Potter’s syndrome
Potter’s syndrome also known as Potter syndrome, Potter sequence or oligohydramnios sequence, is a rare congenital disorder caused by kidney problems and too little amniotic fluid (oligohydramnios) during pregnancy, leading to a range of physical abnormalities in the developing baby in the womb (uterus) 1, 2, 3, 4, 5, 6, 7, 8, 9. Potter’s syndrome was named after Edith Louise Potter (1901-1993), an American pathologist who described the syndrome specifically in association with bilateral renal agenesis 10. In Potter syndrome the main problem is kidney failure in the unborn baby. The unborn baby’s kidneys fail to develop properly as the baby is growing in the womb. The most common cause of Potter syndrome is the absence of both kidneys also called bilateral renal agenesis, but other kidney problems during fetal development such as severe kidney hypoplasia (also known as renal hypoplasia, a condition where one or both kidneys are smaller than normal with fewer nephrons and calyces, but with normal structure), kidney dysplasia (also known as renal dysplasia, a condition where one or both kidneys do not develop properly during fetal development in the womb resulting in malformed kidney tissue and the presence of cysts, which are fluid-filled sacs), obstructive uropathy (a condition where the normal flow of urine is blocked or hindered in the urinary tract, leading to a back-up of urine and potential kidney damage) or infantile polycystic kidney disease (a genetic kidney disorder where numerous fluid-filled cysts grow in the kidneys, leading to kidney enlargement and impaired function) can also lead to lack of amniotic fluid (oligohydramnios) and Potter syndrome. The unborn baby’s kidneys normally produce the protective amniotic fluid as urine that surrounds the unborn baby in the womb to cushion and protect the developing baby. Without the protective amniotic fluid (oligohydramnios), the unborn baby is not cushioned from the walls of the womb (uterus). The pressure of the uterine wall leads to an unusual facial appearance called Potter facies, a specific set of facial features such as wrinkled skin, low-set ears with lack of ear cartilage (Potter ears), a receding chin, and widely spaced eyes (hypertelorism) 10. Babies born with Potter syndrome may also have abnormal limbs or limbs that are held in abnormal positions or contractures. The lack of sufficient amniotic fluid (oligohydramnios) also stops the development of baby’s lungs also known as pulmonary hypoplasia, so baby’s lungs do not work properly at birth leading to a common and serious breathing difficulties. The absence of both kidneys or bilateral renal agenesis is not compatible with life, as the lack of amniotic fluid produced by the kidneys leads to underdeveloped lungs and other complications. 40% of babies with absence of both kidneys or bilateral renal agenesis will be stillborn (an infant born dead), and if born alive, the baby will live only a few hours. Babies with absence of both kidneys (bilateral renal agenesis) will have several unique characteristics: dry loose skin, wide-set eyes (hypertelorism), prominent folds at the inner corner of each eye, sharp nose, and large low-set ears with lack of ear cartilage (Potter ears). They will typically have underdeveloped lungs, absent urinary bladder, anal atresia, esophageal atresia, and unusual genitals. The lack of amniotic fluid causes some of the problems such as undeveloped lungs, sharp nose and clubbed feet and other problems occur because the kidneys and those affected structures are formed at the same time of fetal life such as the ears, genitals and esophagus.
Curry et al 11 performed in 1984 one of the most complete case series on Potter syndrome, where they found that of 80 cases analyzed, 21.25% had bilateral renal agenesis, 47.5% had cystic dysplasia, and 25% had uropathy. However, an important finding to highlight is that 15 of these patients had multiple congenital anomalies such as aneuploidy, autosomal recessive syndromes and causes that at the time were undetermined 11.
A diagnosis of Potter syndrome is based upon identification of characteristic symptoms, a detailed patient history, a thorough clinical evaluation and certain specialized tests. If not detected before birth (prenatally), then lack of urine production, specific (facial) features or difficulty breathing may be signs of Potter syndrome. A routine specialized imaging technique called a prenatal ultrasound (an ultrasound in most pregnant women get in their second trimester at 18 to 20 weeks of pregnancy) may detect Potter syndrome before birth 12. A fetal ultrasound uses reflected sound waves to create an image of the developing fetus and can reveal a lack of amniotic fluid. An ultrasound can also show abnormalities or absence of the kidneys. Swelling of the kidneys due to a buildup of urine (hydronephrosis), which can occur when there is an obstruction of the urinary tract, can also be seen on an ultrasound.
Potter’s syndrome is a very serious condition without any cure. Potter syndrome treatment depends on the severity of your baby’s lung (pulmonary hypoplasia) and heart complications (congenital heart disease) associated with Potter’s syndrome that affects your child. It also depends on the options available to support your child’s kidney (renal) function. Depending on the timeline of your baby’s diagnosis, treatment can begin during pregnancy. You might qualify for trials like amnioinfusion to add fluid into your amniotic cavity to make up for the lack of fluid surrounding your baby. This type of treatment works best before 22 weeks of pregnancy.
Your growing unborn baby needs to be surrounded by amniotic fluid for sufficient lung growth during fetal development. If your baby’s chest is constrained for a prolonged period of fetal development due too little amniotic fluid (oligohydramnios) during pregnancy, there may not be enough lung tissue to sustain life after birth. There is no treatment for Potter syndrome due to absence of both kidneys (bilateral absence of the kidneys), which is not compatible with life. A complete lack of kidney function is also very challenging to treat in a newborn. In some cases, neonatal palliative care measures that limit intensive care interventions and instead focus on the goals of infant-parental bonding and infant comfort are a treatment option.
If your baby does survive birth, treatment focuses on preventing life-threatening complications that could include:
- Using breathing assistance devices (ventilator).
- Supportive medicine to help with lung function.
- Surgery to fix or remove the urinary tract blockage.
- Surgery to improve feeding with intravenous (IV) nutrition therapy, nasogastric tube or a feeding tube.
- Dialysis to remove blood toxins caused by kidney abnormalities. If dialysis isn’t successful after a couple of years of treatment, your baby’s doctor might recommend a kidney transplant.
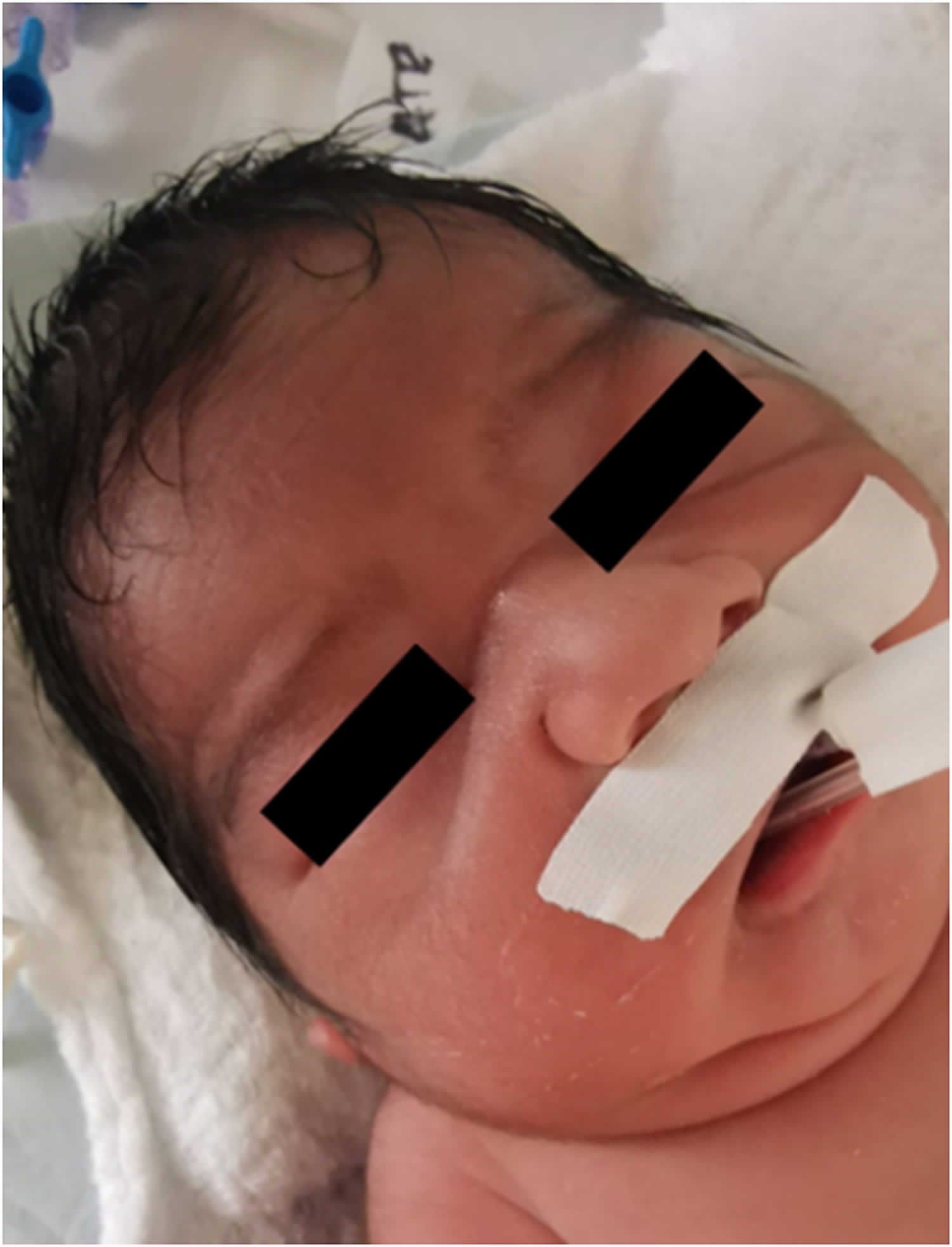
Figure 1. Potter facies
Footnotes: Photograph showing Potter sequence and syndrome facial features (flattened nose, recessed chin, skin folds covering the corners of the eyes and low-set abnormal ears).
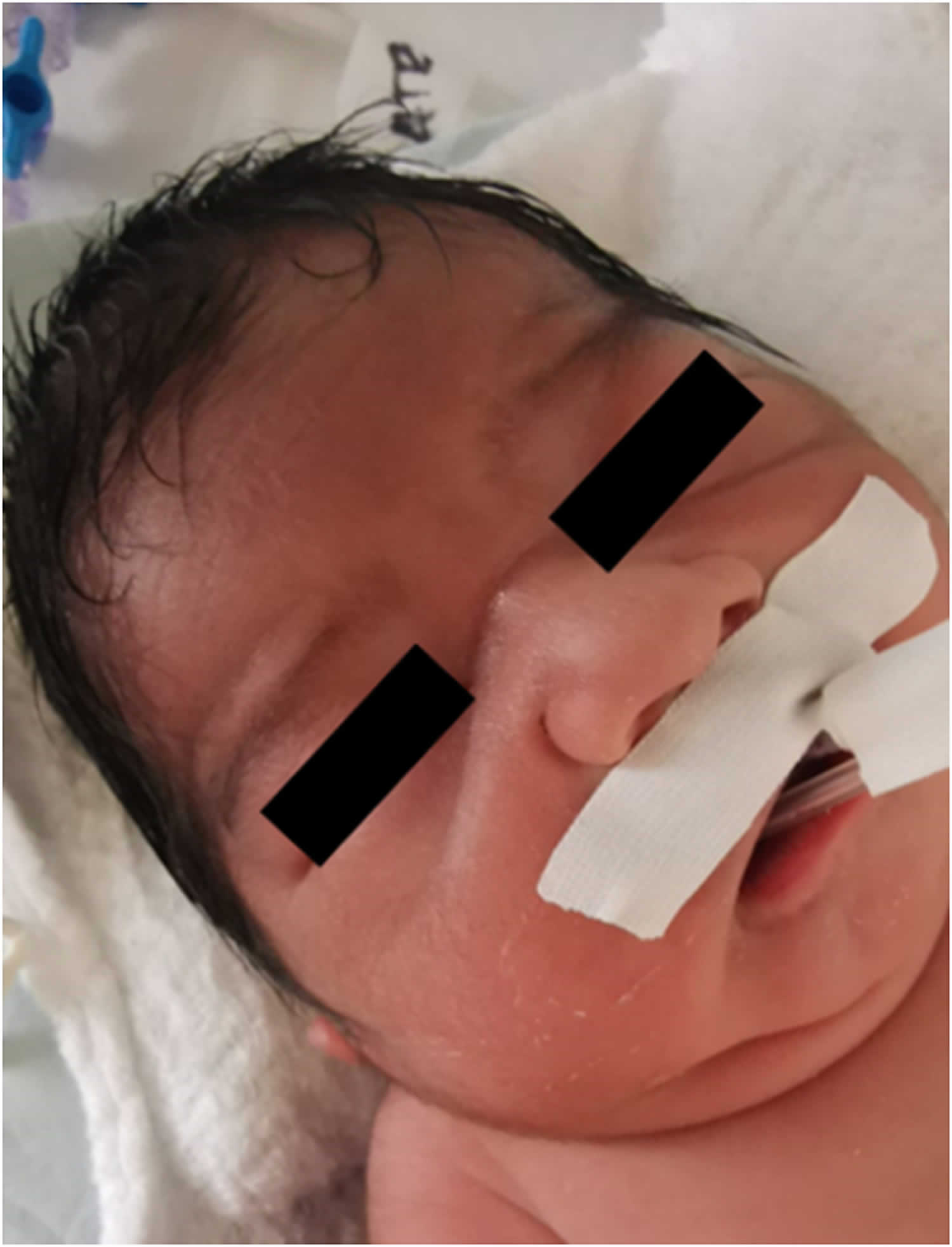
[Source 7 ]Figure 2. Potter syndrome facial features
Footnotes: Potter facies with flattened nose (a), prominent bilateral epicanthal folds (b, arrow) and low set ears with wide pinna (c, arrow).
[Source 1 ]How common is Potter syndrome?
Potter syndrome is a rare disorder and affects an estimated 1 in 4,000 to 1 in 10,000 births. The main cause of Potter syndrome, absence of both kidneys (bilateral renal agenesis), occurs in approximately 1 in 5,000 fetuses and accounts for about 20% of Potter syndrome cases. The incidence or prevalence of other causes are unknown. A couple studies have shown that male newborns are affected more often than female newborns, probably due to the obstructive uropathy that is seen more often in males 13, 14, 15.
What is renal agenesis?
Renal agenesis is the name given to an absence of one kidney (unilateral renal agenesis) or both kidneys (bilateral renal agenesis) that is present at birth (congenital). Usually there is no family history of renal agenesis, but in 20-36% of cases, there is a genetic cause. The absence of both kidneys or bilateral renal agenesis is not compatible with life, as the lack of amniotic fluid produced by the kidneys leads to underdeveloped lungs and other complications. Baby’s kidneys develop between the 5th and 12th week of fetal life, and by the 13th week they are normally producing urine. When the embryonic kidney cells fail to develop, the result is called renal agenesis. It is often detected on fetal ultrasound because there will be a lack of amniotic fluid called oligohydramnios.
When both kidneys are absent (bilateral renal agenesis) this condition is not compatible with life. Absence of both kidneys (bilateral renal agenesis) occurs in 1 of 4500 live births and is usually found in boys. 40% of babies with bilateral renal agenesis (absence of both kidneys) will be stillborn, and if born alive, the baby will live only a few hours. Babies with bilateral renal agenesis will have several unique characteristics: dry loose skin, wide-set eyes, prominent folds at the inner corner of each eye, sharp nose, and large low-set ears with lack of ear cartilage. They will typically have underdeveloped lungs, absent urinary bladder, anal atresia, esophageal atresia, and unusual genitals. The lack of amniotic fluid causes some of the problems such as undeveloped lungs, sharp nose and clubbed feet and other problems occur because the kidneys and those affected structures are formed at the same time of fetal life such as the ears, genitals and esophagus.
Babies with absence of one kidney (unilateral renal agenesis) may have no other symptoms at all. Absence of one kidney (unilateral renal agenesis) occurs in 1 in 1000 to 1 in 2000 live births. Babies with absence of one kidney (unilateral renal agenesis) is more common with intrauterine growth retardation (poor growth during pregnancy) and often results in premature birth. It is also more common when a mother is carrying more than one baby (multiple gestations, such twins, or triplets). Children with absence of one kidney (unilateral renal agenesis) will generally live normal lives with no developmental effects. Many times the single kidney is only detected incidentally when x-rays are done for other purposes. The remaining kidney will enlarge to carry out the function normally done by two kidneys.
Do babies inherit Potter syndrome?
Babies can inherit some causes of Potter syndrome, but Potter syndrome isn’t a genetic condition.
Inherited causes of Potter syndrome include:
- Polycystic kidney disease: Polycystic kidney disease (PKD) is a genetic disorder caused by mutations in PKD1, PKD2, or PKHD1 genes that affect how kidney cells develop and function resulting in fluid-filled cysts developing in the kidneys, leading to kidney enlargement and impaired function. Polycystic kidney disease (PKD) is a common cause of chronic kidney disease and kidney failure. There are two main types of polycystic kidney disease (PKD): autosomal dominant polycystic kidney disease (ADPKD), which is usually diagnosed in adulthood, and autosomal recessive polycystic kidney disease (ARPKD), which can be diagnosed in infancy or childhood.
- Autosomal Dominant polycystic kidney disease (ADPKD): Autosomal dominant polycystic kidney disease (ADPKD) is the most common type, affecting about 1 in every 400 to 1,000 people. It often doesn’t show symptoms until later in life, and is the leading cause of kidney failure in adults.
- Autosomal Recessive polycystic kidney disease (ARPKD): Autosomal Recessive polycystic kidney disease (ARPKD) is a rarer form, affecting about 1 in 20,000 children. It can cause severe complications in infancy or childhood, including liver problems and breathing difficulties.
- Renal agenesis: Renal agenesis is a condition where one or both kidneys fail to develop during fetal development. This can result in a baby being born with only one kidney (unilateral) or no kidneys at all (bilateral). Bilateral renal agenesis or the absence of both kidneys is not compatible with life, as the lack of amniotic fluid produced by the kidneys leads to underdeveloped lungs and other complications. Usually there is no family history of renal agenesis, but in 20-36% of cases there is a genetic cause. A baby can inherit a genetic mutation of the FGF20 or GREB1L gene that causes problems with kidney development. Renal agenesis can be either autosomal dominant or recessive, where one or two copies of the mutated gene need to pass to the child during conception for the child to inherit the condition.
- Sporadic: Some genetic changes that cause Potter syndrome occur randomly, without any history of the genetic condition in a person’s family.
Potter’s syndrome causes
The most common underlying cause of Potter syndrome is absence, underdevelopment or malformation of the kidneys 1. Absence of both kidneys (bilateral renal agenesis) is the most common condition associated with Potter syndrome 15. Abnormal development of your baby’s kidneys causes too little amniotic fluid in the uterus. Many factors could cause Potter syndrome including:
- Underdeveloped or missing kidneys.
- Polycystic kidney disease: Polycystic kidney disease (PKD) is a genetic disorder caused by mutations in PKD1, PKD2, or PKHD1 genes that affect how kidney cells develop and function resulting in fluid-filled cysts developing in the kidneys, leading to kidney enlargement and impaired function. Polycystic kidney disease (PKD) is a common cause of chronic kidney disease and kidney failure. There are two main types of polycystic kidney disease (PKD): autosomal dominant polycystic kidney disease (ADPKD), which is usually diagnosed in adulthood, and autosomal recessive polycystic kidney disease (ARPKD), which can be diagnosed in infancy or childhood.
- Autosomal Dominant polycystic kidney disease (ADPKD): Autosomal dominant polycystic kidney disease (ADPKD) is the most common type, affecting about 1 in every 400 to 1,000 people. It often doesn’t show symptoms until later in life, and is the leading cause of kidney failure in adults.
- Autosomal Recessive polycystic kidney disease (ARPKD): Autosomal Recessive polycystic kidney disease (ARPKD) is a rarer form, affecting about 1 in 20,000 children. It can cause severe complications in infancy or childhood, including liver problems and breathing difficulties.
- Prune belly syndrome (Eagle-Barrett syndrome).
- Blockages of the urinary tract.
- Leaking amniotic fluid caused by premature rupture of the amniotic sac (PROM).
- Unmanaged medical conditions in the mother, like type 1 diabetes.
During pregnancy, your baby floats in a clear to yellow liquid called amniotic fluid. The amniotic fluid surrounds the growing fetus in the womb (uterus) and protects the fetus from injury and temperature changes. The amniotic fluid also allows for freedom of fetal movement and permits musculoskeletal development. Early during your pregnancy, the amniotic fluid is made up of water and nutrients from your body. Your baby drinks the amniotic fluid during your pregnancy. Between 16 and 20 weeks, your baby contributes to the amniotic fluid by peeing (urination). Your baby recycles the amniotic fluid by drinking it and releasing it.
If your baby has Potter syndrome, your baby’s kidneys that create urine didn’t grow properly, is absent or isn’t functioning. Since your baby is unable to urinate, they aren’t able to contribute to the amount of amniotic fluid that protects them, which causes there to be too little amniotic fluid in the uterus or oligohydramnios. Because there is not enough amniotic fluid to protect the unborn baby, the pressure unborn baby undergoes while developing within the uterus that normally does not cause any problems can cause a variety of physical features including distinctive facial features, skeletal abnormalities, and other complications.
Amniotic fluid is also essential for the proper development of your baby’s lungs. Absence of amniotic fluid, especially in the first half of gestation, will result in underdevelopment of the lungs (pulmonary hypoplasia) as well.
Potter syndrome can also result from autosomal recessive polycystic kidney disease (ARPKD), malformation of the kidneys, a rare disorder characterized by absence of the abdominal muscles (prune belly syndrome), certain chromosomal disorders, and obstructive uropathy, in which urine cannot be voided from the body and builds up into the kidneys. Sometimes, Potter syndrome results from prolonged rupture of the amniotic membranes, which allows amniotic fluid to leak out. This usually seen when the rupture occurs early in a pregnancy and goes undetected for a long period of time.
In most instances, Potter syndrome occurs sporadically for no known reason. However, sometimes the underlying cause, such as certain kidney abnormalities, may be genetic. If related to a genetic condition, this can occur spontaneously without a family history of the condition, or the genetic condition could have been inherited and genetic counseling is recommended.
Curry et al 11 performed in 1984 one of the most complete case series on Potter syndrome, where they found that of 80 cases analyzed, 21.25% had bilateral renal agenesis, 47.5% had cystic dysplasia, and 25% had uropathy. However, an important finding to highlight is that 15 of these patients had multiple congenital anomalies such as aneuploidy, autosomal recessive syndromes and causes that at the time were undetermined 11.
Potter syndrome types
Doctors identify different types of Potter syndrome based on symptoms that affect the kidneys including 16, 17, 18:
- Classic Potter syndrome: Classic Potter syndrome is the most common and is the result of the baby being born without both kidneys.
- Potter syndrome type 1: Symptoms of Potter syndrome type 1 occur because of polycystic kidney disease (PKD), where cysts form on the kidneys, caused by the trait passing from both parents. There are two main types of polycystic kidney disease (PKD): autosomal dominant polycystic kidney disease (ADPKD), which is usually diagnosed in adulthood, and autosomal recessive polycystic kidney disease (ARPKD), which can be diagnosed in infancy or childhood. Potter syndrome type 1 is caused by autosomal recessive polycystic kidney disease (ARPKD), which is a rarer type of polycystic kidney disease (PKD) affecting about 1 in 20,000 children. Potter syndrome type 1 can cause severe complications in infancy or childhood, including liver problems and breathing difficulties.
- Potter syndrome type 2: Symptoms of Potter syndrome type 2 are the result of kidney growth abnormalities that occur in the uterus during pregnancy.
- Potter syndrome type 3: Symptoms of Potter syndrome type 3 occur because of polycystic kidney disease similar to type 1, but caused by the trait being passed from only one parent (autosomal dominant). Potter syndrome type 3 is caused by autosomal dominant polycystic kidney disease (ADPKD), which is the most common type of polycystic kidney disease (PKD), affecting about 1 in every 400 to 1,000 people. Potter syndrome type 3 often doesn’t show symptoms until later in life, and is the leading cause of kidney failure in adults.
- Potter syndrome type 4: Symptoms of Potter syndrome type 4 are the result of a blockage of the urinary tract caused by abnormal fetal development in the uterus (obstructive uropathy).
Potter syndrome signs and symptoms
The signs and symptoms of Potter syndrome affect each baby differently and can range in severity. Without the protective amniotic fluid (oligohydramnios), the unborn baby is not cushioned from the walls of the womb (uterus). Symptoms of too little amniotic fluid surrounding the baby in the uterus can affect the length of your pregnancy, which could cause your baby to be born prematurely.
During pregnancy, a clear to yellow fluid (amniotic fluid) surrounds the fetus. This fluid provides protection and space for the fetus to grow by creating a barrier between the uterine wall and the fetus. Unborn babies with Potter syndrome don’t have enough amniotic fluid surrounding them and the pressure from the uterine wall affects how the fetus grows and is often fatal at or shortly after birth. When caused by absence of both kidneys (bilateral agenesis of the kidneys), Potter syndrome is not compatible with life. Potter syndrome due to other causes is also often fatal at or shortly after birth, but there is an increased chance for survival. Infants who do survive the newborn period generally experience chronic lung disease and chronic kidney failure.
Because of the lack of amniotic fluid (oligohydramnios) to protect the developing fetus, normal pressure from the uterine walls can affect the growth and development of the fetus. Such pressure may cause distinctive facial features sometimes referred to as “Potter facies” including a recessed chin; a flattened, depressed bridge of the nose; eyes that are spaced further apart than normal (hypertelorism); low-set ears that lack cartilage (Potter ears); abnormally prominent skins folds in the inner corners of the eyes (prominent epicanthal folds); and a crease beneath the lower lips.
There is usually a lack of urine creation and urine output because of kidney abnormalities. Absence (agenesis) of both kidneys is the most common defect associated with Potter syndrome. The kidneys can also be malformed (dysplastic), or damaged because of a larger syndrome affecting the kidneys such as polycystic kidney disease (PKD), a group of rare disorders characterized by the development of numerous cysts within the kidneys.
The lungs may be underdeveloped (hypoplastic) and most newborns experience severe breathing complications after birth (infant respiratory distress).
Sometimes there are abnormalities in the development of the arms and legs, lack of formation of half of the spine (hemivertebrae), absence of the lower portion of the spine (sacral agenesis), congenital heart defects, or abnormalities of the eyes such as cataracts, or displacement or dislocation (prolapse) of the lenses of the eyes.
Infants with Potter syndrome are often born prematurely and are small for their gestational age, which means they are smaller than would normally be expected for how far along the pregnancy is.
Potter syndrome facial and physical characteristics
Pressure from the lack of amniotic fluid can affect how parts of the fetus develop. This causes distinct facial characteristics, which are called “Potter facies”, including 10:
- Chin that doesn’t grow forward (recessed chin).
- Crease below the lower lip.
- Eyes spaced far apart (hypertelorism).
- Flat bridge of the nose.
- Low-set ears with a small amount of cartilage (Potter ears).
- Skin folds at the corner of the eyes (prominent epicanthal folds).
The pressure from the lack of amniotic fluid can also affect the growth of other parts of the fetus including:
- Short arms and legs.
- Contractures or difficulty fully extending joints.
- Club feet.
- Small for gestational age.
Underdeveloped or malformed organs
Symptoms that affect the organs can be life-threatening. Since Potter syndrome targets fetal development, the internal organs don’t have the instructions or the time to form completely.
Symptoms of Potter syndrome that affect other organs include:
- Congenital heart conditions (ventricular septal defect, endocardial cushion defect, tetralogy of Fallot, and patent ductus arteriosus)
- Eye conditions (cataracts, displaced lenses, angiomatous malformation in the optic disc area, and expulsive hemorrhage).
- Kidney conditions (chronic kidney failure, kidney agenesis, polycystic kidney disease).
- Lung conditions (chronic lung disease, infant respiratory distress).
- Features of prune belly syndrome (Eagle-Barrett syndrome) with deficient abdominal wall, undescended testes, dilated ureters, and a renal pelvis.
Potter syndrome diagnosis
The abnormal development of the kidneys can affect how much urine the newborn can produce. This lack of amniotic fluid surrounding the fetus and kidney abnormalities can be seen by doctors during prenatal ultrasound scan, an ultrasound in most pregnant women get in their second trimester at 18 to 20 weeks of pregnancy 12. A fetal ultrasound uses reflected sound waves to create an image of the developing fetus and can reveal a lack of amniotic fluid. An ultrasound can also show abnormalities or absence of the kidneys. Swelling of the kidneys due to a buildup of urine (hydronephrosis), which can occur when there is an obstruction of the urinary tract, can also be seen on an ultrasound.
Prenatal MRI scans can also provide more detailed images of your baby and his/her organs in equivocal or inconclusive ultrasound findings.
If a diagnosis isn’t made before your baby is born, your doctor will give your child a physical examination, looking for features of Potter syndrome including:
- Minimal urine production.
- Facial characteristics.
- Difficulty breathing.
Prenatal abdominal and transvaginal ultrasound scan
Prenatal abdominal and transvaginal ultrasound scan is the mainstay investigation to accurately evaluate the renal agenesis 12. The absence of urinary bladder and kidneys indicate bilateral renal agenesis 19. Ultrasound signs such as oligohydramnios, retarded intra-uterine growth (R.I.U.G.) and failure to detect the bladder and kidneys constitute fairly clear signs 20. Sometimes, it is difficult to differentiate discoid adrenal glands from kidneys. With the experience of expertise, adrenal glands can be distinguished with the absence of kidneys 21. Ultrasound can detect other causes of oligohydramnios such as polycystic kidney, obstructive uropathy. Doppler ultrasonography visualizes poor angiogenesis in hypoplastic lungs and kidneys. Color doppler ultrasound is adjunct (add-on) to ultrasonography. The absence of renal artery visualization is suggestive of renal agenesis 2.
Tests diagnose Potter syndrome
Your doctor will offer several tests to confirm the Potter syndrome diagnosis:
- A genetic blood test to identify the gene responsible for your baby’s symptoms.
- Imaging tests like an X-ray, MRI or ultrasound of your child’s lungs, kidneys and urinary tract. X-ray examination of your baby’s lungs after birth may show underdevelopment of the lungs.
- Blood or urine tests to check electrolyte and enzyme levels.
- In babies with suspected Potter syndrome, electrolyte abnormalities such as hypernatremia (high blood sodium level, typically above 145 mmol/L), hyperkalemia (high blood potassium level, typically above 5.5 mmol/L), hyponatremia (low blood sodium level, typically below 135 mmol/L ), hypocalcemia (low blood calcium level, typically below 2.1 mmol/L), or metabolic acidosis are evident due to kidney failure.
- Serum creatinine levels are used to assess renal function and the glomerular filtration rate (GFR). The glomerular filtration rate (GFR) can be calculated by using various formulas, such as that reported by Schwartz and colleagues, as follows:
- In low birth weight (LBW) neonates, the formula is (0.33 X height in cm)/serum creatinine level.
- In term infants, the formula is (0.45 X height in cm)/serum creatinine level.
- The serum blood urea nitrogen (BUN) result is not a good indicator of kidney function.
- Obtain a complete blood count (CBC) with differential to evaluate for anemia secondary to erythropoietin deficiency.
- Urinalysis is used to reveal either microhematuria or proteinuria.
- Other tests, such as urine sodium level, urine creatinine level, urine osmolality, and serum osmolality, are indicated if the neonate has kidney failure.
- If sepsis is suspected, obtain cultures of the urine, blood, and cerebrospinal fluid.
- Echocardiogram, which is a test that uses sound waves to create a picture of your baby’s heart, may be conducted to detect congenital heart defects potentially associated with Potter’s syndrome.
Potter syndrome differential diagnosis
Potter syndrome differential diagnosis include:
- Bilateral Renal Agenesis (Mayer-Rokitansky-Kuster-Hauser Syndrome): It is the primary cause of classic Potter syndrome. But bilateral renal agenesis does not always lead to Potter syndrome. Mayer-Rokitansky-Kuster-Hauser syndrome (MRKH) is associated with bilateral renal agenesis. Still, the presence of other features of this syndrome, such as congenital uterine anomalies and other urogenital dysplasia, helps to distinguish it from Potter syndrome 22.
- Multicystic Dysplastic Kidney: Multicystic dysplastic kidney (MCDK) is a congenital condition where one or both kidneys develop abnormally during fetal development, leading to multiple non-communicating cysts formation instead of normal kidney tissue 23. The affected kidney typically does not function, and the healthy kidney compensates for the loss. The dysplastic kidney issue can be recognized in between the cysts. Multicystic dysplastic kidney (MCDK) is considered one of the causes of Potter sequence. Renal insufficiency and low urine output due to multiple cysts kidney lead to the oligohydramnios 23.
- Polycystic kidney disease: Polycystic kidney disease (PKD) is a genetic disorder caused by mutations in PKD1, PKD2, or PKHD1 genes that affect how kidney cells develop and function resulting in fluid-filled cysts developing in the kidneys, leading to kidney enlargement and impaired function. Polycystic kidney disease (PKD) is a common cause of chronic kidney disease and kidney failure. There are two main types of polycystic kidney disease (PKD): autosomal dominant polycystic kidney disease (ADPKD), which is usually diagnosed in adulthood, and autosomal recessive polycystic kidney disease (ARPKD), which can be diagnosed in infancy or childhood.
- Autosomal Dominant polycystic kidney disease (ADPKD): Autosomal dominant polycystic kidney disease (ADPKD) is the most common type, affecting about 1 in every 400 to 1,000 people. It often doesn’t show symptoms until later in life, and is the leading cause of kidney failure in adults.
- Autosomal Recessive polycystic kidney disease (ARPKD): Autosomal Recessive polycystic kidney disease (ARPKD) is a rarer form, affecting about 1 in 20,000 children. It can cause severe complications in infancy or childhood, including liver problems and breathing difficulties.
- Posterior Urethral Valves: The presence of the posterior urethral valve increases the chances of vesicoureteric reflux that prevents amniotic fluid contribution.
- Prune Belly Syndrome: It is more commonly associated with a male child with abdominal wall deformities, skeletal, and renal anomalies, but the urinary bladder is usually dilated. Other genital malformations such as cryptorchidism may be present.
- Sirenomelia: Sirenomelia, also known as mermaid syndrome, is an extremely rare congenital condition characterized by the partial or complete fusion of lower limbs, giving the appearance of a mermaid’s tail 24, 25. Sirenomelia is often associated with severe urogenital and gastrointestinal abnormalities, making it usually incompatible with extrauterine life. Sirenomelia may be associated with oligohydramnios due to bilateral renal agenesis. Other multiple deformities associated with sirenomelia are lower limb fusion, absent external genitalia, or imperforation. Because of oligohydramnios manifestation, it may lead to Potter syndrome 26. Due to renal agenesis, adrenal glands on imaging show a discoid shape that creates confusion with Potter syndrome, but other distinct structural abnormalities help in differentiation 27.
- Ectopic Kidney: An ectopic kidney is a birth defect where one or both kidneys are located outside their normal position in the abdomen, typically in the pelvis, or in the abdomen. This condition results from a failure of the kidneys to ascend to their usual location during fetal development. While often asymptomatic, ectopic kidneys can lead to complications like urinary tract infections, kidney stones, or reduced kidney function if not managed properly. Patients having an ectopic kidney demonstrate empty renal fossa and discoid adrenal resembling an ultrasonographic study of Potter syndrome.
- Melnick-Fraser Syndrome: Melnick-Fraser Syndrome, also known as Branchio-oto-renal syndrome (BOR syndrome), is a rare genetic disorder characterized by a combination of branchial anomalies (branchial cysts or fistulas), ear malformations including hearing loss and preauricular pits, and kidney (renal) abnormalities that can range in severity from renal hypoplasia to agenesis 28, 29. Melnick-Fraser Syndrome is an autosomal dominant condition, meaning a single copy of the mutated gene is sufficient to cause the disorder.
- Fraser Syndrome: Fraser syndrome is a rare, inherited autosomal recessive disorder characterized by a range of developmental anomalies, primarily affecting the eyes, hands, and genitalia. Fraser syndrome is an autosomal recessive condition, meaning individuals need to inherit two copies of the altered gene, one from each parent, to be affected 30, 31. Characteristic features of Fraser syndrome include eyes that are completely covered by skin and usually malformed (cryptophthalmos), fusion of the skin between the fingers and toes (cutaneous syndactyly), and abnormalities of the genitalia and the urinary tract (genitourinary anomalies) 30, 31. Other tissues and organs can also be affected. Depending on the severity of the signs and symptoms, Fraser syndrome can be fatal before or shortly after birth; less severely affected individuals can live into childhood or adulthood.
Potter syndrome treatment
There is no cure for Potter’s syndrome. A neonatologist, pediatric nephrologist, pediatric pulmonologist, pediatric urologist, geneticist, and pediatric surgeon should be consulted as needed. Transfer the patient to a center where pediatric subspecialists are available for consultation. Potter syndrome treatment depends on the severity of your baby’s lung (pulmonary hypoplasia) and heart complications (congenital heart disease) associated with Potter’s syndrome that affects your child. It also depends on the options available to support your child’s kidney (renal) function. Depending on the timeline of your baby’s diagnosis, treatment can begin during pregnancy. You might qualify for trials like amnioinfusion to add fluid into your amniotic cavity to make up for the lack of fluid surrounding your baby. This type of treatment works best before 22 weeks of pregnancy. In the Renal Anhydramnios Fetal Therapy Trial 32, serial amnioinfusions initiated before 26 weeks gestation in singleton pregnancies with bilateral renal agenesis improved neonatal survival and allowed placement of dialysis access; however, only 35% of live-born infants survived past 14 days. Maternal complications included preterm premature rupture of membranes and preterm delivery.
Your growing unborn baby needs to be surrounded by amniotic fluid for sufficient lung growth during fetal development. If your baby’s chest is constrained for a prolonged period of fetal development due too little amniotic fluid (oligohydramnios) during pregnancy, there may not be enough lung tissue to sustain life after birth. There is no treatment for Potter syndrome due to absence of both kidneys (bilateral absence of the kidneys), which is not compatible with life. A complete lack of kidney function is also very challenging to treat in a newborn. In some cases, neonatal palliative care measures that limit intensive care interventions and instead focus on the goals of infant-parental bonding and infant comfort are a treatment option.
If your baby does survive birth, treatment focuses on preventing life-threatening complications that could include:
- Using breathing assistance devices (ventilator).
- Chest tube placement may be required in neonates with spontaneous pneumothorax.
- Supportive medicine to help with lung function.
- Surgery to fix or remove the urinary tract blockage.
- Surgery to improve feeding with intravenous (IV) nutrition therapy, nasogastric tube or a feeding tube.
- Electrolyte abnormalities such as hypocalcemia and hyperphosphatemia can be treated with medications, including calcium carbonate and vitamin D.
- Anemia is treated with oral or parenteral iron and erythropoietin stimulating agents.
- Children may have hypertension from either fluid-related causes or activation of the renin-angiotensin system. Antihypertensives that may be given include diuretics, beta-blockers, calcium channel blockers, and ACE inhibitors.
- Dialysis to remove blood toxins caused by kidney abnormalities. If dialysis isn’t successful after a couple of years of treatment, your baby’s doctor might recommend a kidney transplant.
- Growth hormone is used in children with growth failure associated with chronic renal failure.
In cases of bilateral renal agenesis and recurrence with positive family history, meticulous examinations of fetuses and routine ultrasonographic examinations should be followed in future pregnancies and relevant relatives 15, 33. Genetic counseling of pregnant women about the management of bilateral renal agenesis should be performed 34.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Management of Kidney Failure
Electrolytes imbalances should be corrected, such as hypernatremia (high blood sodium level, typically above 145 mmol/L), hyperkalemia (high blood potassium level, typically above 5.5 mmol/L), hyponatremia (low blood sodium level, typically below 135 mmol/L ), hypocalcemia (low blood calcium level, typically below 2.1 mmol/L), etc. Calcium and phosphate abnormalities can be treated with calcium carbonate and vitamin D. Anemia due to renal tubular insufficiency, and lack of erythropoietin production can be treated with iron and erythropoietin stimulating agents.
Symptomatic treatments of hypertensive children due to activation of the renin-angiotensin system can be achieved with diuretics, angiotensin-converting enzyme (ACE) inhibitors, beta-blockers. Growth hormone supplementation in growth retardation cases and nasogastric feeding may be required for adequate nutrition supplements. Excessive fluids and salt intake are restricted in cases of severe renal failure and hypertensive, respectively.
Pulmonary Hypoplasia
Infants with associated pulmonary hypoplasia show respiratory distress. Ventilatory support is provided with mechanical ventilation and chest tube placement. Oxygenation may be required, but oxygen saturation should be between 90-95%.
Surgical Management
Various structural anomalies of urinary and renal systems necessitate surgical manipulation. Peritoneal or central venous line dialysis is indicated in renal failure. Valve ablation or vesicostomy provides a good alternative in the obstructive posterior urethral valve that decreases the hydronephrosis and improves kidney function 35. Nephrectomy and renal transplantation are indicated in the large kidney with multiple cysts.
Regular follow-up and careful monitoring of renal function, respiratory function, and effects of drugs are required.
Potter syndrome prognosis
Potter syndrome is life-threatening for the baby and many babies have a short life expectancy who either stillborn or die very early 36. A prognostic factor is determined by gestational age at diagnosis, type, and location if associated structural abnormalities. An early diagnosis, often during pregnancy, helps your baby’s doctor plan for the safest delivery of your baby along with treatment to help your baby survive after he/she is born. If the absence of kidneys (renal agenesis) in your baby’s body causes Potter syndrome, the condition is fatal. Children with Potter syndrome due to conditions such as infantile polycystic kidney disease, multicystic dysplastic kidney, hypoplastic kidney, Prune-Belly syndrome, and premature rupture of amniotic membranes during pregnancy have a higher survival rate than children with Potter syndrome due to other conditions. According to the experimental study conducted in John Hopkins hospital, regular saline injected in the mother’s womb has enhanced the lung development. But dialysis is required throughout his/her life. The neonatal mortality rate is 100% if no obstetric interventions are done 34.
Children who experience mild symptoms or who experience low amniotic fluid during pregnancy (oligohydramnios) may survive, but they can develop chronic lung and kidney conditions as they grow. Since treatment begins as soon as your baby is born, not every procedure will be successful and they may face several surgeries early in their life.
Potter syndrome survival rate
Babies with Potter syndrome may have a short life expectancy. Each baby is unique and varies based on the symptoms and severity of Potter syndrome. If your baby’s symptoms are severe and affect the development and function of major organs like the heart, lungs and kidneys, the outlook is poor and most babies won’t survive their first few days of life. Mild cases with less severe symptoms that affect their organs lead to an improved life expectancy.
Your baby’s doctor will discuss the risks of your baby’s diagnosis and offer treatment to prolong your baby’s life. Your baby’s doctor might recommend palliative care, grief or bereavement counseling if your child’s diagnosis is severe to help you cope with a difficult loss of a loved one. Coping with the news that your child might not survive due to a life-threatening diagnosis can be devastating and difficult to navigate. Surround yourself with support from friends and family. Many families find grief and bereavement counseling comforting after a loss of a loved one.
- Shastry SM, Kolte SS, Sanagapati PR. Potter’s Sequence. J Clin Neonatol. 2012 Jul;1(3):157-9. doi: 10.4103/2249-4847.101705[↩][↩][↩]
- Bhandari J, Thada PK, Sergent SR. Potter Syndrome. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK560858[↩][↩]
- Potter Syndrome. https://rarediseases.org/rare-diseases/potter-syndrome[↩]
- Manoj MG, Kakkar S. Potter’s syndrome – a fatal constellation of anomalies. Indian J Med Res. 2014 Apr;139(4):648-9. https://pmc.ncbi.nlm.nih.gov/articles/PMC4078509[↩]
- Prabhu S, Sigamani E, Das P, Sasi A, Safaya R. Potter syndrome with an unusual cardiac anomaly. BMJ Case Rep. 2009;2009:bcr0120091518. doi: 10.1136/bcr.01.2009.1518[↩]
- Kinoshita Y, Sakamoto R, Hattori Y, Furuie K, Kuraoka S, Hidaka Y, Tamura H, Mitsubuchi H, Nakamura K. Elective Cesarean Section during Preterm Prevents Pulmonary Hypoplasia Development in Potter Sequence. Case Rep Pediatr. 2023 Jan 31;2023:3216232. doi: 10.1155/2023/3216232[↩]
- Muñoz-Murillo KL, Muñoz-Murillo WJ, Hernández-López UJ, Aponte-Ceballos LM, Lozada-Martínez ID, Rahman S. First case report of spontaneous perinatal gastric perforation in premature neonate with potter sequence and syndrome. Int J Surg Case Rep. 2021 Sep;86:106297. doi: 10.1016/j.ijscr.2021.106297[↩][↩]
- Gautam U, Kafley R, Chikanbanjar V, Shakya A, Basnet R, Manandhar SR. Rare manifestations of Potter Sequence: A Case Report. JNMA J Nepal Med Assoc. 2020 Mar;58(223):178-180. doi: 10.31729/jnma.4683[↩]
- Fitch N, Lachance RC. The pathogenesis of Potter’s syndrome of renal agenesis. Can Med Assoc J. 1972 Oct 7;107(7):653-6. https://pmc.ncbi.nlm.nih.gov/articles/instance/1940948/pdf/canmedaj01653-0067.pdf[↩]
- Potter, Edith L. Facial Characteristics of Infants with Bilateral Renal Agenesis. American Journal of Obstetrics & Gynecology, Volume 51, Issue 6, 885 – 888. doi:10.1016/s0002-9378(16)39968-9[↩][↩][↩]
- Curry C.J., Jensen K., Holland J., Miller L., Hall B.D. The potter sequence: a clinical analysis of 80 cases. Am. J. Med. Genet. 1984;19(4):679–702. doi: 10.1002/ajmg.1320190408[↩][↩][↩][↩]
- Huber C, Shazly SA, Blumenfeld YJ, Jelin E, Ruano R. Update on the Prenatal Diagnosis and Outcomes of Fetal Bilateral Renal Agenesis. Obstet Gynecol Surv. 2019 May;74(5):298-302. doi: 10.1097/OGX.0000000000000670[↩][↩][↩]
- Khatami F. Potter’s syndrome: A study of 15 patients. Arch. Iran. Med. 2004;7:186–189.[↩]
- Shastry S.M., Kolte S.S., Sanagapati P.R. Potter’s Sequence. J. Clin. Neonatol. 2012;1:157–159. doi: 10.4103/2249-4847.101705[↩]
- Loendersloot EW, Verjaal M, Leschot NJ. Bilateral renal agenesis (Potter’s syndrome) in two consecutive infants. Eur J Obstet Gynecol Reprod Biol. 1978 Jun;8(3):137-42. doi: 10.1016/0028-2243(78)90063-1[↩][↩][↩]
- Kostov S, Slavchev S, Dzhenkov D, Strashilov S, Yordanov A. Discordance for Potter’s Syndrome in a Dichorionic Diamniotic Twin Pregnancy-An Unusual Case Report. Medicina (Kaunas). 2020 Mar 4;56(3):109. doi: 10.3390/medicina56030109[↩]
- Sarkar S., DasGupta S., Barua M., Ghosh R., Mondal K., Chatterjee U., Datta C. Potter’s sequence: A story of the rare, rarer and the rarest. Indian J. Pathol. Microbiol. 2015;58:102–104. doi: 10.4103/0377-4929.151202[↩]
- Ikeda Y., Lister J., Bouton J.M., Buyukpamukcu M. Congenital neuroblastoma, neuroblastoma in situ, and the normal fetal development of the adrenal. J. Pediatr. Surg. 1981;16:636–644. doi: 10.1016/0022-3468(81)90019-1[↩]
- Schmidt W, Kubli F, Schroeder T. Ultrasonographische Befunde beim “Potter-Syndrom” [Ultrasonographic findings in “Potter’s syndrome” (author’s transl)]. Geburtshilfe Frauenheilkd. 1981 May;41(5):374-81. German. doi: 10.1055/s-2008-1036813[↩]
- Garba H, Dechivré J, Broussard P. Diagnostic positif du syndrome de Potter par l’échographie. A propos d’une observation [Positive diagnosis of Potter’s syndrome by ultrasonography. A case report]. Rev Fr Gynecol Obstet. 1993 Mar;88(3):156-61. French.[↩]
- Volberg FM, Dillard R, Sumner T. Ultrasonography of discoid adrenals in Potter’s syndrome: report of three cases. Am J Perinatol. 1989 Jul;6(3):326-8. doi: 10.1055/s-2007-999605[↩]
- Jacquinet A, Boujemla B, Fasquelle C, Thiry J, Josse C, Lumaka A, Brischoux-Boucher E, Dubourg C, David V, Pasquier L, Lehman A, Morcel K, Guerrier D, Bours V. GREB1L variants in familial and sporadic hereditary urogenital adysplasia and Mayer-Rokitansky-Kuster-Hauser syndrome. Clin Genet. 2020 Aug;98(2):126-137. doi: 10.1111/cge.13769[↩]
- Cardona-Grau D, Kogan BA. Update on Multicystic Dysplastic Kidney. Curr Urol Rep. 2015 Oct;16(10):67. doi: 10.1007/s11934-015-0541-7[↩][↩]
- Sirenomelia. https://rarediseases.org/rare-diseases/sirenomelia[↩]
- Samal SK, Rathod S. Sirenomelia: The mermaid syndrome: Report of two cases. J Nat Sci Biol Med. 2015 Jan-Jun;6(1):264-6. doi: 10.4103/0976-9668.149227[↩]
- Al-Haggar M, Yahia S, Abdel-Hadi D, Grill F, Al Kaissi A. Sirenomelia (symelia apus) with Potter’s syndrome in connection with gestational diabetes mellitus: a case report and literature review. Afr Health Sci. 2010 Dec;10(4):395-9. https://pmc.ncbi.nlm.nih.gov/articles/PMC3052805[↩]
- Islam N, Mandal B, Das RN, Bera G, Mukherjee S, Chatterjee U. Sirenomelia associated with discoid adrenal and lumbar meningocoele: An autopsy report. Pathol Res Pract. 2017 Nov;213(11):1450-1453. doi: 10.1016/j.prp.2017.06.010[↩]
- Nasir SB, Ladan SJ, Bemu AN, Jibrin J. Branchiootorenal syndrome: A case report. Niger Postgrad Med J. 2018 Jan-Mar;25(1):60-62. doi: 10.4103/npmj.npmj_203_17[↩]
- Branchio-oto-renal Syndrome (Melnick-Fraser Syndrome). https://medicine.uiowa.edu/iowaprotocols/branchio-oto-renal-syndrome-melnick-fraser-syndrome[↩]
- Fraser syndrome. https://medlineplus.gov/genetics/condition/fraser-syndrome[↩][↩]
- Kalpana Kumari MK, Kamath S, Mysorekar VV, Nandini G. Fraser syndrome. Indian J Pathol Microbiol. 2008 Apr-Jun;51(2):228-9. doi: 10.4103/0377-4929.41664[↩][↩]
- Miller JL, Baschat AA, Rosner M, et al. Neonatal Survival After Serial Amnioinfusions for Bilateral Renal Agenesis: The Renal Anhydramnios Fetal Therapy Trial. JAMA. 2023 Dec 5;330(21):2096-2105. doi: 10.1001/jama.2023.21153[↩]
- Hjort C, Larsen CO, Nathan E. Potter-sekvens. Faenotype, patogenese, aetiologi og arvelige aspekter [Potter’s sequence. Phenotype, pathogenesis, etiology and hereditary aspects]. Ugeskr Laeger. 1992 Feb 17;154(8):488-91. Danish.[↩]
- Thomas AN, McCullough LB, Chervenak FA, Placencia FX. Evidence-based, ethically justified counseling for fetal bilateral renal agenesis. J Perinat Med. 2017 Jul 26;45(5):585-594. doi: 10.1515/jpm-2016-0367[↩][↩]
- Rouzrokh M, Mirshemirani A, Khaleghnejad-Tabari A, Sadeghian N, Mohajerzadeh L, Mohkam M. Protective temporary vesicostomy for upper urinary tract problems in children: a five-year experience. Iran J Pediatr. 2013 Dec;23(6):648-52. https://pmc.ncbi.nlm.nih.gov/articles/PMC4025121[↩]
- Jain M, Agarwal S, Mandal S. Variation in clinical and genitourinary lesions associated with pulmonary hypoplasia in Potter’s syndrome–two autopsy reports. Indian J Pathol Microbiol. 2006 Jul;49(3):416-8.[↩]
{kind=link}