
MECP2 duplication syndrome
MECP2 duplication syndrome also called Xq28 duplication syndrome, trisomy Xq28 or Lubs X-linked mental retardation syndrome, is a ultra-rare genetic neurodevelopmental disorder that occurs almost exclusively in males and is characterized by moderate to severe intellectual disability, early-onset low muscle tone (hypotonia), developmental delays, recurrent respiratory infections, gastrointestinal manifestations including gastroesophageal reflux and constipation, poor speech development, seizures, and progressive spasticity 1. Most people with MECP2 duplication syndrome also have weak muscle tone in infancy, feeding difficulties, poor or absent speech, or muscle stiffness (rigidity). Individuals with MECP2 duplication syndrome have delayed development of motor skills such as sitting and walking. About half of individuals (~50%) have seizures, often of the tonic-clonic type. This type of seizure involves a loss of consciousness, muscle rigidity, and convulsions and may not respond to medication. Some affected individuals experience the loss of previously acquired skills (developmental regression). Approximately half of individuals learn to walk, and about one-third of people with MECP2 duplication syndrome require assistance when walking. Many individuals (in ~75% of affected individuals) with MECP2 duplication syndrome have recurrent respiratory tract infections. These respiratory infections are a major cause of death in affected individuals, with only half surviving past age 25 2.
MECP2 duplication syndrome is caused by the duplication of genetic material on a specific region on the X chromosome (Xq28). This region includes the MECP2 gene and typically several adjacent genes. In most affected individuals, the MECP2 duplication is inherited in an X-linked manner; in rare cases, the duplication may occur randomly for no apparent reason (de novo duplication). MECP2 duplication syndrome is 100% penetrant in males, but females who carry the duplication on one X chromosome (heterozygotes) may exhibit some signs of the disorder. Rarely, females can develop a severe form of the disorder similar to males. Occasionally females have been described with a MECP2 duplication and a range of findings from mild intellectual disability to a phenotype similar to that seen in males. In addition to the core features, autistic behaviors, nonspecific neuroradiologic findings on brain MRI, mottled skin, and urogenital anomalies have been observed in several affected boys.
The prevalence of MECP2 duplication syndrome is unknown; more than 200 affected individuals have been described in the scientific literature. It is estimated that MECP2 duplication syndrome is responsible for 1 to 2 percent of all cases of intellectual disability caused by changes in the X chromosome 2.
Treatment is individualized and based on the signs and symptoms in each person. Because MECP2 duplication syndrome affects many different systems of the body, medical management is often provided by a team of doctors and other healthcare professionals. Treatment may involve routine management of feeding difficulties, infections, developmental delays, spasticity, and seizures 1. For example, babies who have trouble swallowing and/or feeding difficulties may require a feeding tube. Early developmental interventions may be recommended to help affected children reach their potential. This may include physical therapy, speech therapy and/or occupational therapy. Medications may be prescribed to treat seizures or spasticity. Recurrent infections must be treated with appropriate antibiotics.
Please speak with a doctor if you have any questions about your personal medical management plan or that of a family member.
Can a person with MECP2 duplication syndrome have microcephaly?
Yes. Around 40% of people with MECP2 duplication syndrome have microcephaly 3.
MECP2 duplication syndrome causes
MECP2 duplication syndrome is caused by a genetic change in which there is an an extra copy (duplication) of the methyl-CpG-binding protein 2 (MECP2) gene in each cell. This extra copy of the MECP2 gene is caused by a duplication of genetic material on the long (q) arm of the X chromosome. The size of the duplication varies from 100,000 to a few million DNA building blocks (base pairs). The MECP2 gene is always included in this duplication, and other genes may also be involved, depending on the size of the duplicated segment. It is unclear whether extra copies of these other genes affect the severity of the condition. More research is necessary to determine to what extent additional genes influence the development of MECP2 duplication syndrome.
The MECP2 gene provides instructions for making a protein called MeCP2 that is critical for normal brain function and most likely has several different functions in the body. Researchers believe that this protein has several functions, including regulating other genes in the brain by controlling when they are able to participate in protein production. An extra copy of the MECP2 gene leads to the production of excess MeCP2 protein and an increase in protein function. The resulting changes in gene regulation and protein production in the brain lead to abnormal nerve cell (neuronal) function. These neuronal changes disrupt normal brain activity, causing the signs and symptoms of MECP2 duplication syndrome.
Researchers believe that the more excess protein produced, the worse the associated symptoms. Rarely, affected males have MECP2 triplication, in which there are two extra copies of the MECP2 gene for a total of three. In these cases, MeCP2 overexpression is greater and the associated symptoms have been more severe.
Researchers have recently discovered that MeCP2 overexpression can impair the function of the immune system. The immune system is divided into several components, the combined actions of which are responsible for defending against different infectious agents (i.e. invading microscopic life-forms [microorganisms]). One specific part of the immune system affected in this disorder is the T cell system, which helps to fight several viruses, some bacteria and yeast and fungi. Researchers believe that MeCP2 overexpression suppresses the secretion of a protein known as interferon-gamma from certain T cells, as well as lower levels of immunoglobulins IgA, IgG and IgM, leading to a partially immunodeficient state. Low tone, gastrointestinal reflux and swallowing dysfunction may additionally predispose to recurrent respiratory tract infections and pneumonias. More research is necessary to determine the specific pathogens that affect individuals with MECP2 duplication syndrome and how to best treat the abnormal immune system.
MECP2 duplication syndrome inheritance pattern
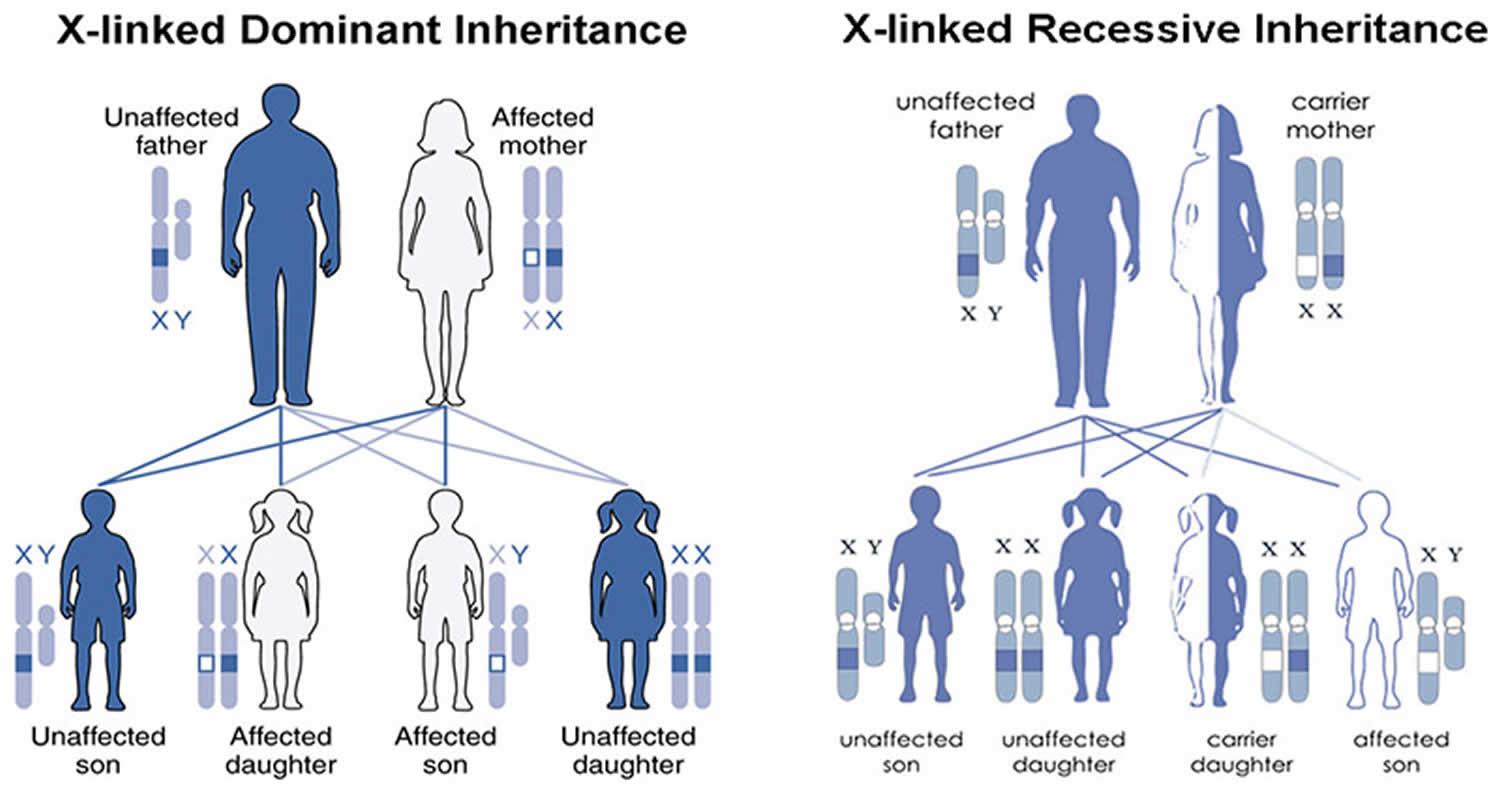
MECP2 duplication syndrome is inherited in an X-linked pattern. The MECP2 gene associated with MECP2 duplication syndrome is located on the X chromosome, which is one of the two sex chromosomes in each cell. In males (who have only one X chromosome), a duplication of the only copy of the MECP2 gene in each cell is sufficient to cause the condition. In females (who have two X chromosomes), a duplication of one of the two copies of the gene typically does not cause the disorder, but can be associated with behavioral and psychiatric symptoms such as depression, anxiety, and features of autism spectrum disorder that affect communication and social interaction.
In most cases, MECP2 duplication syndrome is inherited from a mother who carries the duplication but has no symptoms. Rarely, the condition is not inherited. In these cases it may occur randomly during the formation of the egg or sperm, or shortly after the egg and sperm join together. When this happens, it is called a de novo duplication 4. The duplication can also arise from an unbalanced translocation involving the X chromosome (and the MECP2 gene) 1.
Females with a MECP2 gene duplication tend to be unaffected or less severely affected than males because the X chromosome that contains the duplication may be turned off (inactive) in many of their cells due to a process called X-inactivation. Early in embryonic development in females, one of the two X chromosomes is permanently inactivated in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell. Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body’s cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
Females with a MECP2 gene duplication often have skewed X-inactivation, which results in the inactivation of the X chromosome containing the duplication in most cells of the body. Although this skewed X-inactivation ensures that the chromosome with the normal MECP2 gene is active most often, some of these females develop behavioral and psychiatric symptoms thought to be related to the additional genetic material. It is unclear why these features develop in a small number of females with skewed X-inactivation. Researchers speculate that in these females some cells in the brain may have a different pattern of X-inactivation than the cells in the rest of the body so that the X chromosome with the duplicated MECP2 gene is active, resulting in behavioral and psychiatric symptoms.
Figure 1. MECP2 duplication syndrome X-linked inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
MECP2 duplication syndrome signs and symptoms
MECP2 duplication syndrome is characterized by a wide variety of symptoms. Although researchers have been able to establish a clear syndrome with characteristic or “core” symptoms, much about the disorder is not fully understood. Several factors including the small number of identified patients, the lack of large clinical studies, and the possibility of other genes influencing the disorder hamper physicians from developing a complete picture of associated symptoms and prognosis. Therefore, it is important to note that affected individuals may not have all of the symptoms discussed below. Parents should talk to their children’s physician and medical team about their specific case, associated symptoms and overall prognosis.
Signs and symptoms of MECP2 duplication syndrome may include 1:
- Hypotonia (low muscle tone), which is usually apparent in infancy.
- Delayed development of milestones.
- Moderate to severe intellectual disability.
- Inability to talk, or limited speech ability that may be lost with age.
- Needing assistance to walk or inability to walk.
- Progressive spasticity during childhood, which is generally worse in the legs. This may lead to the development of mild contractures.
- Recurrent respiratory infections (in about 75% of people). Respiratory infections can be life-threatening and are a major cause of death.
- Seizures (in about 50%).
- Feeding difficulties which may require a feeding tube.
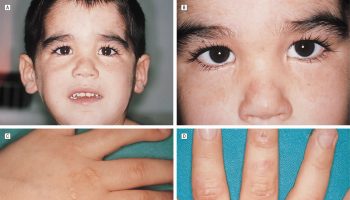
- Distinctive head or facial features, such as brachycephaly (abnormally flat back of the head), midface hypoplasia (underdevelopment of the middle of the face), large ears, deep-set eyes, prominent chin, and a depressed nasal bridge.
Other signs and symptoms may include trouble swallowing, gastroesophageal reflux, failure to thrive, excessive drooling, autistic features, and bowel or bladder problems 4.
One of the initial signs of MECP2 duplication syndrome may be feeding difficulties during the first few weeks of life likely resulting from diminished muscle tone (hypotonia). Affected children may also experience difficulty swallowing, gastroesophageal reflux and excessive drooling. Affected children will often fail to gain weight or grow at the expected rate for age and gender (failure to thrive) and may be at risk of aspiration, however, some infants experience no recognized problems in the newborn (neonatal) period and concern is not raised until other developmental milestones are missed.
Children with MECP2 duplication have moderate to severe intellectual disability and experience delays in attaining developmental milestones including sitting and crawling. Consequently, walking is also delayed and, in some cases, individuals develop an unsteady, uncoordinated gait. This abnormal gait can result in the development of exaggerated inward curvature of the lower spine (lumbar hyperlordosis). As affected individuals grow older, about half may experience neurological regression that ultimately results in the loss of previously acquired skills such as the ability to walk. In many cases the onset of regression is linked to onset of epilepsy.
The majority of affected individuals do not develop the ability to talk. Some individuals may be able to speak a few words during early childhood or have a limited use of speech, but frequently this ability is progressively lost during adolescence. Most affected individuals have better receptive language skills (i.e. understanding what is said to them). Rarely, some boys do retain the use of speech into adulthood.
In many affected children, hypotonia may progress to spasticity, a condition that is generally defined in the medical literature as an abnormal increase in muscle tone or stiffness of affected muscles. Spasticity can be associated with muscle spasms, increased deep tendon reflexes, and fixed joints (contractures). Spasticity affects the legs more severely and, over time, mild to severe contractures of the hips, knees and ankles may develop. A contracture is a condition in which a joint becomes permanently fixed in a bent (flexed) or straightened (extended) position, completely or partially restricting the movement of the affected joint.
Up to half of individuals develop recurrent seizures (epilepsy) in childhood or adolescence. However, the true prevalence of seizures by adulthood may be higher. In some individuals, seizures may not respond to treatment (refractory) and the onset and severity of seizures may correlate with neurological deterioration (e.g. loss of speech, hand use, and ambulation). Seizure types that have been reported in individuals with MECP2 duplication syndrome include head/neck and trunk drop attacks, absence seizures, myoclonic seizures, and generalized or secondarily generalized tonic-clonic or simply tonic seizures (once known as grand mal seizures).
Some individuals with MECP2 duplication syndrome experience dysfunction of the immune system, which causes them to be prone to recurrent infections such as respiratory tract infections. Affected individuals may develop recurrent pneumonia that is sometimes severe requiring mechanical ventilation. Middle ear infections (otitis media) and sinusitis are also common. Additional infections have been reported including meningitis or urinary tract infections. Recurrent infections can cause life-threatening complications, and are a major contributing factor for reduced life-expectancy.
Many individuals with MECP2 duplication syndrome meet formal criteria for diagnosis with an autism spectrum disorder due to poor expressive language skills, abnormal social affect, and restricted/repetitive behaviors. Mood disorders such as anxiety sometimes occur.
Affected individuals often have clinically significant constipation. Bladder dysfunction has also been seen. According to some reports, affected individuals may have chronic intestinal pseudo-obstruction (CIP), a condition characterized by abnormalities affecting the involuntary, coordinated muscular contractions (peristalsis) that propels food and other material through the digestive system. Symptoms may develop due to obstruction of the small bowel and can include nausea, vomiting, abdominal pain, abdominal swelling and constipation. Ultimately, normal nutritional requirements cannot be met leading to unintended weight loss and malnourishment. The acute onset of GI symptoms such as abdominal pain, unusual distension, or vomiting warrants urgent medical evaluation as it can precede life-threatening complications.
Individuals with MECP2 duplication may have distinctive facial features including an abnormally flat back of the head (brachycephaly), underdevelopment of the middle of the face (midface hypoplasia), ear anomalies, large ears, deep-set eyes, prominent chin, pointed nose, and an abnormally flat bridge of the nose.
Additional findings have been reported in some children with MECP2 duplication syndrome including some degree of growth retardation. Early in life, head size appears to be normal, but as the boys get older, some might develop abnormally small head circumference (microcephaly). However, large head circumference (macrocephaly) has also been observed, and overall these are not consistent features of the syndrome.
Some boys have underdeveloped (hypoplastic) genitalia. Affected males may experience failure of the testes to descend (cryptorchidism) and have the urinary opening located on the underside of the penis (hypospadias) rather than at the end.
Affected females
Although MECP2 duplication syndrome is primarily associated with males, females can develop mild symptoms of the disorder or, rarely, a severe form of the disorder. Most females with a MECP2 duplication generally do not have symptoms, although depression, anxiety, and autistic features have been described in some women with the duplication 1. Very rarely, females have severe signs and symptoms, similar to those in males with the syndrome 1.
Some researchers have noted that women who have a MECP2 duplication on one of their X chromosomes (heterozygote or carrier females) may be prone to developing neuropsychiatric features including depression, anxiety, and specific personality traits. In addition, recent reports have identified women (i.e., women with unfavorable X-linked inactivation) who developed some symptoms of the disorder including recurrent infections, poor speech development, or seizures.Rarely, females may have a genetic abnormality, known as a translocation, which involves the X chromosome and a non-sex chromosome (autosome). Females with this specific underlying genetic abnormality may develop a severe form of the disorder similar to that seen in males. For more information, on X-linked inactivation and translocations,
MECP2 duplication syndrome diagnosis
A diagnosis of MECP2 duplication syndrome is based upon the identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and a variety of specialized tests. It should be included in the differential diagnosis of male infants with hypotonia.
Clinical testing and workup
There are a variety of tests that can be used to diagnose MECP2 duplication syndrome including array comparative genomic hybridization (array-CGH). This test can detect the gain or loss of chromosomal material including microduplications. In rare instances, when a current array-CGH does not detect a change in genetic material, whole exome or even whole genome sequencing can be utilized, a sophisticated technique that determines the complete DNA sequence (or, in the case of whole exome, the sequence of the expressed part of the DNA) of an individual.
Additional tests that can confirm CGH results or for direct clinical testing include polymerase chain reaction (PCR), fluorescent is situ hybridization (FISH) analysis, chromosome microarray SNP analysis, and multiplex ligation-dependent probe amplification (MLPA).
PCR is a laboratory technique that has been described as “photocopying”. It enables researchers to enlarge and repeatedly copy sequences of DNA. As a result, they are able to closely analyze DNA and more easily identify genes and genetic changes.
A fluorescent is situ hybridization (FISH) test can be used to determine a person’s karyotype. A karyotype is a visual representation of a person’s chromosomal makeup (i.e. the 46 chromosomes in a cell). The FISH test can detect chromosomal abnormalities such as duplications or translocation.
Chromosome microarray SNP analysis uses probes that can detect chromosomal abnormalities including microduplications, including those that are the underlying cause of many cases of MECP2 duplication syndrome.
The MLPA test is a relatively new method for assessing chromosomes and can detect certain chromosomal abnormalities including those associated with MECP2 duplication syndrome. Pre-implantation genetic diagnosis (PGD) may be an option when a parent has a known genetic abnormality (i.e. carrier mother). Pre-implantation genetic diagnosis can be performed on embryos created through in vitro fertilization (IVF). Pre-implantation genetic diagnosis refers to testing an embryo to determine whether it has the same genetic abnormality as the parent. Families interested such an option should seek the counsel of a certified genetics professional.
MECP2 duplication syndrome treatment
The treatment of MECP2 duplication syndrome is directed toward the specific symptoms that are apparent in each individual. Treatment may require the coordinated efforts of a team of specialists. Pediatricians, surgeons, pediatric neurologists, gastroenterologists, psychiatrists, speech pathologists, and other healthcare professionals may need to systematically and comprehensively plan an affected child’s treatment. Genetic counseling may be of benefit for affected individuals and their families.
Treatment options that may be used to treat individuals with MECP2 duplication syndrome are complex and varied. The specific treatment plan will need to be highly individualized. Decisions concerning the use of specific treatments should be made by physicians and other members of the health care team in careful consultation with an affected child’s parents or with an adult patient based upon the specifics of his or her case; a thorough discussion of the potential benefits and risks, including possible side effects and long-term effects; patient preference; and other appropriate factors.
Early developmental intervention is important to ensure that affected children reach their potential. Most affected children will benefit from occupational, physical and speech therapy. Various methods of rehabilitative and behavioral therapy may be beneficial. Inclusion of novel augmentative communication devices, such as eye-tracking technology based communication devices, in the therapy regimen are encouraged. It is essential that therapies are continued on a year-round basis to promote development of new skills and to prevent regression. Some children who lose skills are able to re-learn them with intensive therapy. Additional medical, social and/or vocational services including special remedial education may be necessary. Psychosocial support for the entire family is essential as well.
Other treatment is symptomatic and supportive. Additional therapies for MECP2 duplication syndrome depend upon the specific abnormalities present and generally follow standard guidelines.
Some general therapies common for infants or children include monitoring feeding and swallowing difficulties. Insertion of a feeding tube through a surgical opening in the stomach (gastrostomy) may be necessary to ensure proper nutritional support and to prevent aspiration.
Drugs may be used to treat a variety of symptoms associated with MECP2 duplication syndrome. Anti-seizure medications (anti-convulsants) are usually effective in treating seizures; however, in a significant subset of cases these medications do not completely control seizures (refractory seizures) and complementary approaches such as some implementation of the ketogenic diet, or implantation of a vagus nerve stimulator may be sought. Drugs may be used to improve spasticity and muscle rigidity, but can also be ineffective or even potentially worsen the condition. Working with a physical medicine and rehabilitation specialist to treat spasticity is recommended. Prompt treatment of spasticity may prevent the development of contractures.
Because of the susceptibility to infections, when affected individuals develop an infection, they should be treated immediately and aggressively with appropriate antibiotics. Recurrent infections, especially respiratory infections, can be severe enough to necessitate hospitalization and require assisted (mechanical) ventilation. Individuals who develop recurrent pneumonia should be evaluated by an immunology specialist since medical interventions including the use of prophylactic antibiotics and/or immunoglobulin therapy are indicated in some cases.
MECP2 duplication syndrome prognosis
The long-term outlook (prognosis) for people with MECP2 duplication syndrome varies. Based on the few documented cases in the medical literature, approximately half of affected people succumb before age 25 years. This shortened life expectancy is largely due to immune system dysfunction and an increased risk for recurrent infections 1.
- Van Esch H. MECP2 Duplication Syndrome. 2008 Jan 18 [Updated 2020 May 21]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1284[↩][↩][↩][↩][↩][↩][↩]
- MECP2 duplication syndrome. https://ghr.nlm.nih.gov/condition/mecp2-duplication-syndrome[↩][↩]
- LUBS X-LINKED MENTAL RETARDATION SYNDROME; MRXSL https://www.omim.org/entry/300260[↩]
- MECP2 Duplication Syndrome. https://rarediseases.org/rare-diseases/mecp2-duplication-syndrome[↩][↩]




{kind=link}