removed from Market) | Acutrim, Dexatrim | Appetite suppressant | Obesity |
| Pilsicainide (Only on Non US Market) | Sunrythm | Anti-arrhythmic | Arrhythmia |
| Pimavanserin | Nuplazid | Antipsychotic, atypical | Psychosis, Parkinson’s Disease |
| Pimozide | Orap | Antipsychotic | Tourette’s Disorder |
| Pipamperone (Only on Non US Market) | Dipiperon (E.U), Propitan (Japan), Dipiperal, Piperonil, Piperonyl | Antipsychotic | Schizophrenia |
| Piperacillin/Tazobactam | Tazosyn and Zosyn | Antibiotic | Bacterial infection |
| Posaconazole | Noxafil, Posamol | Antifungal | Fungal infection |
| Primaquine phosphate | Antimalarial | Malaria | |
| Probucol (Removed from Market) | Lorelco | Antilipemic | Hypercholesterolemia |
| Procainamide | Pronestyl, Procan | Antiarrhythmic | Arrhythmia |
| Promethazine | Phenergan | Antipsychotic / Antiemetic | Nausea, vomiting |
| Propafenone | Rythmol SR, Rytmonorm | Sodium channel blocker | Arrhythmia |
| Propofol | Diprivan, Propoven | Anesthetic, general | Anesthesia |
| Prothipendyl (Only on Non US Market) | Dominal, Largophren, Timoval, Timovan, Tumovan | Antipsychotic | Schizophrenia |
| Pseudoephedrine | PediaCare, Sudafed | Decongestant | Allergic reaction, allergic rhinitis, asthma |
| Quetiapine | Seroquel | Antipsychotic, atypical | Schizophrenia |
| Quinidine | Quinaglute, Duraquin, Quinact, Quinidex, Cin-Quin, Quinora | Antiarrhythmic | Arrhythmia |
| Quinine sulfate | Qualaquin | Antimalarial | Malaria, leg cramps |
| Ranolazine | Ranexa, Ranozex | Antianginal | Angina Pectoris (heart pain) |
| Ribociclib | Kisqali | Anti-cancer | Cancer (breast) |
| Rilpivirine | Edurant, Complera, Eviplera | Antiviral | Viral infection (HIV/AIDS) |
| Risperidone | Risperdal | Antipsychotic, atypical | Schizophrenia |
| Ritodrine (Removed from Market) | Yutopar | Muscle relaxant | Premature labor |
| Romidepsin | Istodax | Histone deacetylase inhibitor | Cancer (lymphoma) |
| Roxithromycin (Only on Non US Market) | Rulide, Xthrocin, Roxl-150, Roxo, Surlid, Rulide, Biaxsig, Roxar, Roximycinv, Roxomycin, Rulid, Tirabicin, Coroxin | Antibiotic | Bacterial infection |
| Salmeterol | Serevent, Advair | Bronchodilator | Asthma |
| Saquinavir | Invirase(combo) | Antiviral | Viral infection (HIV/AIDS) |
| Sertindole (Only on Non US Market) | Serdolect, Serlect | Antipsychotic, atypical | Schizophrenia, anxiety |
| Sertraline | Zoloft, Lustral, Daxid, Altruline, Besitran, Deprax, Elrval, Emergen, Gladem, Implicane, Sedoran, Sealdin, SerivoLowfin, Stimuloton, Tresleen, Sertralin Bluefish | Antidepressant, SSRI | Depression |
| Sevoflurane | Ultane, Sojourn | Anesthetic, general | Anesthesia |
| Sibutramine (Removed from Market) | Meridia | Appetite suppressant | Obesity |
| Solifenacin | Vesicare | Muscle relaxant | Bladder spasm |
| Sorafenib | Nexavar | Anti-cancer | Cancer (liver, renal cell, metastatic thyroid) |
| Sotalol | Betapace, Sotalex, Sotacor | Antiarrhythmic | Arrhythmia |
| Sparfloxacin (Removed from Market) | Zagam | Antibiotic | Bacterial infection |
| Sulpiride (Only on Non US Market) | Dogmatil, Dolmatil, Eglonyl, Espiride, Modal, Sulpor | Antipsychotic, atypical | Schizophrenia |
| Sultopride (Only on Non US Market) | Barnetil, Barnotil, Topral | Antipsychotic, atypical | Schizophrenia |
| Sunitinib | Sutent | Anti-cancer | Cancer (GIST, renal cell, pNET) |
| Tacrolimus | Prograf, Prograf, Advagraf, Protopic | Immunosuppressant | Immune suppression |
| Tamoxifen | Nolvadex(discontinued 6/13), Istubal, Valodex | Anti-cancer | Cancer (breast) |
| Telaprevir | Incivo, Incivek | Antiviral | Viral infection (hepatitis C) |
| Telavancin | Vibativ | Antibiotic | Bacterial infection |
| Telithromycin | Ketek | Antibiotic | Bacterial infection |
| Terbutaline | Brethine, Bricanyl, Brethaire, Terbulin | Bronchodilator | Asthma, premature labor |
| Terfenadine (Removed from Market) | Seldane | Antihistamine | Allergic rhinitis |
| Terlipressin (Only on Non US Market) | Teripress, Glypressin, Terlipin, Remestyp, Tresil, Teriss and others | Vasoconstrictor | Septic shock |
| Terodiline (Only on Non US Market) | Micturin, Mictrol (not bethanechol) | Muscle relaxant | Bladder spasm |
| Tetrabenazine | Nitoman, Xenazine | Vesicular Monoamine Transporter 2 Inhibitor | Chorea (Huntington’s disease) |
| Thioridazine | Mellaril, Novoridazine, Thioril | Antipsychotic | Schizophrenia |
| Tiapride (Only on Non US Market) | Tiapridal, Italprid, Sereprile, Tialaread, Tiaryl, Tiaprim, Tiaprizal, Sereprid, Tiapridex | Selective D2, D3 dopamine antagonist | Alcoholism, withdrawal |
| Tipiracil and Trifluridine | Lonsurf | Anti-cancer | Cancer (Metastatic colorectal cancer) |
| Tizanidine | Zanaflex, Sirdalud | Muscle relaxant | Muscle spasticity |
| Tolterodine | Detrol, Detrusitol | Muscle relaxant | Bladder spasm |
| Toremifene | Fareston | Estrogen agonist/antagonist | Cancer (breast, metastatic) |
| torsemide (torasemide) | Demadex, Diuver, Examide | Diuretic | Hypertension, diuresis |
| Tramadol | Crispin, Ralivia ER, Ralivia Flashtab, Tramadolum, Tramal, Tramodol, Tridural, Ultram, Ultram ER, Zydol | Analgesic | Pain |
| Trazodone | Desyrel (discontinued 6/13), Oleptro, Beneficat, Deprax, Desirel, Molipaxin, Thombran, Trazorel, Trialodine, Trittico, Mesyrel | Antidepressant, SARI | Depression, insomnia |
| Trimethoprim-Sulfamethoxazole | Septra, Bactrim, Sulfatrim, Biseptol, Co-trimoxazole, Cotrim, Septrin, Trisul | Antibiotic | Bacterial infection |
| Trimipramine | Surmontil, Rhotrimine, Stangyl | Antidepressant, Tricyclic | Depression |
| Tropisetron (Only on Non US Market) | Navoban, Setrovel | Antiemetic | Nausea, vomiting |
| Valbenazine | Ingrezza | Vesicular monamine transporter 2 inhibitor | Tardive Dyskinesia |
| Vandetanib | Caprelsa | Anti-cancer | Cancer (thyroid) |
| Vardenafil | Levitra | Phosphodiesterase 5 inhibitor | Erectile dysfunction |
| Vemurafenib | Zelboraf | Anti-cancer | Cancer (melanoma) |
| Venlafaxine | Effexor, Efexor | Antidepressant, SNRI | Depression |
| Vilanterol/fluticasone furoate | Breo Ellipta | Bronchodilator | Asthma |
| Voriconazole | VFend | Antifungal | Fungal infection |
| Vorinostat | Zolinza | Histone deacetylase inhibitor | Cancer (lymphoma) |
| Ziprasidone | Geodon, Zeldox | Antipsychotic, atypical | Schizophrenia |
| Zotepine (Only on Non US Market) | Losizopilon, Lodopin, Setous and Zoleptil | Antipsychotic, atypical | Schizophrenia |
| Zuclopenthixol, Zuclopentixol (Only on Non US Market) | Cisordinol, Clopixol, Acuphase | Antipsychotic | Psychosis |
Long QT syndrome symptoms
Nearly half of patients with long QT syndrome NEVER have any signs or symptoms!
You might be aware of your condition only because of:
- Results of an electrocardiogram (ECG) done for an unrelated reason
- A family history of long QT syndrome
- Genetic testing results
Signs and symptoms of inherited or congenital long QT syndrome might start as a fetus, during the first weeks to months after birth, as late as older age, or never at all. Most people who experience signs or symptoms from long QT syndrome have their first episode by age 40.
Signs and symptoms of long QT syndrome might occur during sleep or arousal from sleep.
The most common symptoms of long QT syndrome include:
- Syncope (fainting): This is the most common symptom of long QT syndrome 56. Long QT syndrome-triggered fainting spells (syncope) are caused by the heart temporarily beating in an erratic way. These fainting spells might happen when you’re excited, angry, scared or during exercise. You may lose consciousness without warning, for instance from being startled by a ringing telephone. If you have a normal fainting spell, you usually will have a warning sign first, such as lightheadedness, heart palpitations, irregular heartbeat, weakness or blurred vision. However, a fainting spell from long QT syndrome can occur with little to no warning.
- Seizures: If the heart continues to beat erratically, the brain will eventually not get enough oxygen, which can cause seizures. Long QT syndrome can be misdiagnosed as epilepsy 57, 58, 59.
- Sudden death: Generally, the heart returns to its normal rhythm. If this doesn’t happen by itself, or if an external defibrillator isn’t used in time to convert the rhythm back to normal, sudden death will occur. In about 1 out of 10 people who have long QT syndrome, sudden cardiac arrest or sudden death is the first sign of the disorder.
- Unexplained drowning or near drowning. This may be due to fainting while swimming.
- Often, people who have LQTS 3 develop an abnormal heart rhythm during sleep. This may cause noisy gasping while sleeping.
The symptoms of long QT syndrome are related to Torsades de Pointes (see Figure 5). During this arrhythmia, the ventricle beats very fast and irregularly. The heart is unable to pump blood effectively to the body. If the brain does not receive an adequate blood supply, syncope (fainting) and seizure-like activity can occur. If the arrhythmia continues, sudden death will occur. If the heart rhythm returns to normal, symptoms will stop.
- If you have potassium channel long QT syndrome, you carry the risk of sudden death if you are startled or awoken suddenly.
- If you have sodium channel long QT syndrome, you have an increased risk of sudden death whilst sleeping.
Symptoms are most common during:
- Exercise (or within a few minutes after)
- Emotional excitement, especially being startled
- During sleep or upon waking suddenly
Some people with congenital long QT syndrome never have symptoms. The diagnosis is made during a routine ECG or during an evaluation because a family member has it. Symptoms usually first appear during the early teen years.
Your doctor will advise you depending on the type of long QT syndrome you have. You may be advised not to take part in competitive sports, which can be difficult – especially for young people. Your doctor will help you arrive at a balance between being active and reducing your risk of making your condition worse.
The risk of sudden death is greater if you have a history of:
- previous cardiac arrest
- blackouts
- a very long QT interval on your ECG [corrected QT (QTc) interval > 500 ms]
- sodium channel mutations.
Strenuous exercise or severe exertion can increase the risk of sudden death for many people with long QT syndrome. Speak to your doctor if you have any questions about the risk of sudden death.
If you are with someone when they collapse suddenly, it is imperative to call your local emergency services number and try to perform CPR until an ambulance or medical help arrives. If you are untrained in CPR, the emergency services operator will be able to talk you through the process.
Silent long QT Syndrome
Sometimes long QT syndrome doesn’t cause any signs or symptoms. This is called silent long QT syndrome. For this reason, doctors often advise family members of people who have long QT syndrome to be tested for the disorder, even if they have no symptoms.
Medical and genetic tests may reveal whether these family members have long QT syndrome and what type of the condition they have.
Long QT syndrome complications
Most of the time, prolonged QT intervals in people with long QT syndrome never cause problems. However, physical or emotional stress might “trip up” a heart that is sensitive to prolonged QT intervals. This can cause the heart’s rhythm to spin out of control, triggering life-threatening, irregular heart rhythms (arrhythmias) including:
Torsades de Pointes — “twisting of the points” in French 60. In this arrhythmia, your heart’s two lower chambers (ventricles) beat fast and chaotically, making the waves on an ECG monitor look twisted (Figure 5). Less blood is pumped out from your heart, so less blood reaches your brain, causing you to faint suddenly and, often, without any warning.
If a Torsades de Pointes episode is short — lasting less than one minute — your heart can correct itself and you regain consciousness on your own. However, if a Torsades de Pointes episode lasts longer, it can result in a sudden fainting spell followed by a full-body seizure. If the dangerous rhythm does not correct itself, then a life-threatening arrhythmia called ventricular fibrillation follows.
Ventricular fibrillation. This condition causes the ventricles to beat so fast that your heart quivers and ceases pumping blood. Unless your heart is shocked back into a normal rhythm by a defibrillator, ventricular fibrillation can lead to brain damage and sudden death.
Sudden cardiac death. It’s now known that long QT syndrome might explain some cases of sudden death in young people who otherwise appear healthy. Long QT syndrome might be responsible for some unexplained events in children and young adults, such as unexplained fainting, drownings or seizures.
- Key risk factors for sudden cardiac death in patients with long QT syndrome include the following:
- QTc interval: QTc interval ≥ 500 ms is a major risk factor, particularly in inherited long QT syndrome type 1 (LQT1) and inherited long QT syndrome type 2 (LQT2) 61
- Genetic subtype: Patients with inherited long QT syndrome type 2 (LQT2) have a higher sudden cardiac death risk compared to other subtypes 62
- Sex and age: Males have higher risk in childhood and females after puberty 63, 64
- History of syncope (fainting): Particularly if recurrent or without trigger 65
- Family history of sudden cardiac death: Especially in young first-degree relatives such as your parents, siblings and your children 66
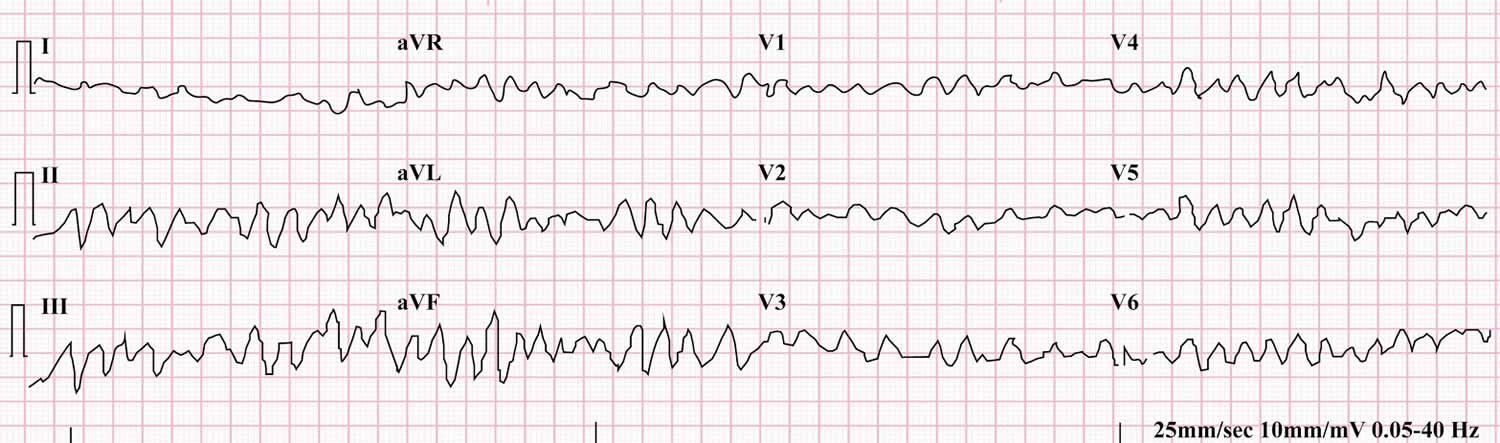
Figure 5. Torsades de Pointes
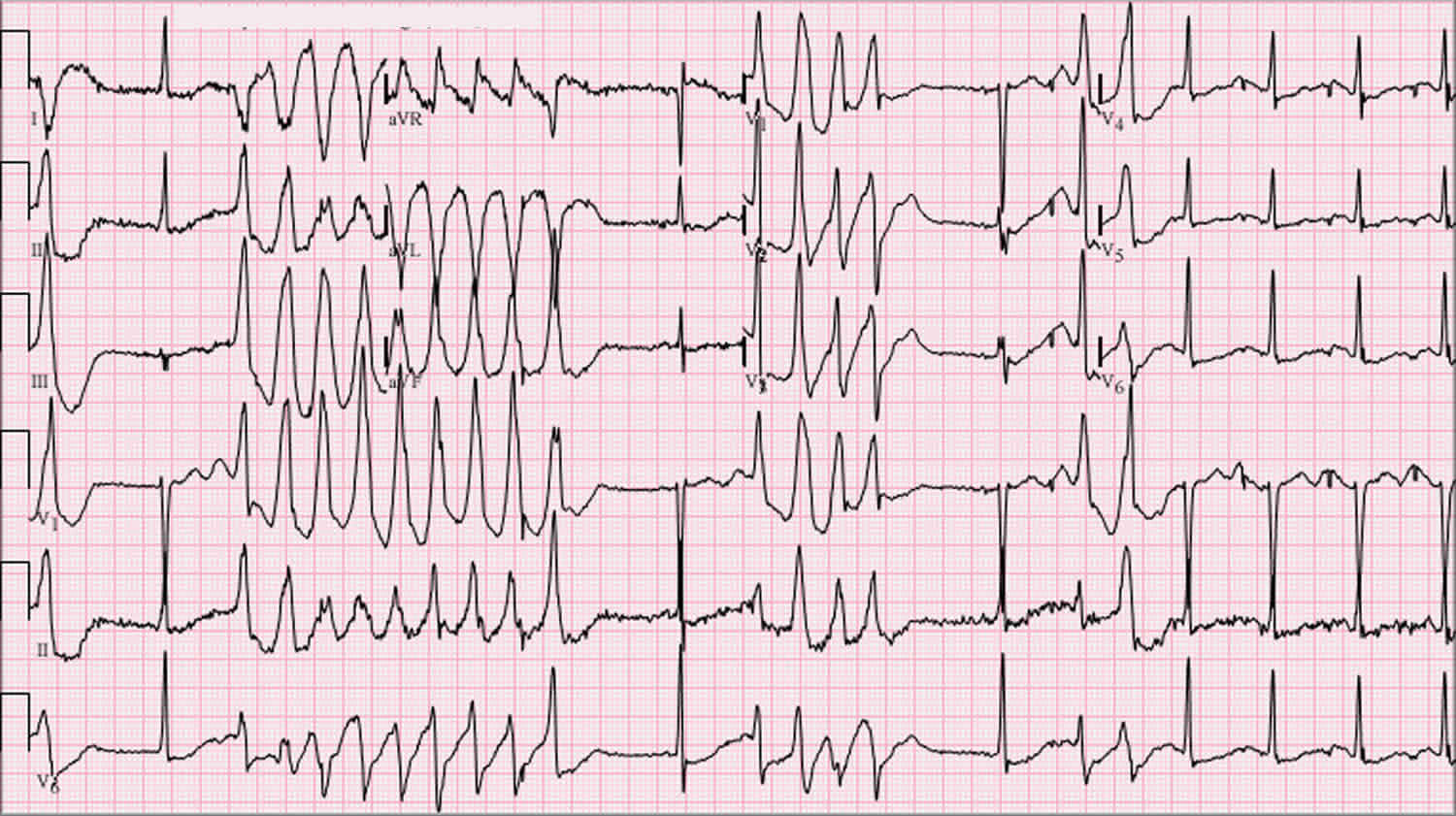
Footnotes: Torsades de Pointes ECG with prolonged QT interval and the “twisting” or oscillating pattern of the QRS complexes, with the amplitude changing around the baseline. 12-lead ECG of Torsades de Pointes in a 56-year-old white female with low blood potassium (2.4 mmol/L) and low blood magnesium (1.6 mg/dL).
[Source 67 ]Figure 6. Ventricular fibrillation
Risk factors for developing abnormal heart rhythms in patients with long QT syndrome
The risk of abnormal heart rhythms in patients with long QT syndrome is influenced by a complex interplay of genetic, clinical, and environmental factors 68.
Major risk factors for abnormal heart rhythms in patients with long QT syndrome include the following:
Genetic subtype:
- Inherited long QT syndrome type 2 (LQT2) and inherited long QT syndrome type 3 (LQT3): Higher risk of syncope and sudden cardiac death compared to inherited long QT syndrome type 1 (LQT1) 69.
- Inherited long QT syndrome type 1 (LQT1): High risk during exercise, especially swimming 70
- Inherited long QT syndrome type 2 (LQT2): Prone to events during rest or sleep 71
- Inherited long QT syndrome type 3 (LQT3): Higher risk during sleep or at rest 72
QTc interval
- QTc interval > 500 ms is associated with a significantly increased risk of sudden cardiac death, particularly in patients with LQT1 and LQT2 73. In addition, the presence of QTc interval > 500 ms in the first year of life is a major risk factor for sudden cardiac death in infants with congenital long QT syndrome 74
Sex
- In childhood, males have a higher risk of sudden cardiac than do females, particularly in LQT1 75. However, after puberty, the risk of sudden cardiac is higher in females, especially in LQT2 76, 64, 77.
History of syncope
- A history of syncope is a major risk factor for sudden cardiac death in patients with long QT syndrome, particularly if the syncope is recurrent or occurs in the absence of a trigger 78. In patients with a history of syncope, the risk of sudden cardiac death is highest in the first year after the syncopal event and decreases thereafter 78. The presence of syncope in the first year of life is also a strong predictor of sudden cardiac death in infants with long QT syndrome 79.
Family history of sudden cardiac death
- Significant risk factor, especially in young relatives (<40 years) or multiple family members 65.
Genotype-specific triggers
- The risk of abnormal heart rhythms in patients with long QT syndrome is influenced by genotype-specific triggers, which can precipitate syncope or sudden cardiac death in susceptible individuals 80.
Electrolyte imbalances
- Electrolyte imbalances, particularly hypokalemia and hypomagnesemia, can prolong the QT interval and increase the risk of arrhythmic events 81.
Medications
Comorbidities
Genotype-phenotype modifiers
- In addition to the primary genetic defect, other genetic factors (such as single nucleotide polymorphisms or modifier genes) can influence the clinical manifestations and arrhythmic risk in patients with Long QT syndrome. For example, the presence of certain polymorphisms in the NOS1AP gene (which encodes a nitric oxide synthase adaptor protein) has been associated with an increased risk of SCD in patients with long QT syndrome, particularly those with LQT1 85, 86.
Long QT syndrome diagnosis
If long QT syndrome is suspected, your doctor will want to ask questions about your medical history, as well as your family’s.
Your doctor will also ask you if you have a:
- Family history of long QT syndrome
- Family history of unexplained fainting, seizures, or cardiac arrest
- History of fainting, seizures or cardiac arrest, especially with exercise
For example, studies of otherwise healthy people with long QT syndrome indicate that they had at least one episode of fainting by age 10. The majority also had a family member with long QT syndrome.
Unexplained fainting episodes or a family history of heart-related death may warrant electrocardiogram (EKG or ECG) testing for you and your closes relatives. Your doctor may also recommend an exercise stress test.
If your doctor thinks you may have long QT syndrome, you might need several tests to confirm the diagnosis. These include:
- Electrocardiogram (ECG). During an ECG, doctors attach sensors to your chest (electrodes) that can detect the electrical activity of your heart. An ECG measures the timing and duration of each electrical phase in your heartbeat. You might have this test while at rest or during an exercise stress test, in which doctors monitor your heart activity as you exercise on a treadmill or a stationary bicycle. Your doctor may also suggest your family members have ECGs.
- The space between the start of the Q wave and the end of the T wave (QT interval) corresponds to the time it takes for your heart to contract and then refill with blood before beginning the next contraction. The QT interval represents the time from the electrical stimulation (depolarization) of the heart’s pumping chambers (ventricles), to the end of the recharging of the electrical system (repolarization) and therefore roughly estimates the duration of an average ventricular action potential 29. The interval between the letters Q and T defines the action of the ventricles. The QT interval is measured in milliseconds (msec) and closely approximates the time from the beginning of the heart ventricles’ contraction until the end of relaxation.
- The QT interval interval can range from 0.20 to 0.40 seconds (200 to 400 msec), depending upon your heart rate. At high heart rates, ventricular action potentials shorten in duration, which decreases the QT interval 29. Long QT syndrome means that time period is too long, even if by fractions of a second. Because prolonged QT intervals can be diagnostic for susceptibility to certain types of heart arrhythmias, it is important to determine if a QT interval is excessively long. The QT interval is often expressed as a corrected QT (QTc) by taking the QT interval and dividing it by the square root of the R-R interval (interval between ventricular depolarizations) using Bazett formula: QTc = QT / √ RR 7. Corrected QT (QTc) allows an assessment of the QT interval that is independent of the heart rate. Normal corrected QTc intervals are 0.44 seconds or less (<440 msec). QTc is prolonged if it is greater than 440 ms in men or greater than 460 ms in women 30. A QTc greater than 500 is associated with an increased risk of Torsade de Pointes. While several equations exist to help correct heart rate variation, the Bazett formula (QTC = QT / √ RR) is the most commonly used. Though the Bazett formula seems relatively accurate in heart rates between 60 to 100 beats/min, it tends to overcorrect with higher heart rates and undercorrect with lower heart rates.
- The hallmark ECG feature of long QT syndrome (LQTS) is a prolonged QT interval, reflecting delayed ventricular repolarization 87. QTc interval > 460 ms in females and QTc interval > 450 ms in males are considered prolonged 30. However, up to 25% of patients with genetically confirmed long QT syndrome may have normal QTc intervals 88, 89.
- Additional ECG findings suggestive of long QT syndrome include T-wave alternans (TWA), T-wave notching, and bradycardia 90.
- Once corrected QT (QTc) is identified as prolonged, the next step in a workup is to look for acquired causes. The most common cause of QT prolongation in an ICU setting is usually drug-related. Serum potassium, calcium, and magnesium levels should be checked, as low serum of each can cause QT prolongation. Also, stimulating thyroid hormone (TSH) levels may be checked in patients with suspected hypothyroidism.
- In the absence of reversible or acquired causes of QT prolongation, long QT syndrome is diagnosed 7. In these patients, obtaining an electrocardiography of the patient and family members may be very helpful. Genetic testing of the patient and family members is the gold standard; however, this testing is limited by cost 7. Drug provocation with epinephrine or isoproterenol is warranted in patients with a borderline presentation 7. The concept of this testing is that patients with Long QT syndrome have an abnormal response to sympathetic stimulation. Their ECG shows the failure of the QT interval to shorten with increased heart rates, or it may even show prolongation. In patients with LQT2, there is marked shortening with exercise, however, exaggerated lengthening of the QT interval as the heart rate declines during late recovery 7.
- Holter monitor. This portable ECG device can be worn for a day or more to record your heart’s activity as you go about your routine.
- Event monitor. This portable ECG device is attached to your body to monitor your heart activity over a few weeks to a few months. When you have symptoms, you press a button. This allows your doctor to check your heart rhythm at the time of your symptoms.
Long QT syndrome is usually diagnosed by measuring the Q-T interval on the ECG.
In some people with suspected long QT syndrome, the ECG doesn’t show an abnormally prolonged QT interval. You may need other tests, such as:
- Exercise stress test. Some people have a long QT interval only while they exercise. For this reason, your doctor may recommend that you have a stress test. During an exercise stress test, you exercise to make your heart work hard and beat fast. An EKG is done while you exercise. If you can’t exercise, you may be given medicine to increase your heart rate.
- A nonexercise (medication) stress test. This is an ECG test done while you’re given a medication such as epinephrine (Adrenalin) that stimulates your heart in a way similar to exercise. Doctors then monitor the effects of the medication on the way your heart recharges. This test can help doctors diagnose people with suspected long QT syndrome and may help determine which genes are associated with the condition. It may also be used to diagnose people with long QT syndrome who have a gene associated with long QT syndrome but who have a normal QT interval (recharging time) at rest.
- Genetic testing. A genetic test for long QT syndrome is available and may be covered by some private and governmental insurance plans. Genetic tests for long QT syndrome can generally find the genetic cause for about 3 out of every 4 cases of inherited long QT syndrome. However, genetic tests can’t detect all cases of long QT syndrome. So, even if you have long QT syndrome, the genetic tests may not show it. Also, some people who test positive for long QT syndrome don’t have any signs or symptoms of the disorder. These people may have silent long QT syndrome. Less than 10 percent of these people will faint or suddenly die from an abnormal heart rhythm. Even if you have silent long QT syndrome, you may be at increased risk of having an abnormal heart rhythm while taking medicines that affect potassium ion channels or blood levels of potassium. If your genetic cause of long QT syndrome is discovered through a positive genetic test, your doctor may recommend that your family members also be tested to determine whether they inherited the same genetic mutation.
- A second opinion. You might want to seek a second opinion before proceeding with treatment if your doctor diagnoses you with long QT syndrome. People can sometimes be misdiagnosed as having long QT syndrome when it’s not actually present.
Schwartz score
The Schwartz’s score is a clinical scoring system used to diagnose congenital long QT syndrome (LQTS) 91. The Schwartz score assigns points to clinical and ECG features to estimate long QT syndrome (LQTS) probability 92. Schwartz’s scores range from 0 to 9, with Schwartz’s score ≤ 1 low probability; 1.5–3 intermediate probability; ≥ 3.5 high long QT syndrome (LQTS) probability.
The clinical features included in the Schwartz score are syncope (1–2 points), congenital deafness (0.5 points), and a family history of LQTS or unexplained sudden death (0.5–1 points). The ECG features included are QTc interval ≥ 480 ms (3 points), 460–479 ms (2 points), or 450–459 ms (1 point); T wave alternans (TWA) (1 point); notched T waves in 3 leads (1 point); and bradycardia (0.5 points).
Table 2. Schwartz Score for Long QT Syndrome (LQTS)
| CRITERIA | POINTS |
|---|---|
| Patient History | |
| Syncope with stress† | 2 |
| Syncope without stress† | 1 |
| Congenital deafness | 0.5 |
| Family History | |
| Family member with known LQTS‡ | 1 |
| Family member with unexplained cardiac death before age 30 ‡ | 0.5 |
| Electrocardiography§ | |
| QTc ≥ 480 msec | 3 |
| QTc 460-479 msec | 2 |
| QTc 450-459 msec (males only) | 1 |
| QTc ≥ 480 msec during 4th minute of recovery from an exercise test | 1 |
| Torsades de pointe arrhythmia§ | 2 |
| T wave alternans (TWA) | 1 |
| Notched T wave in 3 leads | 1 |
| Resting heart rate < 2nd percentile for age (bradycardia) | 0.5 |
Footnotes: * SCORE: ≤ 1 low probability; 1.5–3 intermediate probability; ≥ 3.5 high probability
† Mutually exclusive (ie, if syncope occurs with stress, no points are given for syncope without stress)
‡ Mutually exclusive
§ If torsades de pointe present, do not score syncope
[Source 92 ]Provocative testing
Provocative tests can unmask long QT syndrome in patients with borderline QTc intervals or normal ECGs 93:
- Exercise stress testing: Useful for diagnosing congenital long QT syndrome type 1 (LQT1) 94, 95
- Epinephrine infusion: Helps distinguish between LQT1 and LQT2 96
- Mental stress testing: Potential method for unmasking concealed long QT syndrome 97.
Holter monitoring and event recorders
Ambulatory ECG monitoring can capture transient QT prolongation or T-wave abnormalities 98, 99. Implantable loop recorders may be used for long-term monitoring or risk stratification 100.
Genetic testing
Genetic testing is essential for confirming long QT syndrome diagnosis and identifying specific subtypes 101. Commercial panels screen for mutations in up to 17 long QT syndrome-associated genes, with a 50%–80% diagnostic yield 102, 103. KCNQ1 (LQTS1), KCNH2 (LQTS2), and SCN5A (LQTS3) account for 75% of genetically confirmed cases 104. Genetic testing aids in identifying asymptomatic family members at risk and guiding genotype-specific management 105.
Family screening
Family screening is essential because of the autosomal dominant inheritance of most long QT syndrome subtypes 106. First-degree relatives have a 50% chance of carrying the same mutation and should undergo clinical evaluation, ECG screening, and genetic testing 107. Cascade screening helps identify asymptomatic carriers who may benefit from preventive measures and regular follow-up 108.
Long QT syndrome treatment
Long QT syndrome treatment is aimed at preventing sudden death and controlling symptoms.
Treatment for inherited long QT syndrome involves some simple preventive measures. It can also involve medications, as well as left cardiac sympathetic denervation surgery or implanting medical devices such as a defibrillator (ICD), or both.
The goal of treatment is either to prevent the long QT heart from ever beating out of control or to prevent sudden death. Your doctor will discuss with you the most appropriate treatment options for your condition based on your symptoms, your type of long QT syndrome, and your risk of fainting or sudden cardiac arrest.
For acquired long QT syndrome, treating the cause of the condition may eliminate it. Doctors will also treat heart rhythm disorders (arrhythmias) as needed.
If you are diagnosed with acquired long QT syndrome due to certain medications, your doctor may recommend that you stop taking the medication causing the condition and switching medications. Some people might need additional treatment. Around 100 drugs have been associated with causing acquired long QT syndrome – many of them common including anti-depressants, antibiotics, and diuretics.
If your long QT syndrome is associated with a mineral imbalance such as being deficient in potassium, calcium or magnesium, the first step is to identify and fix the imbalance. You may require no extra treatment.
Immediate treatment
A hemodynamically unstable patient should receive non-synchronized electrical defibrillation or unsynchronized cardioversion that delivers a high-energy shock at any point during the cardiac cycle. Also, the first-line treatment is magnesium sulfate for suppressing early afterdepolarizations and terminating Torsades de Pointes arrhythmia and the benefit is independent of serum magnesium level 109, 22, 110, 111. Magnesium achieves this by decreasing the influx of calcium, thus lowering the amplitude of early afterdepolarizations 112. Magnesium can be given at 1 to 2 g IV initially in 30-60 seconds, which then can be repeated in 5-15 minutes 22. Alternatively, a continuous magnesium infusion can be started at a rate of 3-10 mg/minute 22. Magnesium is effective even in patients with normal magnesium levels. Because of the danger of hypermagnesemia (depression of neuromuscular function), the patient requires close monitoring 22.
Some authorities recommend supplemental potassium to increase the potassium concentration to high normal, which increases the efflux of potassium from myocardial cells, thus causing rapid repolarization 22.
Temporary transvenous overdrive pacing should be considered for those who do not respond to magnesium sulfate 7. Based on the fact that the QT interval shortens with a faster heart rate, pacing can be effective in terminating Torsades de Pointes 22. Pacing is effective in both forms of the long QT syndrome because it facilitates the repolarizing potassium currents and prevents long pauses, suppressing early afterdepolarizations and decreasing the QT interval. Pacing should be instituted at a rate of 90-110 bpm until the QT interval is normalized. Overdrive pacing may be necessary at a rate of up to 140 bpm to control the rhythm 22.
Atrial pacing is the preferred mode because it preserves the atrial contribution to ventricular filling and also results in a narrower QRS complex and hence a shorter QT 22. In patients with AV block, ventricular pacing can be used to suppress Torsades de Pointes 22. This is dependent on intact atrial-to-ventricular conduction at the pacing rate found necessary.
Isoproterenol and Class IB antiarrhythmic drugs, such as lidocaine and phenytoin, may also be used 113, 114, 115, 116.
Mexiletine also may be helpful in suppressing Torsades de Pointes 22. In one study, mexiletine was used in patients with HIV who had acquired long QT interval and Torsades de Pointes 117. Mexiletine effectively suppressed the torsade on a long-term basis 117. Nakashima et al 118 reported Mexiletine successfully preventing refractory Torsades de Pointes and ventricular fibrillation in a 28-year-old patient with congenital type 2 long QT syndrome.
Medications for long-term management of Congenital Long QT syndrome
Medications used to treat long QT syndrome may include:
- Beta-adrenergic antagonists (beta blockers) at maximally tolerated doses are used as a first-line long-term therapy in long QT syndrome 119, 120, 22. These heart drugs include nadolol (Corgard) and propranolol (Inderal LA, InnoPran XL). Propranolol is used most extensively, but other agents such as esmolol or nadolol also can be used 22. They slow the heart rate and make the rhythm associated with long QT syndrome less likely. Beta blockers work by blunting the way your heart reacts to adrenaline. Most patients (even those without symptoms) are treated with a beta-blocker.
- However, beta-blockers should be avoided in those congenital long QT syndrome in which bradycardia is a prominent feature 22. Beta-blockers are also contraindicated in acquired long QT syndrome because bradycardia produced by these agents can precipitate Torsades de Pointes 22.
- Patients with congenital long QT syndromes are thought to have an abnormality of sympathetic balance or tone and are treated with beta-blockers 22. If the patient experiences breakthrough Torsades de Pointes, a short-acting beta-blocker, such as esmolol, can be tried 121.
- Isoproterenol can be used in bradycardia-dependent Torsades de Pointes that usually is associated with acquired long QT syndrome (pause-dependent) 22. Isoproterenol should be administered as a continuous IV infusion to keep the heart rate above 90 bpm 22. Isoproterenol accelerates AV conduction and decreases the QT interval by increasing the heart rate and reducing temporal dispersion of repolarization 22. Beta-adrenergic agonists such as isoproterenol are contraindicated in the congenital form of long QT syndrome (adrenergic-dependent) 22. Because of precautions, contraindications, and adverse effects associated with its use, Isoproterenol is used as an interim agent until overdrive pacing can be started 22.
- One approach to assess the adequacy of beta-blockade is by exercise testing. One investigator recommends aiming for at least a 20% reduction in maximum heart rate compared to that of the baseline (pre-beta blocker therapy). Another approach is to check the blood levels of beta blockers (eg, propranolol) when possible 122.
- Other medications may be used to shorten the Q-T interval. Your doctor will discuss what medications are best for you.
- Mexiletine. Taking this anti-arrhythmic drug in combination with beta blockers might help shorten the QT interval and decrease the likelihood of a long QT syndrome-triggered faint, seizure or sudden death. This medication is often used for the third-most common subtype of inherited long QT syndrome, LQT3 123, 42, 124. However, it may also be used for the two most common subtypes, LQT1 and LQT2.
- Ranolazine, a sodium current (INaL) inhibitor, shows benefits in LQT3 and along QT syndrome 119, 125, 126. These medications are currently off-label and reserved for patients failing first-line therapies 127.
- Spironolactone and potassium. For certain forms of long QT syndrome, spironolactone (Aldactone), a medication used to help the body hold on to potassium, or potassium supplements, or both, might improve the heart’s recharging system.
- Magnesium and potassium supplements are used as adjunctive (add-on) therapies in long QT syndrome, particularly for recurrent arrhythmic events despite beta-blocker therapy 128, 129. Magnesium does not shorten QT interval but can reduce Torsades de Pointes risk in along QT syndrome 130. Potassium supplements help maintain normal serum levels, preventing QT prolongation and arrhythmic events 119, 131. Routine use is not currently recommended; their use should be individualized on the basis of electrolyte status and clinical response.
- Fish oil. In general, current evidence does not support supplementation with heart-healthy fish oil (omega-3 fatty acid) or eating fish high in omega-3 fatty acids to decrease the risk of cardiac events or death from abnormal heart rhythms. However, if you have inherited long QT syndrome, fish oil may be a reasonable complementary alternative medication to take in addition to other therapies. Talk to your doctor before starting fish oil or any other supplements or medications.
Your doctor might suggest treatment for long QT syndrome even if you don’t often experience signs or symptoms.
If you do need treatment, take the medications your doctor prescribes as directed. While medications won’t cure the condition, they provide some protection against possibly fatal disruptions of your heart rhythm. You might need to take a medication such as a beta blocker indefinitely.
Long-term treatment in acquired long QT syndrome usually is not required because the QT interval returns to normal once the inciting factor or predisposing condition has been corrected 22. Pacemaker implantation is effective in cases that are associated with heart block or bradycardia 22. Implantable cardioverter-defibrillators (ICDs) are indicated in cases that cannot be managed by avoidance of the offending agent.
Medical Devices
- Patients who have a history of cardiac arrest or symptoms, in spite of beta-blocker therapy, may receive an implantable cardioverter defibrillator (ICD). This device detects life-threatening arrhythmias and automatically shocks the heart to prevent sudden death. Most new implantable cardioverter defibrillators (ICDs) can do the same job as a pacemaker, as well as detect dangerous heart rhythms. Once these are detected, the ICD can deliver a shock to restore your heart back to its normal rhythm. The implantable cardioverter-defibrillator (ICD) device is implanted under the skin of your chest, can stop a potentially fatal arrhythmia. An implantable cardioverter defibrillator (ICD) continuously monitors your heartbeat. If it detects an abnormal heart rhythm, it delivers electrical shocks to reset the heart to a normal rhythm. Implanting an implantable cardioverter defibrillator (ICD) is a major procedure and can result in inappropriate shocks and other complications. Therefore, the decision to implant an implantable cardioverter defibrillator, especially in children, needs to be carefully considered. Most people with inherited long QT syndrome do not need an implantable cardioverter defibrillator. Importantly, an implantable cardioverter defibrillator should not be implanted solely because of the tragic occurrence of a long QT syndrome-triggered sudden death in a relative. But the implantable cardioverter-defibrillator (ICD) device may be suggested for some athletes to help them return to competitive sports. The decision to place an implantable cardioverter-defibrillator (ICD), especially in children, needs to be carefully considered. Sometimes the device may send out shocks that aren’t needed. Talk with your heart specialist about the benefits and risks of an implantable cardioverter-defibrillator (ICD).
- Pacemaker. Patients who have an abnormally slow heart rate may receive a pacemaker. A pacemaker delivers electrical impulses to control the rhythm of your heart, but it can’t deliver a shock to correct an arrhythmia. Pacemakers are devices that can be placed in your body, usually by surgery, to support the electrical system in your heart. Pacemaker can stabilize abnormal heart rhythms and prevent problems that can disrupt or endanger your life.
Left cardiac sympathetic denervation surgery
In left cardiac sympathetic denervation (LCSD) or a sympathectomy operation procedure, surgeons cut specific nerves along the left side of your spine in your chest. These nerves are part of the body’s sympathetic nervous system, which helps regulate heart rhythm that prompt your heart to beat faster in response to physical or emotional stress. This type of surgery keeps the heart beating at a steady pace and lowers the risk of dangerous heart rhythms in response to stress or exercise. The surgery also significantly reduces the risk of sudden death.
The left cardiac sympathetic denervation (LCSD) surgery is generally reserved for people who are considered at high risk of sudden death and are experiencing appropriate implantable cardioverter defibrillator (ICD) shocks, people who are continuing to experience fainting or seizures while taking their medications, or people unable to tolerate their medications because of side effects. However, accidental ablation of ocular efferent sympathetic nerves may result in Horner syndrome 22.
Lifestyle changes
In addition to medications or surgery, your doctor might recommend lifestyle changes to reduce your chances of a long QT syndrome-related fainting spell or sudden cardiac arrest. These could include:
- Family testing – All first-line relatives (brothers, sisters, parents and children) should have EKG testing. Any other family members who have a history of seizures or fainting should also undergo testing.
- Avoiding strenuous exercise or contact sports – If you have congenital long QT syndrome, sometimes, fatal arrhythmias occur with exercise 132. The decision to participate in competitive sports should be managed by a heart rhythm expert and certain precautions may be suggested. Or have a safety plan in place if you continue in competitive sports. In general, people with long QT syndrome should never swim alone. Physical activities are likely to be okay if you take someone along who can help if you faint.
- Buddy system – Your family and friends should be told you have long QT syndrome. They should be told to call for emergency help if you begin to have symptoms or faint.
- Avoiding medications that could cause prolonged QT intervals
- Avoid diuretics and medications causing electrolyte disturbances
- Getting plenty of liquids during illnesses that are causing vomiting or diarrhea
- Maintain normal potassium and magnesium levels 135
- Lowering your temperature if a fever occurs
- Reducing your exposure to loud or startling noises. Turn down the volume on doorbells and other devices such as phones that may startle you, especially during sleep. Things that startle you can cause you to pass out 66.
- Staying away from situations that could make you excited or angry. Being very excited, angry or surprised can trigger heartbeat changes in some people with long QT syndrome 136, 137, 138. Getting more exercise, practicing mindfulness and connecting with others in support groups are some ways to reduce stress.
Work with your doctor to balance these lifestyle recommendations against the clear, heart-healthy benefits of an active lifestyle. It may be possible to stay fully active in sports, including competitive sports, after carefully reviewing the risks and benefits with your doctor.
If your symptoms are mild or don’t occur very often, your doctor might recommend only simple preventive measures or lifestyle changes, and he or she might not prescribe any daily medications for you.
Future treatments will be geared toward more gene specific therapies. For example, certain types of long QT syndrome are more likely to initiate events during exercise, while others are more related to startling or emotional distress. Your doctor will be able to give you activity guidelines based on the specific type of long QT syndrome gene you carry. Therapies may be directed to treat the specific gene, rather than prevention of future complications.
Long QT syndrome life expectancy
More than half of the people who have untreated, inherited types of long QT syndrome die within 10 years with up to 21 percent of people with Long QT syndrome may die within a year after they start fainting 28. The prognosis for people with Long QT syndrome that is being treated is good with the risk of suffering cardiac arrest dropping to less than 1% over 20 years. Lifestyle changes and medicines can help people who have long QT syndrome prevent complications and live longer. Women with long QT syndrome can live a full life and give birth to children.
If you have long QT syndrome, talk with your doctor about which lifestyle changes and treatments are best for you.
Once you’ve been diagnosed with long QT syndrome, several steps can help you avoid serious consequences, including:
- Avoiding strenuous physical activity or startling noises.
- Adding more potassium to your diet (as your doctor advises).
- Taking heart medicines called beta blockers. These medicines help prevent sudden cardiac arrest.
- Having an implanted medical device, such as a pacemaker or implantable cardioverter defibrillator. These devices help control abnormal heart rhythms.
- Don’t overexert yourself. You might not need to give up sports if you have long QT syndrome. Your doctor might permit recreational activities as long as you have a buddy along in case you have a fainting episode. In general, people with long QT syndrome should never swim alone. Strenuous exercise might be dangerous and isn’t recommended for some people with long QT syndrome. However, others might have a lower risk of complications and may be able to continue strenuous exercise and even competitive sports. Discuss this issue with your doctor in detail.
- Know your symptoms. Be fully aware of symptoms that can warn you of irregular heart rhythms and decreased blood flow to your brain, such as feeling like you may faint.
- Inform other people. Make family, friends, teachers, neighbors and anyone else who has regular contact with you aware of your heart condition. Wear a medical alert identification to notify health care providers of your condition.
- Have plans in case of an emergency cardiac event. Family members may want to learn cardiopulmonary resuscitation (CPR) so they can provide immediate resuscitation if you ever need it. In some situations, it might be appropriate to have or be able to rapidly access an automatic external defibrillator. However, if your inherited long QT syndrome has been evaluated and treated carefully, you are generally unlikely to ever need CPR or an automatic external defibrillator.
- Control startling events as much as possible. Turn down the volume on doorbells and turn off the telephone ringer or your cellphone at night.
- Visit your doctor. Your cardiologist will likely recommend that you have regular follow-up appointments with him or her. Let your doctor know if you have symptoms of long QT syndrome or any changes in your condition. Your doctor may make changes to your treatment plan or suggest additional treatments for you.
Sexual intercourse doesn’t appear to increase the risk of long QT syndrome. Pregnancy and delivery aren’t associated with an increased risk of symptoms in women with long QT syndrome.
Still, if you have inherited long QT syndrome, your doctor will want to monitor you closely both during your pregnancy and after. Women with long QT syndrome, especially a form called LQT2, are at increased risk during the period following delivery and need careful monitoring.
Living With long QT syndrome
Long QT syndrome usually is a lifelong condition. The risk of having an abnormal heart rhythm that leads to fainting or sudden cardiac arrest may lessen as you age. However, the risk never completely goes away.
You’ll need to take certain steps for the rest of your life to prevent abnormal heart rhythms. You can:
- Avoid things that trigger abnormal heart rhythms
- Let others know you might faint or your heart might stop beating, and tell them what steps they can take
- Have a plan in place for how to handle abnormal heart rhythms
If an abnormal heart rhythm does occur, you’ll need to seek treatment right away.
Avoid Triggers
If exercise triggers an abnormal heart rhythm, your doctor may tell you to avoid any strenuous exercise, especially swimming. Ask your doctor what types and amounts of exercise are safe for you.
If you have a pacemaker or implantable cardioverter defibrillator, avoid contact sports that may dislodge these devices. You may want to exercise in public or with a friend who can help you if you faint.
Avoid medicines that can trigger an abnormal heart rhythm. This includes some medicines used to treat allergies, infections, high blood pressure, high blood cholesterol, depression, and arrhythmias. Talk with your doctor before taking any prescription, over-the-counter, or other medicines or drugs.
Seek medical care right away for conditions that lower the sodium or potassium level in your blood. These conditions include the eating disorders anorexia nervosa and bulimia, excessive vomiting or diarrhea, and certain thyroid disorders.
If you have long QT syndrome 2, try to avoid unexpected noises, such as loud or jarring alarm clock buzzers and telephone ringers.
Inform Others
You may want to wear a medical ID necklace or bracelet that states that you have long QT syndrome. This will help alert medical personnel and others about your condition if you have an emergency.
Let your roommates, coworkers, or other people with whom you have regular contact know that you have a condition that might cause you to faint or go into cardiac arrest. Tell them to call your local emergency services number right away if you faint.
Consider asking a family member and/or coworker to learn cardiopulmonary resuscitation (CPR) in case your heart stops beating.
You also may want to keep an automated external defibrillator with you at home or at work. This device uses electric shocks to restore a normal heart rhythm.
Someone at your home and/or workplace should be trained on how to use the automated external defibrillator, just in case your heart stops beating. If a trained person isn’t available, an untrained person also can use the automated external defibrillator to help save your life.
If you have long QT syndrome 3 and you sleep alone, you may want to have an intercom in your bedroom that’s connected to someone else’s bedroom. This will let others detect the noisy gasping that often occurs if you have an abnormal heart rhythm while lying down.
Ongoing Health Care Needs
You should see your cardiologist (heart specialist) regularly. He or she will adjust your treatment as needed. For example, if you still faint often while using less aggressive treatments, your doctor may suggest other treatment options.
Emotional Issues and Support
Living with long QT syndrome may cause fear, anxiety, depression, and stress. Talk about how you feel with your health care team. Talking to a professional counselor also can help. If you’re very depressed, your doctor may recommend medicines or other treatments that can improve your quality of life.
Joining a patient support group may help you adjust to living with long QT syndrome. You can see how other people have coped with the condition. Talk with your doctor about local support groups or check with an area medical center.
Support from family and friends also can help relieve stress and anxiety. Let your loved ones know how you feel and what they can do to help you.
Some people learn they have long QT syndrome because they’re tested after a family member dies suddenly from long QT syndrome. Grief counseling may help you cope if this has happened to you. Talk with your doctor about finding a grief counselor.
- Zhu W, Bian X, Lv J. From genes to clinical management: A comprehensive review of long QT syndrome pathogenesis and treatment. Heart Rhythm O2. 2024 Jul 15;5(8):573-586. doi: 10.1016/j.hroo.2024.07.006[↩][↩][↩][↩]
- Yu Y, Deschenes I, Zhao MT. Precision medicine for long QT syndrome: patient-specific iPSCs take the lead. Expert Rev Mol Med. 2023 Jan 4;25:e5. doi: 10.1017/erm.2022.43[↩]
- Odening KE, van der Linde HJ, Ackerman MJ, Volders PGA, Ter Bekke RMA. Electromechanical reciprocity and arrhythmogenesis in long-QT syndrome and beyond. Eur Heart J. 2022 Aug 21;43(32):3018-3028. doi: 10.1093/eurheartj/ehac135[↩]
- Ponce-Balbuena D, Deschênes I. Long QT syndrome – Bench to bedside. Heart Rhythm O2. 2021 Jan 22;2(1):89-106. doi: 10.1016/j.hroo.2021.01.006[↩]
- Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. doi: 10.1016/S0140-6736(99)02107-8[↩]
- Long QT syndrome. https://rarediseases.info.nih.gov/diseases/6922/long-qt-syndrome[↩]
- Al-Akchar M, Siddique MS. Long QT Syndrome. [Updated 2022 Dec 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441860[↩][↩][↩][↩][↩][↩][↩][↩]
- Schwartz PJ, Ackerman MJ, George AL Jr, Wilde AAM. Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol. 2013 Jul 16;62(3):169-180. doi: 10.1016/j.jacc.2013.04.044[↩]
- Giudicessi JR, Ackerman MJ. Calcium Revisited: New Insights Into the Molecular Basis of Long-QT Syndrome. Circ Arrhythm Electrophysiol. 2016 Jul;9(7):e002480. doi: 10.1161/CIRCEP.116.002480[↩]
- Eggeling T., Höher M., Osterhues H.H., Kochs M., Weismüller P., Hombach V. The arrhythmogenic substrate of the long QT syndrome: genetic basis, pathology, and pathophysiologic mechanisms. Eur Heart J. 1993;14:73–79. doi: 10.1093/eurheartj/14.suppl_e.73[↩]
- Krahn A.D., Laksman Z., Sy R.W., et al. Congenital long QT syndrome. JACC Clin Electrophysiol. 2022;8:687–706. doi: 10.1016/j.jacep.2022.02.017[↩]
- Schwartz P.J., Ackerman M.J. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J. 2013;34:3109–3116. doi: 10.1093/eurheartj/eht089[↩]
- Alders M, Bikker H, Christiaans I. Long QT Syndrome. 2003 Feb 20 [Updated 2018 Feb 8]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1129[↩]
- Evaluation of drug-induced QT interval prolongation: implications for drug approval and labelling. Malik M, Camm AJ. Drug Saf. 2001;24:323–351. doi: 10.2165/00002018-200124050-00001[↩]
- Clinical practice. Long-QT syndrome. Roden DM. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513[↩]
- The long QT syndrome. Prospective longitudinal study of 328 families. Moss AJ, Schwartz PJ, Crampton RS, et al. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136[↩]
- Krahn AD, Laksman Z, Sy RW, Postema PG, Ackerman MJ, Wilde AAM, Han HC. Congenital Long QT Syndrome. JACC Clin Electrophysiol. 2022 May;8(5):687-706. doi: 10.1016/j.jacep.2022.02.017[↩][↩]
- Lankaputhra M, Voskoboinik A. Congenital long QT syndrome: a clinician’s guide. Intern Med J. 2021 Dec;51(12):1999-2011. doi: 10.1111/imj.15437[↩][↩]
- Long QT Syndrome. https://rarediseases.org/rare-diseases/romano-ward-syndrome/[↩][↩]
- Wilde A.A.M., Amin A.S., Postema P.G. Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart. 2022;108:332–338. doi: 10.1136/heartjnl-2020-318259[↩][↩]
- Neves R., Bains S., Bos J.M., MacIntyre C., Giudicessi J.R., Ackerman M.J. Precision therapy in congenital long QT syndrome. Trends Cardiovasc Med. 2024;34:39–47. doi: 10.1016/j.tcm.2022.06.006[↩][↩]
- Treatment of Torsades de Pointes. https://emedicine.medscape.com/article/1950863-overview#a9[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Chivers S, Ovadia C, Regan W, Zidere V, Vigneswaran T, Sharland G, Rosenthal E, Seed PT, Simpson JM, Williamson C. Systematic review of long QT syndrome identified during fetal life. Heart Rhythm. 2023 Apr;20(4):596-606. doi: 10.1016/j.hrthm.2022.12.026[↩][↩][↩]
- Itoh H., Crotti L., Aiba T., et al. The genetics underlying acquired long QT syndrome: impact for genetic screening. Eur Heart J. 2016;37:1456–1464. doi: 10.1093/eurheartj/ehv695[↩][↩][↩]
- El-Sherif N., Turitto G., Boutjdir M. Acquired long QT syndrome and torsade de pointes. Pacing Clin Electrophysiol. 2018;41:414–421. doi: 10.1111/pace.13296[↩][↩][↩]
- Long QT syndrome. https://www.mayoclinic.org/diseases-conditions/long-qt-syndrome/symptoms-causes/syc-20352518[↩][↩]
- Long Q-T Syndrome (LQTS). https://my.clevelandclinic.org/health/diseases/17183-long-q-t-syndrome-lqts[↩]
- Long QT Syndrome. https://www.nhlbi.nih.gov/health-topics/long-qt-syndrome[↩][↩]
- Electrocardiogram (EKG, ECG). https://cvphysiology.com/arrhythmias/a009[↩][↩][↩][↩]
- Fukuyama M., Horie M., Aoki H., et al. School-based routine screenings of electrocardiograms for the diagnosis of long QT syndrome. Europace. 2022;24:1496–1503. doi: 10.1093/europace/euab320[↩][↩][↩]
- Presentation of 1st case in Italian pediatric literature)] Clin Pediatr (Bologna) 1963;45:656–683.[↩]
- Ward O.C. A new familial cardiac syndrome in children. J Ir Med Assoc. 1964;54:103–106.[↩]
- Abriel H., Zaklyazminskaya E.V. Cardiac channelopathies: genetic and molecular mechanisms. Gene. 2013;517:1–11. doi: 10.1016/j.gene.2012.12.061[↩]
- Timothy syndrome. https://medlineplus.gov/genetics/condition/timothy-syndrome[↩]
- Weiss J.N., Qu Z., Shivkumar K. Electrophysiology of hypokalemia and hyperkalemia. Circ Arrhythm Electrophysiol. 2017;10 doi: 10.1161/CIRCEP.116.004667[↩]
- Antzelevitch C., Burashnikov A. Overview of basic mechanisms of cardiac arrhythmia. Card Electrophysiol Clin. 2011;3:23–45. doi: 10.1016/j.ccep.2010.10.012[↩]
- Gupta A., Lawrence A.T., Krishnan K., Kavinsky C.J., Trohman R.G. Current concepts in the mechanisms and management of drug-induced QT prolongation and torsade de pointes. Am Heart J. 2007;153:891–899. doi: 10.1016/j.ahj.2007.01.040[↩]
- Mubagwa K., Gwanyanya A., Zakharov S., Macianskiene R. Regulation of cation channels in cardiac and smooth muscle cells by intracellular magnesium. Arch Biochem Biophys. 2007;458:73–89. doi: 10.1016/j.abb.2006.10.014[↩]
- Eryol N.K., Colak R., Ozdoğru I., et al. Effects of calcium treatment on QT interval and QT dispersion in hypocalcemia. Am J Cardiol. 2003;91:750–752. doi: 10.1016/s0002-9149(02)03423-9[↩]
- Etchegoyen C.V., Keller G.A., Mrad S., Cheng S., Di Girolamo G. Drug-induced QT interval prolongation in the intensive care unit. Curr Clin Pharmacol. 2017;12:210–222. doi: 10.2174/1574884713666180223123947[↩]
- Chevalier P., Bellocq C., Millat G., et al. Torsades de pointes complicating atrioventricular block: evidence for a genetic predisposition. Heart Rhythm. 2007;4:170–174. doi: 10.1016/j.hrthm.2006.10.004[↩]
- Li G., Zhang L. The role of mexiletine in the management of long QT syndrome. J Electrocardiol. 2018;51:1061–1065. doi: 10.1016/j.jelectrocard.2018.08.035[↩][↩]
- Tomaselli G.F., Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6[↩]
- Kenigsberg D.N., Khanal S., Kowalski M., Krishnan S.C. Prolongation of the QTc interval is seen uniformly during early transmural ischemia. J Am Coll Cardiol. 2007;49:1299–1305. doi: 10.1016/j.jacc.2006.11.035[↩]
- Colzani R.M., Emdin M., Conforti F., Passino C., Scarlattini M., Iervasi G. Hyperthyroidism is associated with lengthening of ventricular repolarization. Clin Endocrinol (Oxf) 2001;55:27–32. doi: 10.1046/j.1365-2265.2001.01295.x[↩]
- Choi S.Y., Cho K.I., Han Y.J., et al. Impact of pheochromocytoma on left ventricular hypertrophy and QTc prolongation: comparison with Takotsubo cardiomyopathy. Korean Circ J. 2014;44:89–96. doi: 10.4070/kcj.2014.44.2.89[↩]
- van der Bilt I.A.C., Hasan D., Vandertop W.P., et al. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: a meta-analysis. Neurology. 2009;72:635–642. doi: 10.1212/01.wnl.0000342471.07290.07[↩]
- Koppikar S., Baranchuk A., Guzmán J.C., Morillo C.A. Stroke and ventricular arrhythmias. Int J Cardiol. 2013;168:653–659. doi: 10.1016/j.ijcard.2013.03.058[↩]
- Lee W., Vandenberk B., Raj S.R., Lee S.S. Prolonged QT interval in cirrhosis: twisting time? Gut Liver. 2022;16:849–860. doi: 10.5009/gnl210537[↩]
- Liu P., Wang L., Han D., Sun C., Xue X., Li G. Acquired long QT syndrome in chronic kidney disease patients. Ren Fail. 2020;42:54–65. doi: 10.1080/0886022X.2019.1707098[↩]
- Sachs K.V., Harnke B., Mehler P.S., Krantz M.J. Cardiovascular complications of anorexia nervosa: a systematic review. Int J Eat Disord. 2016;49:238–248. doi: 10.1002/eat.22481[↩][↩]
- Lazzerini P.E., Laghi-Pasini F., Bertolozzi I., et al. Systemic inflammation as a novel QT-prolonging risk factor in patients with torsades de pointes. Heart. 2017;103:1821–1829. doi: 10.1136/heartjnl-2016-311079[↩]
- Aromolaran A.S., Srivastava U., Ali A., et al. Interleukin-6 inhibition of hERG underlies risk for acquired long QT in cardiac and systemic inflammation. PLoS One. 2018;13 doi: 10.1371/journal.pone.0208321[↩]
- Lazzerini P.E., Laghi-Pasini F., Boutjdir M., Capecchi P.L. Anti-Ro/SSA antibodies and the autoimmune long-QT syndrome. Front Med (Lausanne) 2021;8 doi: 10.3389/fmed.2021.730161[↩]
- https://www.crediblemeds.org/[↩]
- Colman N., Bakker A., Linzer M., Reitsma J.B., Wieling W., Wilde A.A. Value of history-taking in syncope patients: in whom to suspect long QT syndrome? Europace. 2009;11:937–943. doi: 10.1093/europace/eup101[↩]
- Eddy C.A., MacCormick J.M., Chung S.K., et al. Identification of large gene deletions and duplications in KCNQ1 and KCNH2 in patients with long QT syndrome. Heart Rhythm. 2008;5:1275–1281. doi: 10.1016/j.hrthm.2008.05.033[↩]
- Anderson J.H., Bos J.M., Cascino G.D., Ackerman M.J. Prevalence and spectrum of electroencephalogram-identified epileptiform activity among patients with long QT syndrome. Heart Rhythm. 2014;11:53–57. doi: 10.1016/j.hrthm.2013.10.010[↩]
- Gonzalez A., Aurlien D., Larsson P.G., et al. Seizure-like episodes and EEG abnormalities in patients with long QT syndrome. Seizure. 2018;61:214–220. doi: 10.1016/j.seizure.2018.08.020[↩]
- Kahlon SS, Sikandar R, Tejovath S, Nair S, Hassan D, K Patel K, Peddemul A, Mostafa JA. Diagnosing Torsades De Pointes Based on Correlation to QT Interval: A Systematic Review. Cureus. 2022 Aug 9;14(8):e27833. doi: 10.7759/cureus.27833[↩]
- Koponen M., Marjamaa A., Hiippala A., et al. Follow-up of 316 molecularly defined pediatric long-QT syndrome patients: clinical course, treatments, and side effects. Circ Arrhythm Electrophysiol. 2015;8:815–823. doi: 10.1161/CIRCEP.114.002654[↩]
- Wedekind H., Burde D., Zumhagen S., et al. QT interval prolongation and risk for cardiac events in genotyped LQTS-index children. Eur J Pediatr. 2009;168:1107–1115. doi: 10.1007/s00431-008-0896-6[↩]
- Diez-Escute N., Arbelo E., Martinez-Barrios E., et al. Sex differences in long QT syndrome. Front Cardiovasc Med. 2023;10 doi: 10.3389/fcvm.2023.1164028[↩]
- Furukawa T., Kurokawa J. Regulation of cardiac ion channels via non-genomic action of sex steroid hormones: implication for the gender difference in cardiac arrhythmias. Pharmacol Ther. 2007;115:106–115. doi: 10.1016/j.pharmthera.2007.04.008[↩][↩]
- Kaufman E.S., McNitt S., Moss A.J., et al. Risk of death in the long QT syndrome when a sibling has died. Heart Rhythm. 2008;5:831–836. doi: 10.1016/j.hrthm.2008.02.029[↩][↩]
- Goldenberg I., Horr S., Moss A.J., et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J Am Coll Cardiol. 2011;57:51–59. doi: 10.1016/j.jacc.2010.07.038[↩][↩]
- https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png[↩]
- Giudicessi J.R., Ackerman M.J. Genotype- and phenotype-guided management of congenital long QT syndrome. Curr Probl Cardiol. 2013;38:417–455. doi: 10.1016/j.cpcardiol.2013.08.001[↩]
- Priori S.G., Schwartz P.J., Napolitano C., et al. Risk stratification in the long-QT syndrome. N Engl J Med. 2003;348:1866–1874. doi: 10.1056/NEJMoa022147[↩]
- Lanfranchi P.A., Ackerman M.J., Kara T., et al. Gene-specific paradoxical QT responses during rapid eye movement sleep in women with congenital long QT syndrome. Heart Rhythm. 2010;7:1067–1074. doi: 10.1016/j.hrthm.2010.05.012[↩]
- Dahlberg P., Axelsson K.J., Rydberg A., Lundahl G., Gransberg L., Bergfeldt L. Spatiotemporal repolarization dispersion before and after exercise in patients with long QT syndrome type 1 versus controls: probing into the arrhythmia substrate. Am J Physiol Heart Circ Physiol. 2023;325:H1279–H1289. doi: 10.1152/ajpheart.00335.2023[↩]
- Schwartz P.J., Priori S.G., Spazzolini C., et al. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89[↩]
- Brink P.A., Crotti L., Corfield V., et al. Phenotypic variability and unusual clinical severity of congenital long-QT syndrome in a founder population. Circulation. 2005;112:2602–2610. doi: 10.1161/CIRCULATIONAHA.105.572453[↩]
- Spazzolini C., Mullally J., Moss A.J., et al. Clinical implications for patients with long QT syndrome who experience a cardiac event during infancy. J Am Coll Cardiol. 2009;54:832–837. doi: 10.1016/j.jacc.2009.05.029[↩]
- Locati E.H., Zareba W., Moss A.J., et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: findings from the International LQTS Registry. Circulation. 1998;97:2237–2244. doi: 10.1161/01.cir.97.22.2237[↩]
- Daubert J.P., Zareba W., Rosero S.Z., Budzikowski A., Robinson J.L., Moss A.J. Role of implantable cardioverter defibrillator therapy in patients with long QT syndrome. Am Heart J. 2007;153:53–58. doi: 10.1016/j.ahj.2007.01.025[↩]
- Bjelic M., Zareba W., Peterson D.R., et al. Sex hormones and repolarization dynamics during the menstrual cycle in women with congenital long QT syndrome. Heart Rhythm. 2022;19:1532–1540. doi: 10.1016/j.hrthm.2022.04.029[↩]
- Hobbs J.B., Peterson D.R., Moss A.J., et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA. 2006;296:1249–1254. doi: 10.1001/jama.296.10.1249[↩][↩]
- Kelle A.M., Bos J.M., Etheridge S.P., et al. Cardiac transplantation in children and adolescents with long QT syndrome. Heart Rhythm. 2017;14:1182–1188. doi: 10.1016/j.hrthm.2017.04.023[↩]
- Keller D.I., Grenier J., Christe G., et al. Characterization of novel KCNH2 mutations in type 2 long QT syndrome manifesting as seizures. Can J Cardiol. 2009;25:455–462. doi: 10.1016/s0828-282x(09)70117-5[↩]
- Roden D.M. Clinical practice: long-QT syndrome. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513[↩]
- Roden D.M. Drug-induced prolongation of the QT interval. N Engl J Med. 2004;350:1013–1022. doi: 10.1056/NEJMra032426[↩]
- Barra S., Agarwal S., Begley D., Providencia R. Post-acute management of the acquired long QT syndrome. Postgrad Med J. 2014;90:348–358. doi: 10.1136/postgradmedj-2013-132398[↩]
- Lubberding A.F., Zhang J., Lundh M., et al. Age-dependent transition from islet insulin hypersecretion to hyposecretion in mice with the long QT-syndrome loss-of-function mutation Kcnq1-A340V. Sci Rep. 2021;11 doi: 10.1038/s41598-021-90452-8[↩]
- Crotti L., Monti M.C., Insolia R., et al. NOS1AP is a genetic modifier of the long-QT syndrome. Circulation. 2009;120:1657–1663. doi: 10.1161/CIRCULATIONAHA.109.879643[↩]
- Earle N., Yeo Han D., Pilbrow A., et al. Single nucleotide polymorphisms in arrhythmia genes modify the risk of cardiac events and sudden death in long QT syndrome. Heart Rhythm. 2014;11:76–82. doi: 10.1016/j.hrthm.2013.10.005[↩]
- Moss A.J., Kass R.S. Long QT syndrome: from channels to cardiac arrhythmias. J Clin Invest. 2005;115:2018–2024. doi: 10.1172/JCI25537[↩]
- Immanuel S.A., Sadrieh A., Baumert M., et al. T-wave morphology can distinguish healthy controls from LQTS patients. Physiol Meas. 2016;37:1456–1473. doi: 10.1088/0967-3334/37/9/1456[↩]
- Doldi F., Plagwitz L., Hoffmann L.P., et al. Detection of patients with congenital and often concealed long-QT syndrome by novel deep learning models. J Pers Med. 2022;12:1135. doi: 10.3390/jpm12071135[↩]
- Lane C.M., Bos J.M., Rohatgi R.K., Ackerman M.J. Beyond the length and look of repolarization: defining the non-QTc electrocardiographic profiles of patients with congenital long QT syndrome. Heart Rhythm. 2018;15:1413–1419. doi: 10.1016/j.hrthm.2018.04.033[↩]
- Schwartz P.J., Moss A.J., Vincent G.M., Crampton R.S. Diagnostic criteria for the long QT syndrome: an update. Circulation. 1993;88:782–784. doi: 10.1161/01.cir.88.2.782[↩]
- Schwartz PJ, Crotti L. QTc behavior during exercise and genetic testing for the long-QT syndrome. Circulation. 2011 Nov 15;124(20):2181-4. doi: 10.1161/CIRCULATIONAHA.111.062182[↩][↩]
- Abrahams T., Davies B., Laksman Z., et al. Provocation testing in congenital long QT syndrome: a practical guide. Heart Rhythm. 2023;20:1570–1582. doi: 10.1016/j.hrthm.2023.07.059[↩]
- Chattha I.S., Sy R.W., Yee R., et al. Utility of the recovery electrocardiogram after exercise: a novel indicator for the diagnosis and genotyping of long QT syndrome? Heart Rhythm. 2010;7:906–911. doi: 10.1016/j.hrthm.2010.03.006[↩]
- Horner J.M., Horner M.M., Ackerman M.J. The diagnostic utility of recovery phase QTc during treadmill exercise stress testing in the evaluation of long QT syndrome. Heart Rhythm. 2011;8:1698–1704. doi: 10.1016/j.hrthm.2011.05.018[↩]
- Ackerman M.J., Khositseth A., Tester D.J., Hejlik J.B., Shen W.K., Porter C.B. Epinephrine-induced QT interval prolongation: a gene-specific paradoxical response in congenital long QT syndrome. Mayo Clin Proc. 2002;77:413–421. doi: 10.4065/77.5.413[↩]
- Etienne P., Huchet F., Gaborit N., et al. Mental stress test: a rapid, simple, and efficient test to unmask long QT syndrome. Europace. 2018;20:2014–2020. doi: 10.1093/europace/euy078[↩]
- Katritsis D.G., Siontis G.C., Camm A.J. Prognostic significance of ambulatory ECG monitoring for ventricular arrhythmias. Prog Cardiovasc Dis. 2013;56:133–142. doi: 10.1016/j.pcad.2013.07.005[↩]
- Anys S., Arnaud M., Minois D., et al. Dose response to nadolol in congenital long QT syndrome. Heart Rhythm. 2021;18:1377–1383. doi: 10.1016/j.hrthm.2021.04.021[↩]
- Solbiati M., Casazza G., Dipaola F., et al. The diagnostic yield of implantable loop recorders in unexplained syncope: a systematic review and meta-analysis. Int J Cardiol. 2017;231:170–176. doi: 10.1016/j.ijcard.2016.12.128[↩]
- Ackerman M.J., Priori S.G., Willems S., et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Europace. 2011;13:1077–1109. doi: 10.1093/europace/eur245[↩]
- Chung S.K., MacCormick J.M., McCulley C.H., et al. Long QT and Brugada syndrome gene mutations in New Zealand. Heart Rhythm. 2007;4:1306–1314. doi: 10.1016/j.hrthm.2007.06.022[↩]
- Horigome H., Nagashima M., Sumitomo N., et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: a nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol. 2010;3:10–17. doi: 10.1161/CIRCEP.109.88215[↩]
- Barsheshet A, Goldenberg I. Cardiovascular syncope: diagnostic approach and risk assessment. Minerva Med. 2011 Jun;102(3):223-38. https://www.minervamedica.it/en/journals/minerva-medica/article.php?cod=R10Y2011N03A0223[↩]
- Hofman N., Wilde A.A., Kaab S., et al. Diagnostic criteria for congenital long QT syndrome in the era of molecular genetics: do we need a scoring system? Eur Heart J. 2007;28:575–580. doi: 10.1093/eurheartj/ehl355[↩]
- Baruteau A.E., Baruteau J., Joomye R., et al. Role of congenital long-QT syndrome in unexplained sudden infant death: proposal for an electrocardiographic screening in relatives. Eur J Pediatr. 2009;168:771–777. doi: 10.1007/s00431-009-0951-y[↩]
- Bokil N.J., Baisden J.M., Radford D.J., Summers K.M. Molecular genetics of long QT syndrome. Mol Genet Metab. 2010;101:1–8. doi: 10.1016/j.ymgme.2010.05.011[↩]
- Hofman N., Tan H.L., Alders M., van Langen I.M., Wilde A.A. Active cascade screening in primary inherited arrhythmia syndromes: does it lead to prophylactic treatment? J Am Coll Cardiol. 2010;55:2570–2576. doi: 10.1016/j.jacc.2009.12.063[↩]
- Thomas SH, Behr ER. Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br J Clin Pharmacol. 2016;81(3):420-427. doi: 10.1111/bcp.12726[↩]
- Jaiswal A, Goldbarg S. Dofetilide induced torsade de pointes: mechanism, risk factors and management strategies. Indian Heart J. 2014;66(6):640-648. doi: 10.1016/j.ihj.2013.12.021[↩]
- Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002 Jan;143(1):7-14. doi: 10.1067/mhj.2002.120295[↩]
- Baker WL. Treating arrhythmias with adjunctive magnesium: identifying future research directions. Eur Heart J Cardiovasc Pharmacother. 2017 Apr 1;3(2):108-117. doi: 10.1093/ehjcvp/pvw028[↩]
- Beach SR, Celano CM, Sugrue AM, Adams C, Ackerman MJ, Noseworthy PA, Huffman JC. QT Prolongation, Torsades de Pointes, and Psychotropic Medications: A 5-Year Update. Psychosomatics. 2018 Mar-Apr;59(2):105-122. doi: 10.1016/j.psym.2017.10.009[↩]
- Anderson HN, Bos JM, Haugaa KH, Morlan BW, Tarrell RF, Caraballo PJ, Ackerman MJ. Prevalence and Outcome of High-Risk QT Prolongation Recorded in the Emergency Department from an Institution-Wide QT Alert System. J Emerg Med. 2018 Jan;54(1):8-15. doi: 10.1016/j.jemermed.2017.08.073[↩]
- Gromova OA, Torshin IY, Kalacheva AG, Grishina TR. [On Synergism of Potassium and Magnesium in Maintenance of Myocardial Function]. Kardiologiia. 2016 Mar;56(3):73-80. Russian. doi: 10.18565/cardio.2016.3.73-80[↩]
- Chouchoulis K, Chiladakis J, Koutsogiannis N, Davlouros P, Kaza M, Alexopoulos D. Impact of QT interval prolongation following antiarrhythmic drug therapy on left ventricular function. Future Cardiol. 2017 Jan;13(1):13-22. doi: 10.2217/fca-2016-0052[↩]
- Shimabukuro-Vornhagen A, Rybniker J, Zoghi S, Faetkenheuer G, Michels G, Erdmann E, von Bergwelt-Baildon M, Kochanek M. Acquired Long QT Syndrome and Torsade de Pointes Associated with HIV Infection. Case Rep Med. 2010;2010:278427. doi: 10.1155/2010/278427[↩][↩]
- Nakashima R, Takase S, Kai K, Sakamoto K, Tsutsui H. Mexiletine effectively prevented refractory Torsades de Pointes and ventricular fibrillation in a patient with congenital type 2 long QT syndrome. J Cardiovasc Electrophysiol. 2022 Jul;33(7):1592-1595. doi: 10.1111/jce.15517[↩]
- Barsheshet A., Dotsenko O., Goldenberg I. Congenital long QT syndromes: prevalence, pathophysiology and management. Paediatr Drugs. 2014;16:447–456. doi: 10.1007/s40272-014-0090-4[↩][↩][↩]
- Zipes D.P., Camm A.J., Borggrefe M., et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death) J Am Coll Cardiol. 2006;48:e247–e346. doi: 10.1016/j.jacc.2006.07.010[↩]
- Kaye AD, Volpi-Abadie J, Bensler JM, Kaye AM, Diaz JH. QT interval abnormalities: risk factors and perioperative management in long QT syndromes and Torsades de Pointes. J Anesth. 2013 Aug;27(4):575-87. doi: 10.1007/s00540-013-1564-1[↩]
- Shah M, Carter C. Long QT syndrome: A therapeutic challenge. Ann Pediatr Cardiol. 2008 Jan;1(1):18-26. doi: 10.4103/0974-2069.41051[↩]
- Alhourani N., Wolfes J., Konemann H., et al. Relevance of mexiletine in the era of evolving antiarrhythmic therapy of ventricular arrhythmias. Clin Res Cardiol. 2024;113:791–800. doi: 10.1007/s00392-024-02383-9[↩]
- Badri M., Patel A., Patel C., et al. Mexiletine prevents recurrent torsades de pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin Electrophysiol. 2015;1:315–322. doi: 10.1016/j.jacep.2015.05.008[↩]
- Huang H., Priori S.G., Napolitano C., O’Leary M.E., Chahine M. Y1767C, a novel SCN5A mutation, induces a persistent Na+ current and potentiates ranolazine inhibition of Nav1.5 channels. Am J Physiol Heart Circ Physiol. 2011;300:H288–H299. doi: 10.1152/ajpheart.00539.2010[↩]
- Cano J., Zorio E., Mazzanti A., et al. Ranolazine as an alternative therapy to flecainide for SCN5A V411M long QT syndrome type 3 patients. Front Pharmacol. 2020;11 doi: 10.3389/fphar.2020.580481[↩]
- Yu S., Li G., Huang C.L., Lei M., Wu L. Late sodium current associated cardiac electrophysiological and mechanical dysfunction. Pflugers Arch. 2018;470:461–469. doi: 10.1007/s00424-017-2079-7[↩]
- El-Sherif N., Turitto G., Boutjdir M. Acquired long QT syndrome and electrophysiology of torsade de pointes. Arrhythm Electrophysiol Rev. 2019;8:122–130. doi: 10.15420/aer.2019.8.3[↩]
- Hoshino K., Ogawa K., Hishitani T., Isobe T., Eto Y. Optimal administration dosage of magnesium sulfate for torsades de pointes in children with long QT syndrome. J Am Coll Nutr. 2004;23:497S–500S. doi: 10.1080/07315724.2004.10719388[↩]
- Hoshino K., Ogawa K., Hishitani T., Isobe T., Etoh Y. Successful uses of magnesium sulfate for torsades de pointes in children with long QT syndrome. Pediatr Int. 2006;48:112–117. doi: 10.1111/j.1442-200X.2006.02177.x[↩]
- Etheridge S.P., Compton S.J., Tristani-Firouzi M., Mason J.W. A new oral therapy for long QT syndrome: long-term oral potassium improves repolarization in patients with HERG mutations. J Am Coll Cardiol. 2003;42:1777–1782. doi: 10.1016/j.jacc.2003.07.006[↩]
- Behere S.P., Shubkin C.D., Weindling S.N. Recent advances in the understanding and management of long QT syndrome. Curr Opin Pediatr. 2014;26:727–733. doi: 10.1097/MOP.0000000000000161[↩]
- Choi G., Kopplin L.J., Tester D.J., Will M.L., Haglund C.M., Ackerman M.J. Spectrum and frequency of cardiac channel defects in swimming-triggered arrhythmia syndromes. Circulation. 2004;110:2119–2124. doi: 10.1161/01.CIR.0000144471.98080.CA[↩]
- Furst M.L., Aziz P.F. The evolution of sports participation guidelines and the influence of genotype-phenotype correlation in long QT syndrome. Trends Cardiovasc Med. 2016;26:690–697. doi: 10.1016/j.tcm.2016.04.013[↩]
- El-Sherif N., Turitto G. Electrolyte disorders and arrhythmogenesis. Cardiol J. 2011;18:233–245.[↩]
- Hintsa T., Puttonen S., Toivonen L., Kontula K., Swan H., Keltikangas-Jarvinen L. A history of stressful life events, prolonged mental stress and arrhythmic events in inherited long QT syndrome. Heart. 2010;96:1281–1286. doi: 10.1136/hrt.2009.190868[↩]
- Hintsa T., Maattanen I., Hintsanen M., et al. Work stress and the long QT syndrome: high job strain and effort-reward imbalance at work associated with arrhythmic risk in the long QT syndrome. J Occup Environ Med. 2013;55:1387–1393. doi: 10.1097/JOM.0000000000000026[↩]
- Ingles J., Yeates L., Hunt L., et al. Health status of cardiac genetic disease patients and their at-risk relatives. Int J Cardiol. 2013;165:448–453. doi: 10.1016/j.ijcard.2011.08.083[↩]


{kind=link}