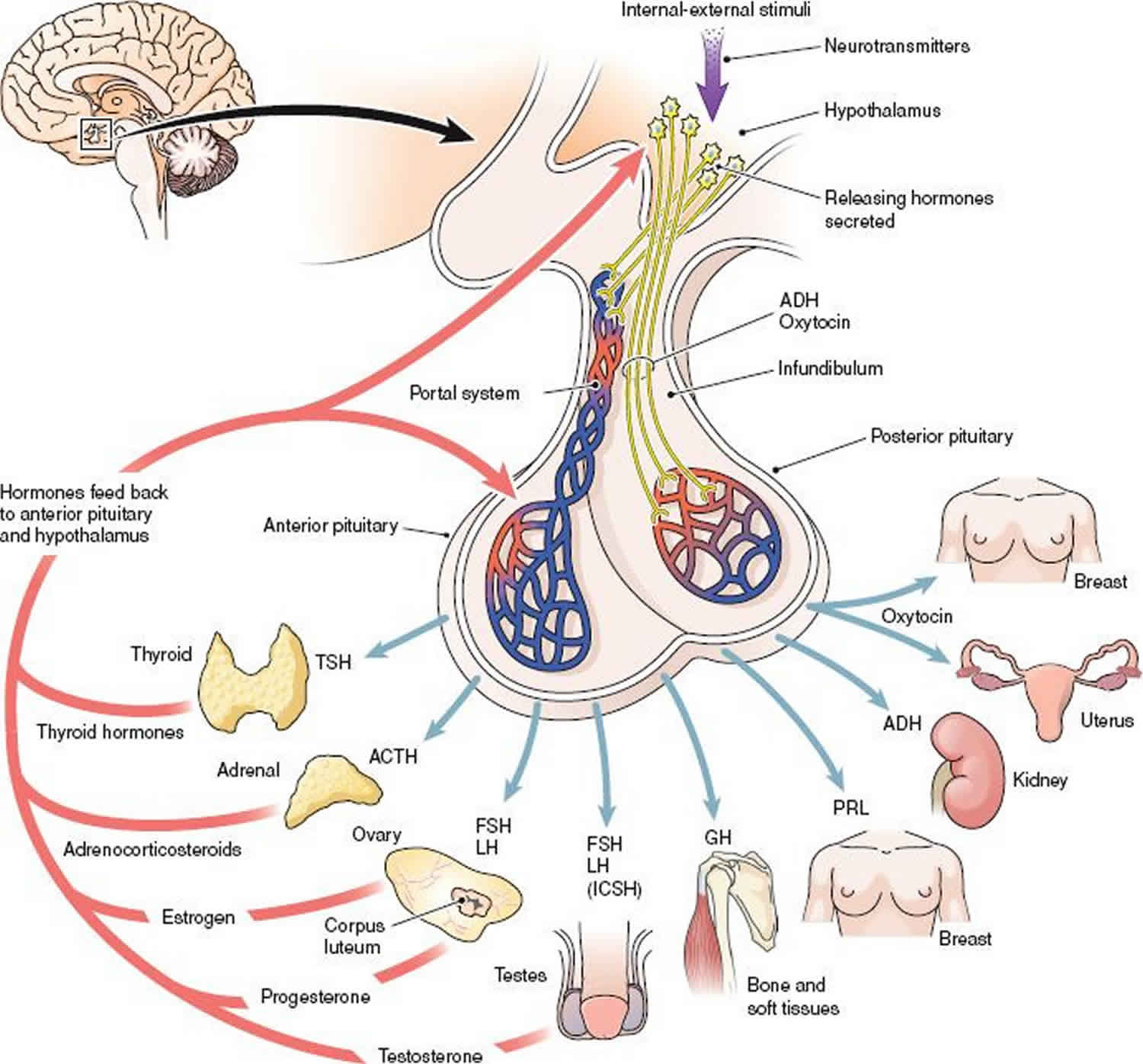
Panhypopituitarism
Panhypopituitarism also known as hypopituitarism, is defined as a deficiency of one or more of the hormones produced by the pituitary gland 1. The pituitary gland is called the master endocrine gland of the body because it controls the function of other endocrine organs. The pituitary gland consists of an anterior and a posterior lobe. The anterior pituitary produces the hormones thyrotropin (thyroid-stimulating hormone [TSH]), corticotropin (adrenocorticotropic hormone [ACTH]), luteinizing hormone (LH), follicle-stimulating hormone (FSH), growth hormone (GH), and prolactin (PRL). The anterior pituitary is controlled by specific hypothalamic-releasing hormones. The posterior pituitary produces vasopressin (antidiuretic hormone [ADH]) and oxytocin.
The presence of hypopituitarism is associated with increased mortality due to increased cardiovascular and respiratory diseases, and early diagnosis is important to prevent further morbidity due to the subtle presentation 2.
There is limited data available regarding the incidence and prevalence of panhypopituitarism. One study from Spain reported the annual incidence of anterior pituitary hormone deficiency to be 4.21 cases per 100,000 population.
Panhypopituitarism causes
The causes of panhypopituitarism can be attributed to either pathology of the hypothalamus affecting the production of trophic hormones that act on the pituitary or direct pathology of the pituitary gland itself. The most common cause of panhypopituitarism (61%) is the presence of pituitary tumors (both non-secretory and secretory). Pituitary tumors may cause the increased production of one hormone with resultant deficiency of the other pituitary hormones as in acromegaly (excess growth hormone with panhypopituitarism from the macroadenoma). Most pituitary tumors are benign and may be secretory or non-secretory. Secondary metastases originating from, for example, breast, colon and prostate cancers do occur less commonly. Hypothalamic and para-pituitary tumors such as suprasellar meningiomas, gliomas and craniopharyngiomas may also be associated with panhypopituitarism. Other causes of panhypopituitarism include injury to the pituitary gland following traumatic brain injury or iatrogenically during surgery or cranial irradiation.
Inflammatory conditions of the pituitary may also be responsible for the occurrence of panhypopituitarism. The infectious agents that can have been related to pituitary insufficiency include Mycobacterium tuberculosis and non-mycobacterial agents such as histoplasmosis, syphilis, viruses, and protozoa. Lymphocytic hypophysitis usually presents in the post-partum period as a mass lesion on magnetic resonance imaging (MRI) due to infiltration of the pituitary with lymphocytes and plasma cells and is responsive to steroid therapy.
Infiltrative diseases such as hemochromatosis, sarcoidosis, and histiocytosis may be associated with the development of panhypopituitarism.
Pituitary apoplexy is a medical emergency and is due to acute ischaemic infarction or hemorrhage of the pituitary gland. Pituitary apoplexy may occur in the presence of a pituitary adenoma but may also occur in the normal pituitary gland. Sheehan syndrome refers to infarction of the hyperplastic pituitary gland during pregnancy due to severe blood loss (post-partum hemorrhage). Because of the rich and complex vascular supply pituitary adenomas have an increased risk of bleeding when compared to other brain tumors.
Congenital absence of the pituitary gland is related to midline and craniofacial defects. Genetic mutations in transcription factors such as HESX1, PROP1, and Pit-1 can lead to congenital panhypopituitarism. Empty Sella syndrome is a rare disorder which is characterized by enlargement or malformation of the sella turcica resulting in a herniation of the arachnoid membrane into the pituitary fossa dislodging the pituitary to the floor of the fossa. It is associated with a small or absent pituitary gland. Empty Sella syndrome may be idiopathic or occur secondarily to a treated pituitary tumor, head trauma, or a condition known as idiopathic intracranial hypertension (pseudotumor cerebri). Kallmann syndrome is a rare genetic condition associated with an inability to smell (hyposmia/anosmia) and hypogonadotropic hypogonadism (decreased FSH decreased LH, and decreased testosterone/estradiol) due to a mutation in the Kal1 gene as the commonest genetic abnormality in males.
Panhypopituitarism symptoms
It appears that that 75% of the pituitary needs to be damaged to result in panhypopituitarism. Clinical features of panhypopituitarism are diverse and may be subtle and ill-defined or severe with the acute presentation. Conditions such as Sheehan’s syndrome/pituitary apoplexy, pituitary infection, hypophysitis and traumatic brain injury present with acute findings 3.
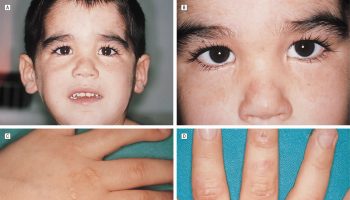
Presenting signs and symptoms may be linked to that of a deficiency of pituitary hormone, mass effects in the presence of pituitary tumors, and/or features of the causative disease. Mass effects include visual field defects known as bitemporal hemianopsia. Visual field defects may also occur unilaterally. Patients may also present with headaches, secondary to the mass lesions. Manifestations that suggest congenital anterior hypopituitarism include micropenis, midline defects, optic atrophy, hypoglycemia, and poor growth 1. The presence of a secretory pituitary tumor may result in features of hormone excess for the particular hormone produced by a tumor while other pituitary hormones may be deficient 4.
Figure 1. Panhypopituitarism symptoms
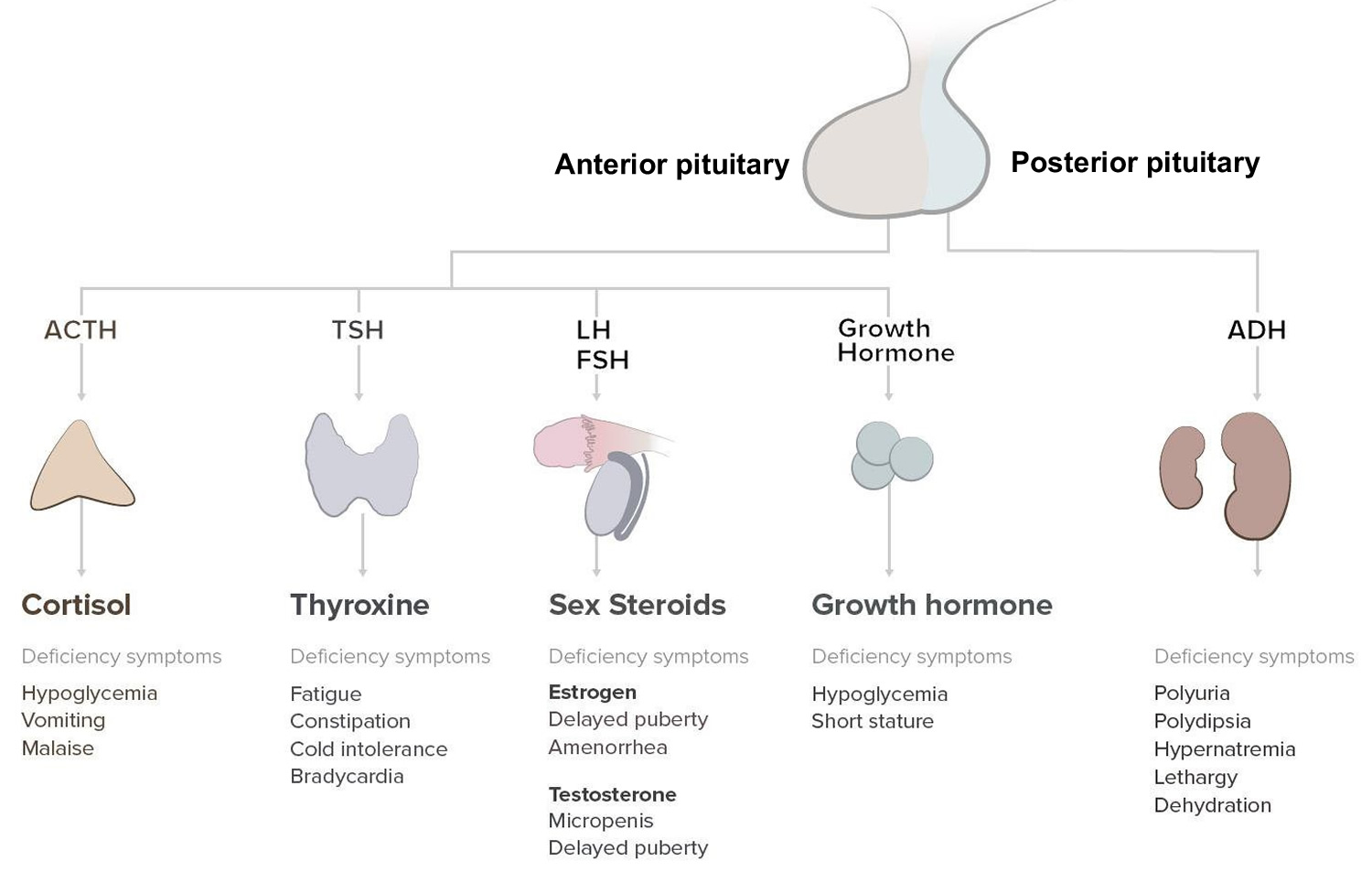
Panhypopituitarism diagnosis
Laboratory investigations
Initial testing involves baseline levels of pituitary hormones and hormones produced by target hormones. Due to the variation of hormone levels related to the time of day, season and pulsatile secretion of certain pituitary hormones, baseline levels may not be helpful. In this instance, dynamic function testing may be performed to confirm biochemical deficiency or excess of a particular pituitary hormone. In dynamic function testing for the investigation of a hormone deficiency a stimulatory agent that would normally increase secretion of the hormone is given to the patient and blood levels are measured before administration of the agent and at defined time points after that to determine if there has been an adequate response to stimulation.
Insulin Tolerance Test
This is the best provocative test that is used to assess the presence of the deficiency of both growth hormone and cortisol. Following an overnight fast, baseline samples are obtained for the following hormones cortisol, growth hormone, and glucose. An insulin dose of 0.1 U/kg or 0.05 U/kg is administered intravenously (IV). Further samples for analysis of the hormones measured in the baseline samples are then taken at several other time points after administration. Should not be performed in those with cardiac disease or epilepsy. The plasma glucose should fall to 40 mg /dl within 30 to 45 minutes or by 50% of baseline. The test is terminated by giving IV dextrose and assessing the patient’s status for at least 90 minutes. A normal/adequate response is indicated by a cortisol of more than 20 ug/dL and growth hormone more than 5 ng/mL to 10 ng/mL.
Modern combined test
Patient is given GHRH, CRH, GnRH, and TRH as the provocative stimuli and growth hormone, TSH, ACTH, Cortisol, LH, and FSH are measured at baseline and at specified time intervals after that. Doses of each stimulating hormone are as follows GHRH (1.0 ug/kg); CRH (1.0 ug/Kg); GnRH (100 ug) and TRH (200 ug). However, this testing is rarely required.
Radiological investigations
Imaging studies of the pituitary using magnetic resonance imaging (MRI) with gadolinium enhancement be used to visualize the pituitary, in particular, to detect the presence of a mass lesion. Visual field defects need to be assessed if a pituitary mass is the cause of the hypopituitarism.
Panhypopituitarism treatment
Management is dependent on the cause of panhypopituitarism. Initial treatment is to address the underlying cause of panhypopituitarism. Mass lesions may be removed surgically and other medical conditions treated accordingly. Many patients may require hormone replacement therapy.
Adrenocorticotropic hormone (ACTH) deficiency: Corticosteroid replacement should be initiated before replacement of thyroid hormone to avoid precipitating an adrenal crisis. Hydrocortisone at a dose of 10 mg to 20 mg in the morning and 5 mg to 10 mg in the evening. Prednisone may be considered advantageous because of twice-daily dosing (at about 20-25% of the dose for hydrocortisone). However, growth suppression is a more common problem with prednisone, which should generally be avoided 5.
Increased dosages of corticosteroids are given during periods of stress and during surgery and in pregnancy
Thyroid-stimulating hormone (TSH) deficiency: Thyroid hormone (L-thyroxine) replacement, in particular for the elderly and those with the cardiac disease It is important to start with a low dose of 25 ug/daily and the up-titrate as required regarding biochemical findings and clinical signs and symptoms.
Evaluate and treat cortisol deficiency before starting T4 replacement to avoid precipitating an adrenal crisis.
FSH/LH deficit: In secondary hypogonadism, testosterone can be delivered by gel, patch, or intramuscular (IM) injections every 2 weeks with a careful monitoring of prostate-specific antigen (PSA) and Testosterone levels.
In women: estrogen/progesterone hormone replacement therapy via oral, intramuscular or transdermal routes can be given.
If fertility is desired, then one starts off with human chorionic gonadotropin (HCG) to augment testosterone levels and improve semen quality. If this is not successful after a 1-year, consider human menopausal gonadotropin (HMG)/recombinant FSH concomitant therapy to enhance fertility further
Growth hormone (GH) deficiency: Unlike in children with short stature due to growth hormone deficiency, the role of growth hormone replacement in the treatment of adult growth hormone deficiency has not been well established. Synthetic growth hormone replacement is used for somatotrophin. Replacement therapy is titrated against IGF-1 levels. Administer growth hormone replacement in doses of 0.18-0.3 mg/kg/week subcutaneously, divided in 6-7 doses. Higher doses of up to 0.7 mg/kg/week may be beneficial in puberty. The goal of treatment is to ensure that adult height is obtained. Further evaluation is made post-puberty to determine whether growth hormone replacement should continue into adulthood.
ADH deficit: Replacement of ADH with intranasal desmopressin (synthetic vasopressin) helps stabilize water balance and polyuria.
- Gounden V, Jialal I. Hypopituitarism (Panhypopituitarism) [Updated 2019 Feb 19]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK470414[↩][↩]
- Sun S, Liu A, Zhang Y. Long-Term Follow-Up Studies of Gamma Knife Radiosurgery for Postsurgical Nonfunctioning Pituitary Adenomas. World Neurosurg. 2019 Jan 17.[↩]
- Abdelmannan D, Aron DC. Incidentally discovered pituitary masses: pituitary incidentalomas. Expert Rev Endocrinol Metab. 2010 Mar;5(2):253-264.[↩]
- Delgado-López PD, Pi-Barrio J, Dueñas-Polo MT, Pascual-Llorente M, Gordón-Bolaños MC. Recurrent non-functioning pituitary adenomas: a review on the new pathological classification, management guidelines and treatment options. Clin Transl Oncol. 2018 Oct;20(10):1233-1245.[↩]
- DeVile CJ, Stanhope R. Hydrocortisone replacement therapy in children and adolescents with hypopituitarism. Clin Endocrinol (Oxf). 1997 Jul. 47(1):37-41.[↩]


{kind=link}