
Ohtahara syndrome
Ohtahara syndrome also called early infantile epileptic encephalopathy or type 4 of Early Infantile Epileptic Encephalopathy with suppression bursts, is an uncommon type of epilepsy characterized by hard to control seizures and developmental delays 1. Ohtahara syndrome affects infants, usually within the first three months of life (most often within the first 10 days) in the form of epileptic seizures 2, 3. Infants have primarily tonic seizures (stiffening of the muscles, upward eye gaze, dilated pupils, and altered breathing), but may also experience focal seizures (involving only one area or side of the brain), and rarely, myoclonic seizures (involving sudden muscle jerks) 4. Ohtahara syndrome can affect both boys and girls. Ohtahara syndrome is classically caused by very abnormal brain structure that may be due to damage or abnormal development. It also can be due to metabolic disorders or genetic epilepsy syndromes, although the cause or causes for many cases can’t be determined. Recent studies suggest that there is often an identifiable genetic cause of Ohtahara syndrome. Electroencephalography recordings of brain activity of infants with Ohtahara syndrome reveal a characteristic pattern of high voltage abnormal brain activity alternating with periods of very little activity. This EEG pattern is known as “burst suppression” (see Figure 1 below). Ohtahara syndrome has a characteristic burst suppression pattern on an EEG (burst activity followed by very little brain activity) 5.
Seizures that develop due to Ohtahara syndrome are very difficult to control even with combination of anticonvulsive drugs 6. Individuals with Ohtahara syndrome also suffer from slow mental development resulting in severe intellectual disability and requiring constant care.
Antiseizure drugs are used to control seizures, but are unfortunately not usually very effective for this disorder. Corticosteroids (prednisolone or ACTH) are occasionally helpful and sometimes the ketogenic diet (high fat, low carbohydrate) is appropriate. In cases where there is a focal brain lesion (damage or abnormal development of one area/side of the brain), epilepsy surgery may be beneficial. Other therapies are symptomatic and supportive; these can include tratments for abnormal muscle tone, irritability, or stomach or lung problems. Physical, speech, and occupational therapies are usually offered. Consultation with a palliative care team is often helpful in order to define goals of care and ensure the infant’s family and medical team are working toward the same set of goals.
Figure 1. Suppression-burst pattern on EEG
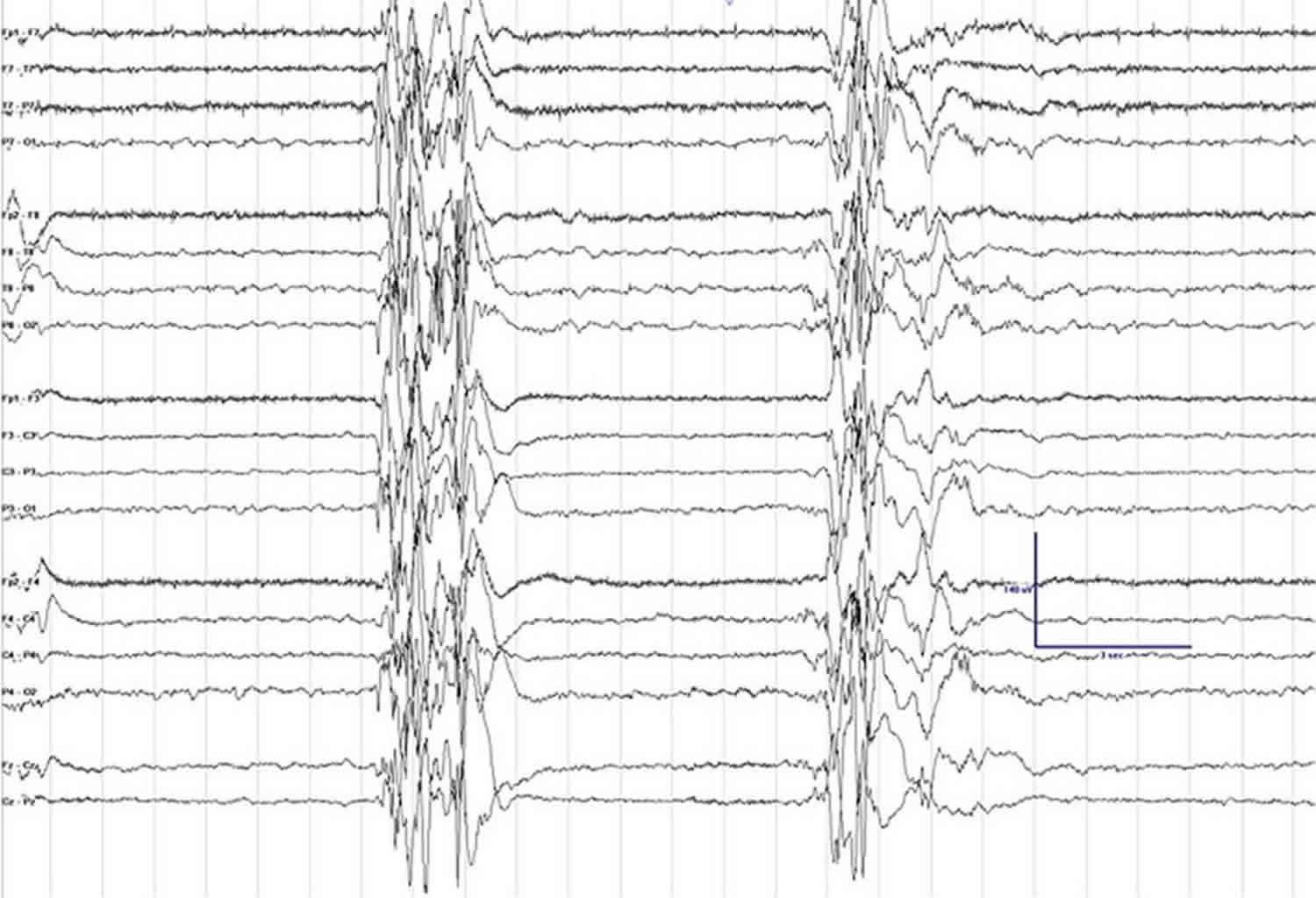
Footnote: Electroencephalogram (EEG) clip showing suppression-burst background. Sensitivity is set at 7 μV, low frequency filter at 1 Hz, high frequency filter at 70 Hz, and notch filter is turned on.
[Source 7 ]Ohtahara syndrome causes
Most cases of Ohtahara syndrome are due to brain malformation or certain gene mutations 8. Metabolic causes (e.g., mitochondrial disorders, non-ketotic hyperglyinemia, pyridoxine/pyridoxal-5-phosphate disorders, carnitine palmitoyl transferase deficiency and others) are less likely. In some cases, no clear cause is found.
Brain malformations may be diffuse (affecting both sides of the brain) or focal (affecting only one area or side).
Ohtahara syndrome genetics
Gene mutations refer to changes in a gene or group of genes that may affect how the gene affects brain development or function. Some genes that have been associated with Ohtahara syndrome include 9:
- ARX
- CDKL5
- SLC25A22
- STXBP1
- SPTAN1
- KCNQ2
- ARHGEF9
- PCDH19
- PNKP
- SCN2A
- PLCB1
- SCN8A
- ST3GAL3
- TBC1D24
- BRAT1
Ohtahara syndrome have mutations most frequently in the gene encoding syntaxin-binding protein 1 (STXBP1) also known as munc18-1 10. The exact molecular mechanism of how these munc18-1 mutations cause impaired cognition, remains elusive. The leading haploinsufficiency hypothesis posits that mutations in munc18-1 render the protein unstable leading to its degradation. Expression driven by the healthy allele is not sufficient to maintain the physiological function resulting in haploinsufficiency.
Proteins from the Sec1/Munc18 and SNARE (soluble NSF attachment protein receptor) families drive membrane fusion, a conserved mechanism that is important in intracellular membrane trafficking and exocytosis. In neurons action potentials induce neurotransmission by exocytosis of synaptic vesicles. Transmitter release involves a series of steps that include vesicle docking to the plasma membrane, priming to a release ready state, and calcium-triggered membrane fusion. The calcium signal-triggered neurotransmitter release provides the basic mechanism for communication among neurons, which is essential for Information processing in the brain. At neuronal synapses munc18-1 interacts with these three SNAREs to drive vesicular release of transmitters: syntaxin-1, SNAP-25, and synaptobrevin/vesicle associated membrane protein. The three SNAREs produce fusion by forming tight four-helix bundles called SNARE complexes that bring the synaptic vesicle and plasma membranes together enabling transmitter release. Munc18-1 was initially linked to synaptic vesicle fusion based on its tight binding to syntaxin-1 in the so-called closed conformation that blocks its participation in SNARE complex formation 11. Therefore, it was postulated that munc18 had an inhibitory role in exocytosis. Recent findings in models with munc18-1 deletion contradicted this view. In munc18-1 KO mice synaptic release is completely abrogated 12, just the opposite of what one would expect with disinhibition caused by eliminating an inhibitory factor. The expression level of munc18-1 is reduced to zero in the null mutant and to 50% in the heterozygous KO brain at E18 13.
Munc18-1 binding to the SNARE complex is essential for SNARE associated exocytosis from the presynaptic terminal (Synaptic assembly of the brain in the absence of neurotransmitter secretion. Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Science. 2000 Feb 4; 287(5454):864-9.)). Deletion of munc18-1 in animals is embryonic lethal due to the complete lack of neurotransmitter secretion 14. Individuals with Ohtahara syndrome were found to have deletions and mostly de novo heterozygous missense mutations along the gene that encodes Munc18-1 15. These mutations are suspected to lead to unstable products which are degraded or aggregated 16 leading to haploinsufficiency, where the remaining unaffected STXBP1 allele is unable to produce enough protein leading to aberrant neurophysiology 17. The mechanism by which these mutations cause Ohtahara syndrome is not clear but the current theories suggest impairments in neurotransmission may be the primary cause of Ohtahara syndrome. Looking at one of the major symptoms of Ohtahara syndrome, intellectual disability, it is possible that seizures associated with the disorder are destroying vital areas of the brain for learning and memory. Importantly, encephalopathy cases with cognitive impairment have been reported, which are caused by mutations in munc18-1 gene, however, some patients do not seize 18.
It is well established that synaptic plasticity is the basic process responsible for learning and memory in the brain 19 via modification of synaptic transmission and information processing in neuronal networks. The effects of reduced munc18-1 levels on learning and memory remain elusive.
Ohtahara syndrome symptoms
Ohtahara syndrome symptoms include:
- Seizures begin before age 3 months. Infants have primarily tonic seizures (stiffening of the muscles, upward eye gaze, dilated pupils, and altered breathing), but may also experience focal seizures (involving only one area or side of the brain), and rarely, myoclonic seizures (involving sudden muscle jerks).
- Babies typically show severe developmental challenges and abnormal neurological examination, even before seizures start.
- Motor and cognitive problems can get progressively worse as seizures increase.
Types of seizures are seen in Ohtahara syndrome
Many seizure types may occur, but tonic seizures (stiffening of the arms or legs) are seen most often.
- These seizures last only seconds and can occur alone or in clusters.
- They may affect one side of the body more prominantly.
- They are seen both when the baby is awake and sleeping.
- Other seizure types that may also occur in Ohtahara syndrome include focal (start in one area of the brain), atonic, myoclonic or generalized tonic-clonic seizures.
- Infants with Ohtahara syndrome may also develop infantile spasms.
Ohtahara syndrome diagnosis
Diagnosing a baby with Ohtahara syndrome is based on clinical features (what signs or symptoms the baby has) and EEG (electroencephalogram) findings.
The EEG is the most important test in making a diagnosis of Ohtahara syndrome.
The EEG is very abnormal with a burst suppression pattern (high amplitude spikes followed by little brain activity or flattening of the brain waves) 20. These changes can be seen during sleep and when the infant is awake.
An MRI (magnetic resonance imaging) scan is required to look for structural changes in the brain that could cause Ohtahara syndrome. If the MRI is initially normal, follow up MRIs can show atrophy (shrinkage) of the brain.
Genetic testing with chromosomal microarray followed by an epilepsy gene panel or whole exome sequencing should be done if no cause is found on MRI.
Blood and urine tests to evaluate for metabolic disorders should also be considered.
Ohtahara syndrome treatment
Anti-seizure medications are routinely used, but seizures with Ohtahara syndrome are usually drug resistant. Medications that are often tried include vigabatrin (Sabril), ACTH or prednisone, clobazam (Onfi), clonazepam (Klonopin), topiramate (Topamax), zonisamide (Zonegran), phenobarbital, valproate, or felbamate (Felbatol).
Epilepsy surgery may be an option in children with seizures starting in one area or involving one side of the brain. In these situations, a focal resection (removal of one area) or a hemispherectomy (removing most of one side of the brain) should be considered early. Successful surgery may be associated with improved developmental outcome.
Ketogenic diet (high fat, low carbohydrate, and restricted protein) may be considered if seizures are drug resistant and the child is not a candidate for epilepsy surgery 7.
When a child is in the early preschool years, a vagus nerve stimulator could be considered.
Correcting metabolic problems. Sometimes a metabolic disorder that affects how the brain works may lead to Ohtahara syndrome. While these currently can’t be reversed, sometimes treating the underlying disorder can help.
Ohtahara syndrome prognosis
The outlook for children with Ohtahara syndrome may be grim. The course of Ohtahara syndrome is severely progressive. Children who are candidates for epilepsy surgery should be considered for that procedure early, as this may lead to improved developmental outcome. If epilepsy surgery is not feasible, seizures become more frequent and are accompanied by delays in physical and cognitive development 4. Some children will die in infancy; others will survive but usually have severe handicaps. As they grow, some children will progress into other epilepsy syndromes such as infantile spasms (West Syndrome) and Lennox-Gastaut syndrome 4.
Ohtahara syndrome life expectancy
Some children with Ohtahara syndrome will die in infancy and others may die within the first 2 years of life 21. Those who survive are typically left with severe physical and cognitive disabilities 6.
- Beal JC, Cherian K, Moshe SL. Early-Onset Epileptic Encephalopathies: Ohtahara Syndrome and Early Myoclonic Encephalopathy. Pediatric Neurology. 2012;47(5):317–323. doi: http://dx.doi.org/10.1016/j.pediatrneurol.2012.06.002[↩]
- Saitsu H, Kato M, Matsumoto N. In: Haploinsufficiency of STXBP1 and Ohtahara syndrome. Jasper’s Basic Mechanisms of the Epilepsies. Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Bethesda (MD): National Center for Biotechnology Information (US); 2012.[↩]
- Ohtahara Syndrome. https://www.epilepsy.com/learn/types-epilepsy-syndromes/ohtahara-syndrome[↩]
- https://www.ninds.nih.gov/Disorders/All-Disorders/Ohtahara-Syndrome-Information-Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Ohtahara-Syndrome-Information-Page[↩][↩][↩]
- Diagnosis and management of epileptic encephalopathies in children. Jain P, Sharma S, Tripathi M. Epilepsy Res Treat. 2013; 2013():501981.[↩]
- Early-onset epileptic encephalopathies: Ohtahara syndrome and early myoclonic encephalopathy. Beal JC, Cherian K, Moshe SL. Pediatr Neurol. 2012 Nov; 47(5):317-23.[↩][↩]
- Sivaraju A, Nussbaum I, Cardoza CS, Mattson RH. Substantial and sustained seizure reduction with ketogenic diet in a patient with Ohtahara syndrome. Epilepsy Behav Case Rep. 2015;3:43–45. Published 2015 May 15. doi:10.1016/j.ebcr.2015.03.003 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4436508[↩][↩]
- Ohtahara syndrome associated with hemimegalencephaly and intracranial lipoma. Gowda VK, Bhat A, Bhat M, Ramaswamy P. J Pediatr Neurosci. 2015 Apr-Jun; 10(2):185-7.[↩]
- Ohtahara syndrome with emphasis on recent genetic discovery. Pavone P, Spalice A, Polizzi A, Parisi P, Ruggieri M. Brain Dev. 2012 Jun; 34(6):459-68.[↩]
- Orock A, Logan S, Deak F. Munc18-1 haploinsufficiency impairs learning and memory by reduced synaptic vesicular release in a model of Ohtahara syndrome. Mol Cell Neurosci. 2018;88:33–42. doi:10.1016/j.mcn.2017.12.002 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5893365[↩]
- Membrane fusion: grappling with SNARE and SM proteins. Südhof TC, Rothman JE. Science. 2009 Jan 23; 323(5913):474-7.[↩]
- Synaptic assembly of the brain in the absence of neurotransmitter secretion. Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Südhof TC. Science. 2000 Feb 4; 287(5454):864-9.[↩]
- Munc18-1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. Toonen RF, de Vries KJ, Zalm R, Südhof TC, Verhage M. J Neurochem. 2005 Jun; 93(6):1393-400.[↩]
- Munc18-1 binding to the neuronal SNARE complex controls synaptic vesicle priming. Deák F, Xu Y, Chang WP, Dulubova I, Khvotchev M, Liu X, Südhof TC, Rizo J. J Cell Biol. 2009 Mar 9; 184(5):751-64.[↩]
- De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, Uruno K, Kumada S, Nishiyama K, Nishimura A, Okada I, Yoshimura Y, Hirai S, Kumada T, Hayasaka K, Fukuda A, Ogata K, Matsumoto N. Nat Genet. 2008 Jun; 40(6):782-8.[↩]
- Munc18-1 is a molecular chaperone for α-synuclein, controlling its self-replicating aggregation. Chai YJ, Sierecki E, Tomatis VM, Gormal RS, Giles N, Morrow IC, Xia D, Götz J, Parton RG, Collins BM, Gambin Y, Meunier FA. J Cell Biol. 2016 Sep 12; 214(6):705-18.[↩]
- Munc18-1 haploinsufficiency results in enhanced anxiety-like behavior as determined by heart rate responses in mice. Hager T, Maroteaux G, Pont Pd, Julsing J, van Vliet R, Stiedl O. Behav Brain Res. 2014 Mar 1; 260():44-52.[↩]
- Epilepsy is not a mandatory feature of STXBP1 associated ataxia-tremor-retardation syndrome. Gburek-Augustat J, Beck-Woedl S, Tzschach A, Bauer P, Schoening M, Riess A. Eur J Paediatr Neurol. 2016 Jul; 20(4):661-5.[↩]
- Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Neves G, Cooke SF, Bliss TV. Nat Rev Neurosci. 2008 Jan; 9(1):65-75.[↩]
- Ohtahara S., Yamatogi Y. Epileptic encephalopathies in early infancy with suppression-burst. J Clin Neurophysiol. 2003;20:398–407.[↩]
- Age-dependent epileptic encephalopathy: a longitudinal study. Yamatogi Y, Ohtahara S. Folia Psychiatr Neurol Jpn. 1981; 35(3):321-32.[↩]


{kind=link}