
Torsades de Pointes
Torsades de Pointes (TdP), which mean “twisting of the point” in French, is a type of polymorphic ventricular tachycardia (fast heart rate originating in the ventricles) characterized on electrocardiogram (ECG) by a gradual change in the amplitude and twisting of the QRS complexes around the isoelectric line that can potentially lead to sudden cardiac death if not diagnosed and treated 1, 2, 3, 4, 5, 6, 7. Torsades de Pointes is often associated with QTc interval prolongation (the time it takes for the ventricles to depolarize and repolarize), which is the heart rate adjusted lengthening of the QT interval. The rate of Torsades de Pointes ranges from 160 to 240 beats per minute, which is slower than ventricular fibrillation 6. Torsades de Pointes rhythm may terminate spontaneously or may degenerate into ventricular fibrillation (VFib) and therefore can be fatal if not diagnosed and managed 8, 6. Torsades de Pointes is usually self-terminating and may cause palpitations (the sensation of your heart beating rapidly, irregularly, or with a forceful beat), dizziness, or fainting (syncope). Fainting (syncope) is typically precipitous and without warning.
Torsades de Pointes (TdP) can be caused by either congenital due to inherited Long QT syndrome or acquired due to electrolyte disturbances such as low blood potassium (hypokalemia), low blood calcium (hypocalcemia) and low blood magnesium (hypomagnesemia), and can be triggered by certain medications 9, 3, 10, 11, 12. More often, certain medications such as antiarrhythmics, antipsychotics, antibiotics, and antidepressants are the most frequent cause Torsades de Pointes 13.
The prevalence of Torsades de Pointes is unknown. Torsades de Pointes is a life-threatening arrhythmia and may present as sudden cardiac death in patients with structurally normal hearts 14. In the United States, 300,000 sudden cardiac deaths occur per year. Torsades de Pointes probably accounts for fewer than 5% 15.
Torsades de Pointes is 2-3 times more common in women than in men 15. Women have longer QT intervals as well as have more QT prolongation secondary to drug therapy 16.
Long QT syndrome usually falls into 2 groups 17, 18, 19:
- Congenital long QT syndrome. You’re born with this type of long QT syndrome. It’s estimated to affect about 1 in 2,000 people 20, 21. It’s caused by changes in DNA that are passed down through families. That means it is inherited. Congenital long QT syndrome has been shown to be caused by mutations in one of at least 15 different ion-channel genes 22: the KCNQ1 gene causing LQTS1; KCNH2 causing LQT2; SCN5A causing LQT3; ANK2 causing LQTS4; KCNE1 causing LQTS5; KCNE2 causing LQT6; KCNJ2 causing LQTS7; CACNA1c causing LQTS8; CAV3 causing LQTS9; SCN4B causing LQTS10; AKAB9 causing LQTS11; SNTA1 causing LQTS12; KCNJ5 causing LQTS13; CALM1 causing LQTS14; and CALM2 causing LQTS15. Mutations in KCNQ1, KCNH2, and SCN5A correlate to Long QT types 1-3 and account for the majority (60-75%) of genetically identifiable cases.
- There are 2 types of congenital long QT syndrome:
- Romano-Ward syndrome. This more common type happens in people who get only a single gene change from one parent. Receiving a changed gene from one parent is known as an autosomal dominant inheritance pattern.
- Jervell and Lange-Nielsen syndrome. This rare form of long QT syndrome usually happens very early in life and is severe. Children with this type of long QT syndrome also are deaf. In Jervell and Lange-Nielsen syndrome, children get the gene change from both parents. This is called an autosomal recessive inheritance pattern.
- There are 2 types of congenital long QT syndrome:
- Acquired long QT syndrome. Acquired long QT syndrome is caused by another health condition or medicine. It usually can be reversed when the specific cause is found and treated.
- If a medicine causes acquired long QT syndrome, the disorder may be called drug-induced long QT syndrome. More than 100 medicines can cause prolonged QT intervals in otherwise healthy people. Medicines that can cause long QT syndrome include 23:
- Some antibiotics, such as erythromycin (Eryc, Erythrocin, others), azithromycin (Zithromax) and others.
- Some antifungal medicines used to treat yeast infections.
- Water pills, also called diuretics, that cause the body to remove too much potassium or other minerals.
- Heart rhythm medicines called anti-arrhythmics, which can make the QT interval longer.
- Some medicines used to treat mental health conditions such as anxiety and depression.
- Some medicines used to treat upset stomach.
- Always tell your healthcare professional about all the medicines you take, including those you buy without a prescription.
- Health conditions that can cause acquired long QT syndrome include 23:
- Body temperature below 95 degrees Fahrenheit (35 degrees Celsius), a condition called hypothermia.
- Low calcium, also called hypocalcemia.
- Low magnesium, also called hypomagnesemia.
- Low potassium, also called hypokalemia.
- A tumor of the adrenal gland that usually is not cancer, called pheochromocytoma.
- Stroke or brain bleed.
- Underactive thyroid, also called hypothyroidism.
- If a medicine causes acquired long QT syndrome, the disorder may be called drug-induced long QT syndrome. More than 100 medicines can cause prolonged QT intervals in otherwise healthy people. Medicines that can cause long QT syndrome include 23:
Congenital Long QT syndrome is autosomal in genetic transmission but shows a greater frequency of expression and a greater lengthening of the QT interval in women than in men. You should avoid competitive sports if you have congenital long QT syndrome 24. If you inherited Long QT syndrome from a parent, you should talk to a heart rhythm specialist before your start exercising. In congenital long QT syndrome, the mortality rate for untreated patients is 50% in 10 years, which can be reduced to 3-4% with therapeutic intervention. A systematic review of babies born with long QT syndrome from 83 studies comprising 265 newborns with postnatal confirmation of long QT syndrome found that a longer fetal QTc was more predictive of death than any other antenatal factor, and the mortality risk was significantly raised when the fetal QTc was longer than 600 ms 25. Other factors that were highly predictive of death included the combination of ventricular tachycardia/Torsade de Pointes or functional 2:1 heart block and lack of a family history of long QT syndrome 25. However, fetal heart rate and heart z-score did not predict death 25.
Torsades de Pointes occurs in patients of a wide age range, from newborns to the very elderly 15. The highest frequency is in patients aged 35-50 years. Torsades de Pointes that occurs at an early age usually is due to congenital long QT syndrome. In older persons, it usually is due to acquired long QT syndrome 15. In a systematic review, investigators found that slightly over half (50.8%) of elderly patients age ≥80 years with drug-induced Torsades de Pointes experienced it as the result of “reckless administration” of a QT-interval prolonging agent 26. The most common occurrences of reckless administration of a QT-interval prolonging drugs were in conjunction with another such agent (51.6%) or despite a known QT-interval prolongation (25.8%) 26. Several European centers estimate that the annual reporting rate of drug-induced Torsades de Pointes is between 0.8 and 1.2 per million person-years 3. The incidence of drug-induced Torsades de Pointes also varies based on the drug in question and the population being studied 27, 28, 29.
Torsades de Pointes is an electrocardiographic (ECG) diagnosis, and obtaining an ECG is essential. Frequent ECG monitoring is indicated for patients who are at risk due to chronic conditions or drug therapy. When the patient is in sinus rhythm, examine the QT interval. Usually, a prolonged QT interval and pathological U waves are present, reflecting abnormal ventricular repolarization 30. The most consistent indicator of QT prolongation is a QT of 0.60 second or longer or a QTc (corrected for heart rate) of 0.45 second or longer 30.
Tests to diagnose Torsades de Pointes include:
- Electrocardiogram (EKG).
- Blood tests to check electrolyte levels.
- Echocardiogram.
- A heart monitor you wear at home.
Treatment of Torsades de Pointes can be divided into short-term or immediate management and long-term management 24. The goal of short-term management or immediate management is to assess for, achieve, and maintain hemodynamic stability. Short-term management or immediate management of Torsades de Pointes is the same in both acquired and congenital long QT syndrome, except that beta1-adrenergic stimulation may be tried in the acquired form but is contraindicated in the congenital form 24. If you inherited Long QT syndrome from your parents, your treatment will include more long-term solutions than someone who got Long QT syndrome from a medication.
In an otherwise stable patient, direct current (DC) cardioversion is kept as a last resort, because Torsades de Pointes is paroxysmal in nature and is characterized by its frequent recurrences following cardioversion 24. Although Torsades de Pointes frequently is self-terminating, it may degenerate into ventricular fibrillation, which requires direct current (DC) defibrillation 24.
Any offending agent should be withdrawn. Predisposing conditions such as hypokalemia, hypomagnesemia, and bradycardia should be identified and corrected 24.
Figure 1. Torsades de Pointes

Footnotes: In 1966, the French cardiologist Dessertenne 1 described the electrocardiographic (ECG) pattern of ventricular tachycardia (VT), which he named “torsades de pointes” or “twisting of the points” and noted its association with a markedly prolonged QT interval.
[Source 13 ]Figure 2. Onset of Torsades de Pointes
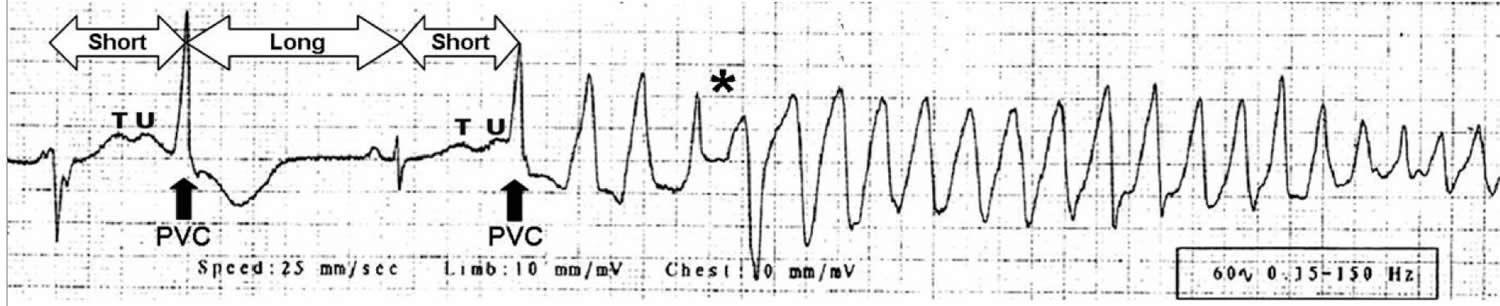
Footnotes: Onset of Torsades de Pointes during the recording of a standard 12-lead ECG in a young male with a history of drug addiction treated with chronic methadone therapy who presented to a hospital emergency department after ingesting an overdose of prescription and over-the-counter drugs from his parent’s drug cabinet. Classic ECG features evident in this rhythm strip include a prolonged QT interval with distorted T-U complex, initiation of the arrhythmia after a short-long-short cycle sequence by a Premature Ventricular Contraction (PVC) that falls near the peak of the distorted T-U complex, “warm-up” phenomenon with initial R-R cycles longer than subsequent cycles, and abrupt switching of QRS morphology from predominately positive to predominately negative complexes (asterisk).
[Source 6 ]Figure 3. Torsade de Pointes ECG
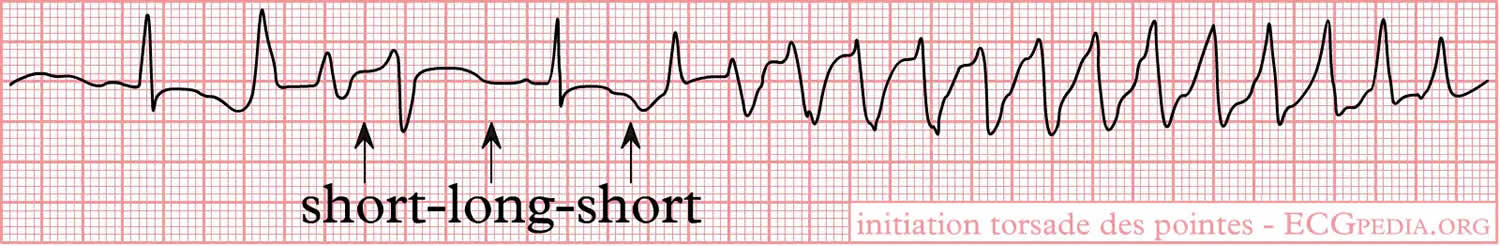
Footnotes: A normal sinus beat is followed by a ventricular extrasystole (shortly after the sinus beat), the compensatory pause results in a longer interval to the next beat. This longer beat therefore has a longer QT interval. The next beat follows shortly thereafter, within the QT interval. Not all ventricular cells have been repolarized by that time and a ventricular arrhythmia results. This short-long-short sequence is typical for Torsades de Pointes.
[Source 31 ]Figure 4. Torsades de Pointes ECG
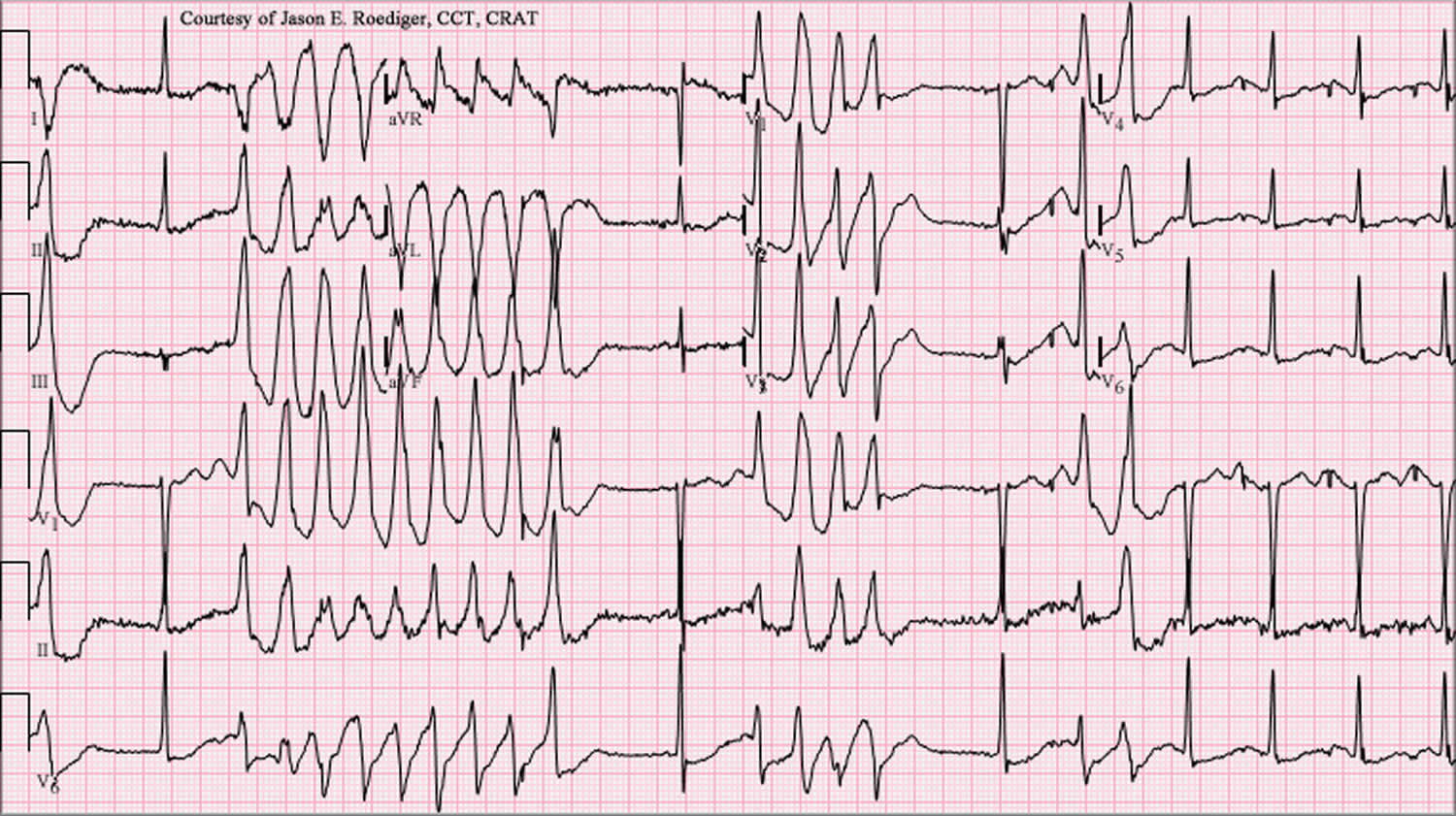
Footnotes: Torsades de Pointes ECG with prolonged QT interval and the “twisting” or oscillating pattern of the QRS complexes, with the amplitude changing around the baseline. 12-lead ECG of Torsades de Pointes in a 56-year-old white female with low blood potassium (2.4 mmol/L) and low blood magnesium (1.6 mg/dL).
[Source 32 ]What is the QT interval?
An ECG measures electrical impulses as five distinct waves (Figure 5). Doctors label these five waves using the letters P, Q, R, S and T. The waves labeled Q through T show electrical activity in your heart’s lower chambers (ventricles). The QT interval is a time interval on the ECG.
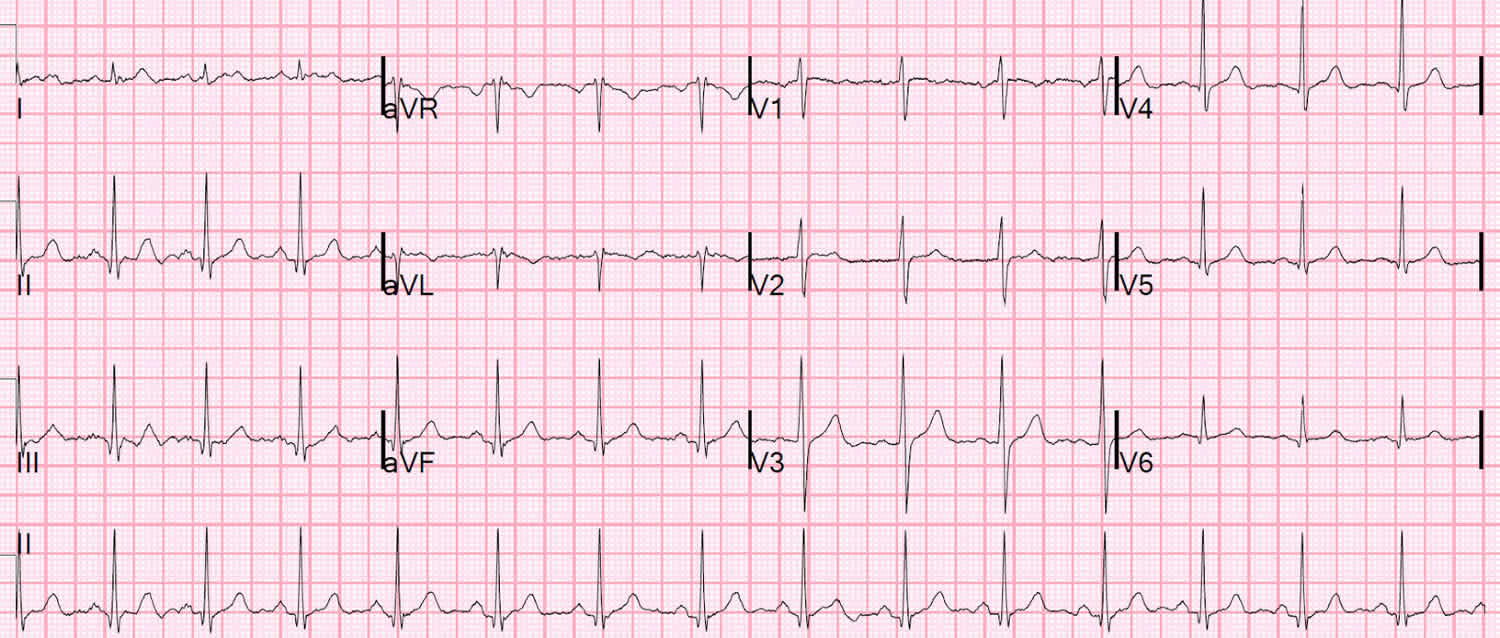
A typical ECG tracing is shown in Figure 5 below. The different waves that comprise the ECG represent the sequence of depolarization and repolarization of the atria and ventricles. The ECG is recorded at a speed of 25 mm/second (5 large squares/second or 5 dark red boxes), and the voltages are calibrated so that 1 mV = 10 mm (2 large squares) in the vertical axis. Therefore, each large dark red 5 mm square represents 0.2 sec (20 msec [millisecond]) and each small 1 mm square represents 0.04 sec (40 msec [millisecond]) in time and 0.10 mV in voltage. Because the recording speed is standardized, you can calculate the heart rate from the intervals between different waves. One method is to divide 1500 by the number of small squares between two R waves. For example, the rate between beats 1 and 2 in Figure 7 below is 1500/22, which equals 68 beats/min. Alternatively, you can divide 300 by the number of large squares, which is 300/4.4 (68 beats/min). Another method, which gives a rough approximation, is the “count off” method. Count the number of large squares between R waves with the following rates: 300 – 150 – 100 – 75 – 60. For example, if there are three large boxes between R waves, then the rate is 100 beats/min. You must extrapolate, however, between boxes. Atrial rate can be determined like the ventricular rate, but using the P waves. Remember, if the heart is in sinus rhythm and there is a one-to-one correspondence between P waves and QRS completes, then the atrial rate will be the same as the ventricular rate. The rate is normal if the interval lies between 5 and 3 large squares (60 – 100 beats/min). Intervals less than 3 large squares or greater than 5 large squares represent tachycardia or bradycardia, respectively.
The space between the start of the Q wave and the end of the T wave (QT interval) corresponds to the time it takes for your heart to contract and then refill with blood before beginning the next contraction. The QT interval represents the time from the electrical stimulation (depolarization) of the heart’s pumping chambers (ventricles), to the end of the recharging of the electrical system (repolarization) and therefore roughly estimates the duration of an average ventricular action potential 33. The interval between the letters Q and T defines the action of the ventricles. The QT interval is measured in milliseconds (msec) and closely approximates the time from the beginning of the heart ventricles’ contraction until the end of relaxation.
The QT interval interval can range from 0.20 to 0.40 seconds (200 to 400 msec), depending upon your heart rate. At high heart rates, ventricular action potentials shorten in duration, which decreases the QT interval 33. Long QT syndrome means that time period is too long, even if by fractions of a second. Because prolonged QT intervals can be diagnostic for susceptibility to certain types of heart arrhythmias, it is important to determine if a QT interval is excessively long. The QT interval is often expressed as a corrected QT (QTc) by taking the QT interval and dividing it by the square root of the R-R interval (interval between ventricular depolarizations) using Bazett formula: QTc = QT / √ RR 34. Corrected QT (QTc) allows an assessment of the QT interval that is independent of the heart rate. Normal corrected QTc intervals are 0.44 seconds or less (<440 msec). QTc is prolonged if it is greater than 440 ms in men or greater than 460 ms in women 35. A QTc greater than 500 is associated with an increased risk of Torsade de Pointes. While several equations exist to help correct heart rate variation, the Bazett formula (QTC = QT / √ RR) is the most commonly used. Though the Bazett formula seems relatively accurate in heart rates between 60 to 100 beats/min, it tends to overcorrect with higher heart rates and undercorrect with lower heart rates.
An occasional prolonged QT interval can be precipitated by everyday circumstances, including:
- When startled by a noise
- Physical activity or exercise
- Intense emotion (such as fright, anger or pain)
In these instances, the heartbeat usually regains its normal contraction rhythm quickly.
Long QT syndrome results from abnormalities in the heart’s electrical recharging system. However, the heart’s structure is normal. Abnormalities in your heart’s electrical system might be inherited. Or, they may be acquired due to an underlying medical condition or a medication.
Figure 5. Normal ECG pattern
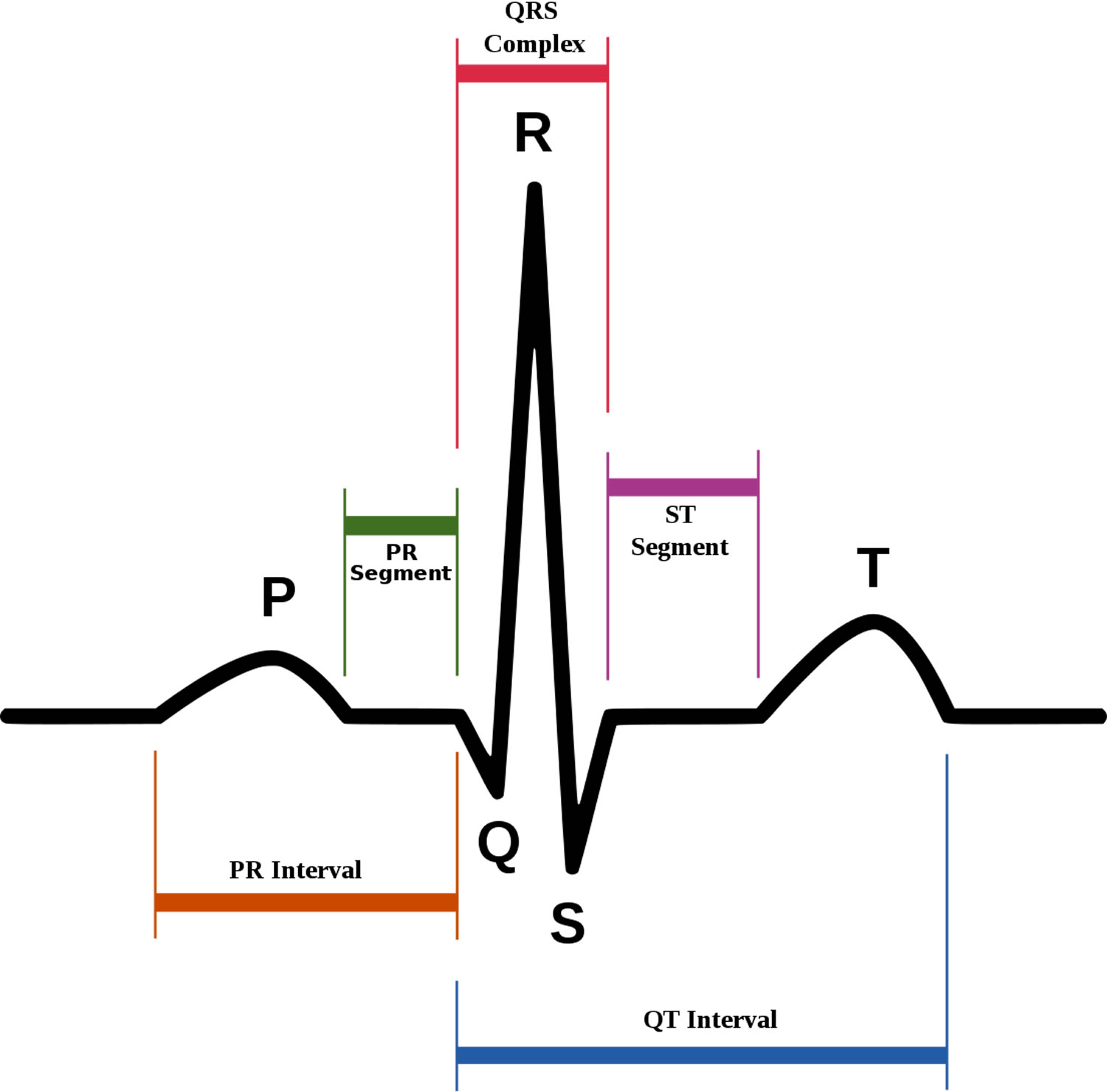
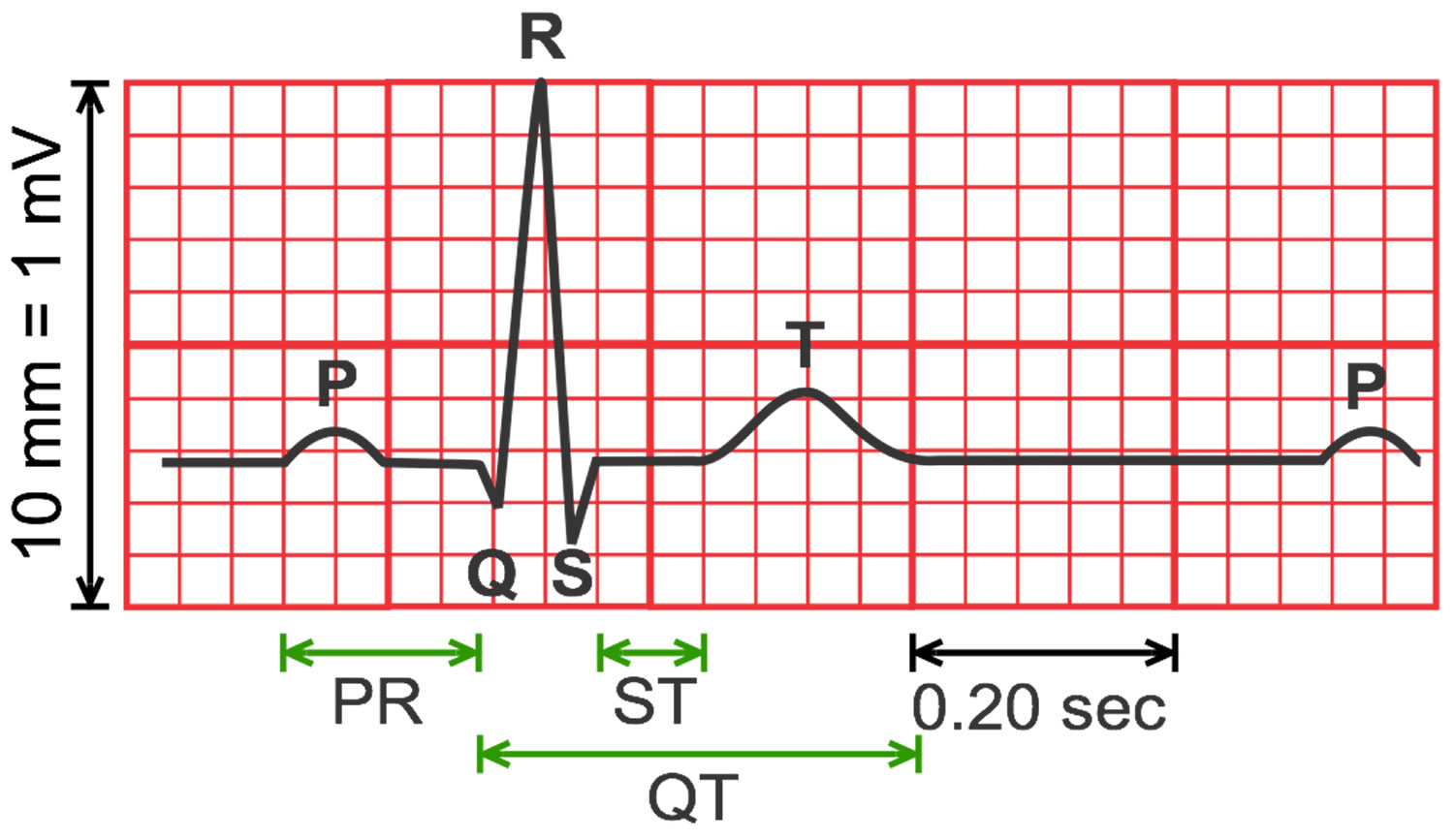
Footnotes:
- P wave represents the wave of depolarization that spreads from the SA node throughout the atria and is usually 0.08 to 0.10 seconds (80-100 ms) in duration. There is no distinctly visible wave representing atrial repolarization in the ECG because it occurs during ventricular depolarization. Because the wave of atrial repolarization is relatively small in amplitude (i.e., has low voltage), it is masked by the much larger ventricular-generated QRS complex.
- QRS complex represents ventricular depolarization. Ventricular rate can be calculated by determining the time interval between QRS complexes. The duration of the QRS complex is normally 0.06 to 0.10 seconds.
- ST segment is the isoelectric period following the QRS and ending at the beginning of the T wave. This represents the period at which both ventricles are completely depolarized. This segment roughly corresponds to the plateau phase of the ventricular action potentials. The ST segment is crucial in the diagnosis of ventricular ischemia or hypoxia because under those conditions, the ST segment can become either depressed or elevated.
- T wave represents ventricular repolarization. The T wave exhibits a positive deflection. This is because the last cells to depolarize in the ventricles are the first to repolarize. This occurs because the last cells to depolarize are in the subepicardial region of the ventricles, and these cells have shorter action potentials than found in the subendocardial regions of the ventricular wall. Therefore, although the depolarization of the subepicardial cells occurs after subendocardial depolarization, the subepicardial cells undergo phase 3 repolarization before the subendocardial cells. Therefore, repolarization waves generally are oriented opposite of depolarization waves and repolarization waves moving away from a positive recording electrode produce a positive voltage.
- QT interval represents the time for both ventricular depolarization and repolarization to occur, and therefore roughly estimates the duration of an average ventricular action potential. This interval can range from 0.20 to 0.40 seconds, depending upon heart rate. At high heart rates, ventricular action potentials shorten in duration, which decreases the QT interval.
Figure 6. Long QT syndrome
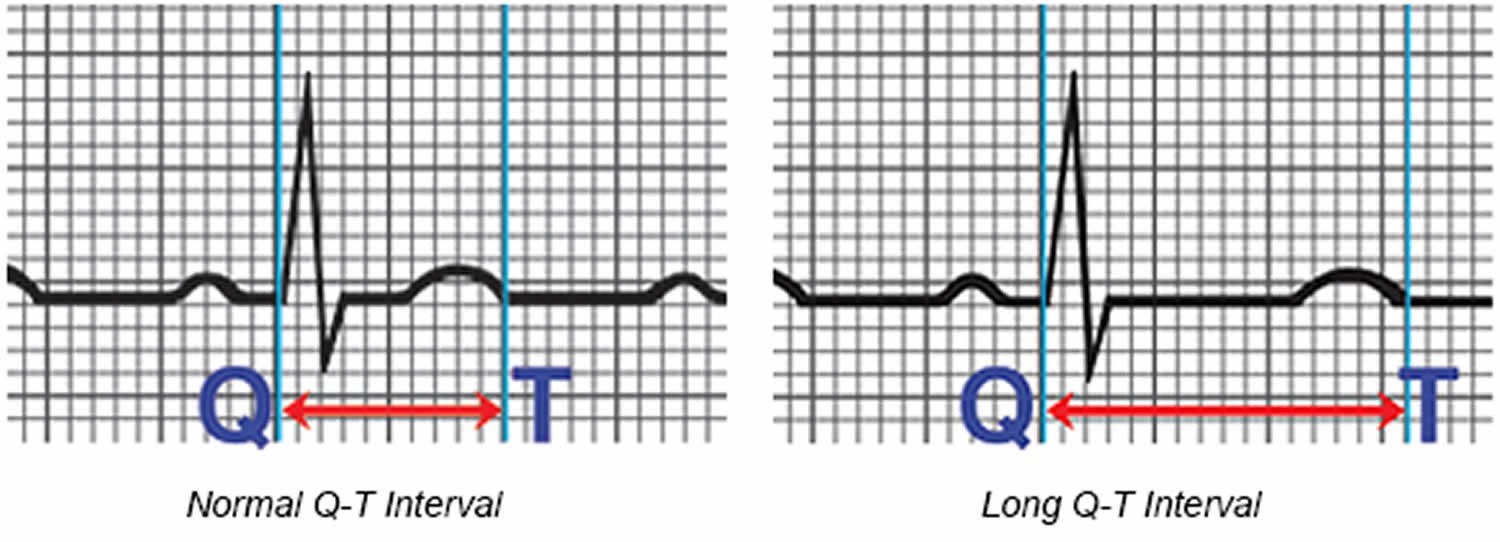
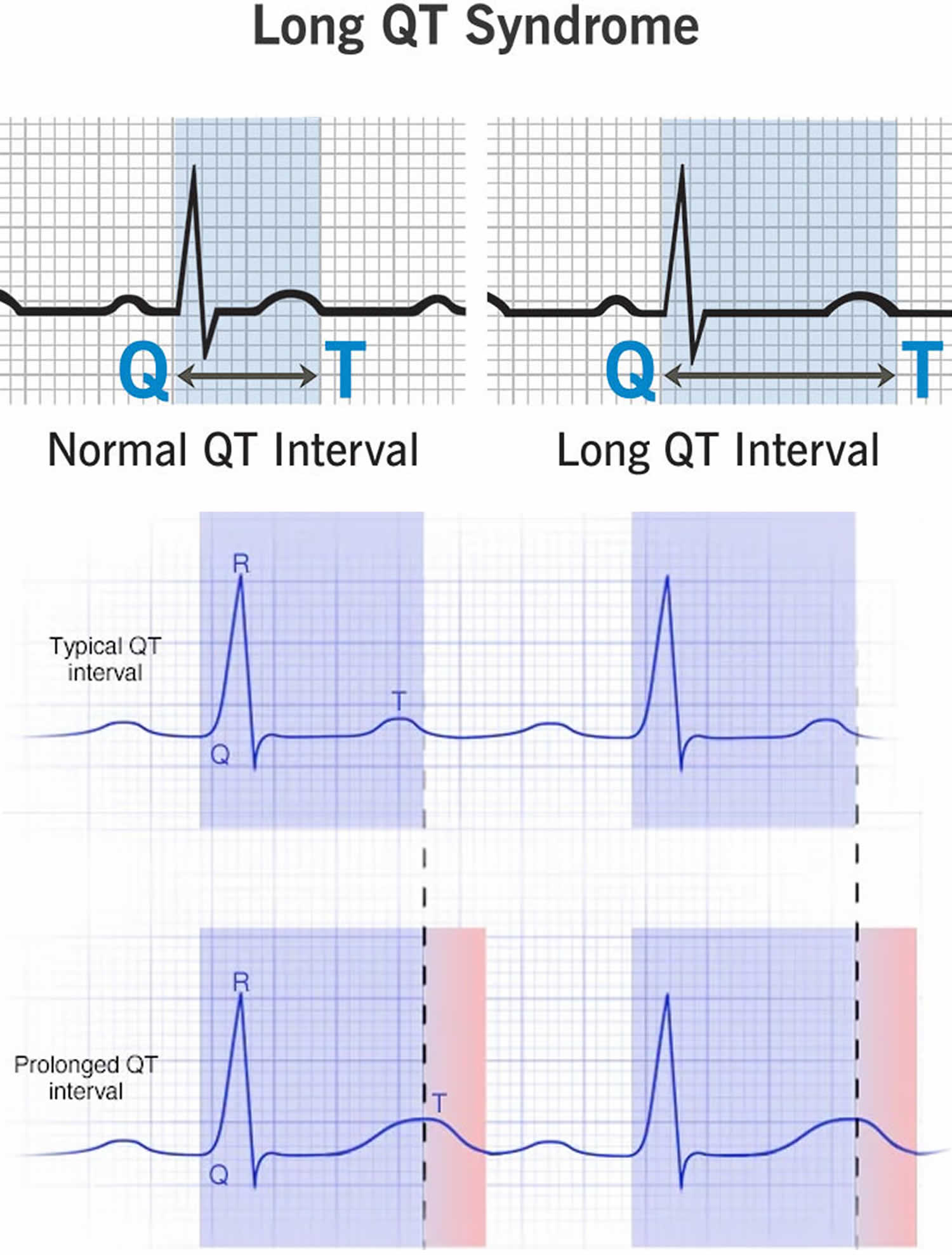
Figure 7. Normal ECG
What is the difference between Torsades de Pointes and ventricular fibrillation?
Torsades de Pointes (TdP) is actually ventricular tachycardia that happens in the setting of Long QT interval. During Torsades de Pointes, your electrocardiogram (ECG) have a specific pattern of ventricular tachycardia (VT) that looks like “twisting” or “oscillating” points or peaks on an electrocardiogram (ECG). Torsades de Pointes can lead to ventricular fibrillation, a dangerous heart rhythm where the heart’s lower chambers quiver erratically instead of pumping blood effectively, that can lead to sudden cardiac arrest and death if not treated immediately.
How common is Torsades de Pointes?
Researchers aren’t sure how many people get Torsades de Pointes because of side effects from medicines, but getting Long QT syndrome from medicines is common. This can put you at risk for Torsades de Pointes. Anywhere from one in 2,000 people to one in 20,000 people may have been born with a genetic problem that can lead to Torsades de Pointes.
Torsades de Pointes causes
Some people are born with Long QT syndrome, which can lead to Torsades de Pointes 36, 37. Prolongation of the QT interval may be congenital, as seen in the Jervell and Lange-Nielsen syndrome (ie, congenitally long QT associated with congenital deafness) and the Romano Ward syndrome (ie, isolated prolongation of QT interval) 10. Both of Jervell and Lange-Nielsen syndrome and the Romano Ward syndrome are associated with sudden death due to either primary ventricular fibrillation or Torsades de Pointes that degenerates into ventricular fibrillation.
Brugada syndrome is characterized by a coved ST segment in the right precordial leads 10. Brugada syndrome may cause sudden death due to polymorphic ventricular tachycardia resembling Torsades de Pointes.
Takotsubo cardiomyopathy, also known as “broken heart syndrome” or stress-induced cardiomyopathy, is a temporary heart condition triggered by intense emotional or physical stress. Takotsubo cardiomyopathy causes a part of the heart to weaken and change shape, similar to an octopus trap (tako-tsubo in Japanese), leading to symptoms like chest pain and shortness of breath and Torsades de Pointes 38, 39.
The acquired conditions that predispose someone to Torsades de Pointes include electrolyte disturbances that either decrease the outward potassium current or interfere with the inward sodium and calcium currents, or fluxes. The electrolyte disturbances that have been reported to precipitate torsade include low blood potassium (hypokalemia) and low blood magnesium (hypomagnesemia) 10. These electrolyte abnormalities cause a delay in phase III (ie, reprolongation) and form the substrate for emergence of the irregular heartbeat 10. Close observation is required in predisposed patients, such as those with cirrhosis of the liver or hypothyroidism (underactive thyroid).
Medications that are used to treat and prevent irregular heart rhythms or arrhythmias called antiarrhythmic drugs have been reported to cause Torsades de Pointes include class 1A agents (eg, quinidine, procainamide, disopyramide), class 1C agents (eg, flecainide [encainide was withdrawn in 1991]), and class 3 agents (eg, sotalol, amiodarone) 10.
Other drugs that prolong the QT interval and have been implicated in cases of Torsades de Pointes include phenothiazines (first-generation typical antipsychotic medications used for the treatment of psychosis, schizophrenia, bipolar disorders, control nausea and vomiting), tricyclic antidepressants, lithium carbonate, ziprasidone, cisapride, highly active antiretroviral drugs, high-dose methadone, anthracycline chemotherapeutic agents (eg, doxorubicin, daunomycin), some fluoroquinolones, and any other medication using the CYP3A metabolic pathway 12, 40, 10. Ranolazine, an antiangina agent, also prolongs the QTc, but Torsades is a rare complication of this therapy 41, 42, 43.. Often, multiple agents act synergistically. Because QT prolongation is highly variable among and even in a single individual, the specific “causative” agent is often speculative 10. Accordingly, lists of agents suspected of increasing the QT interval may include some drugs that do not have this particular effect 10.
Medicines that can cause Torsades de Pointes include 13, 44, 27, 28, 29:
- Antifungals.
- Antibiotics.
- Antipsychotics.
- Antiemetics (for nausea and vomiting).
- Antiarrhythmics.
- Cancer medicines.
A single-center, retrospective (2008-2019), observational study of the effects of 272 medications on 310,335 electrocardiograms (ECGs) from 159,397 individuals found that drugs associated with the greatest QT interval prolongation were dofetilide, mexiletine, amiodarone, rifaximin, and sotalol 44. However, the investigators also noted that several top drugs not previously known to prolong QT included rifaximin, lactulose, cinacalcet, and lenlidomide 44.
A 2023 case report described emergency department administration of a small dose (4 mg) of intravenous ondansetron resulting in QT prolongation, Torsades de pointes, and cardiac arrest in a middle-aged woman with alcohol disuse disorder who was not on any medications or supplements 45. Another 2023 case report described a rare presentation of levetiracetam-induced Torsades de pointes and cardiac arrest in a middle-age man with a history of a seizure disorder but noncompliance with valproic acid 46. The patient received a total of 1000 mg intravenous levetiracetam as well as 4 mg of intramuscular lorazepam for two sets of episodes of tonic-clonic seizures in the emergency department 46.
Risk factors for developing Torsades de Pointes
Risk factors for developing Torsades de Pointes include 6:
- Having Long QT syndrome that you inherited from your parents.
- Having a parent, brother, sister or child with long QT syndrome.
- Having heart disease.
- A history of cardiac arrest.
- Being a woman.
- Being older than 65.
- Using medicines known to cause prolonged QT intervals.
- Taking diuretics.
- Having profound bradycardia (very slow heart rate).
- Having low levels of calcium (hypocalcemia), magnesium (hypomagnesemia) or potassium (hypokalemia).
- Eating disorders such as anorexia nervosa, which also cause changes in the levels of body minerals.
- Having excessive diarrhea and vomiting, which can cause changes in body minerals such as potassium.
Torsades de Pointes Prevention
You can reduce your risk of Torsades de Pointes:
- Avoid medications that can cause Long QT interval and can put you at risk for Torsades de Pointes.
- Stop taking medicines that can cause Long QT syndrome. Your doctor can find an alternative.
- Increase your levels of calcium, magnesium and potassium if your doctor recommends it.
If Long QT syndrome runs in your family, your doctor can test your family members to see if they have it. With proper treatment, you can manage and prevent the dangerous heartbeats that can lead to Long QT syndrome complications.
Regular health checkups and good communication with your doctor also may help prevent causes of some types of acquired long QT syndrome. It’s especially important not to take medicines that can affect the heart rhythm and cause a prolonged QT interval.
Torsades de Pointes signs and symptoms
Half of the people with Torsades de Pointes don’t have any symptoms.
People with Torsades de Pointes who get symptoms can experience:
- Dizziness.
- Palpitations (feeling your heart pounding).
- Lightheadedness.
- Syncope (fainting).
- Cardiac arrest.
- Sudden cardiac death – an unexpected death due to cardiac causes that occurs in a short time period (generally within 1 hour of symptom onset) in a person with known or unknown cardiac disease.
Torsades de Pointes Complications
Complications of Torsades de Pointes include:
- Ventricular fibrillation. This type of irregular heartbeat causes the lower heart chambers to beat so fast that the heart trembles and stops pumping blood. Unless a device called a defibrillator is quickly used to correct the heart’s rhythm, brain damage and death can happen.
- Syncope (fainting).
- Sudden cardiac death. This is the swift and not expected ending of all heart activity. Torsades de Pointes has been linked to sudden cardiac death in young people who otherwise appear healthy. Long QT syndrome might be responsible for some unexplained events in children and young adults, such as unexplained fainting, drownings or seizures.
Torsades de Pointes diagnosis
Doctor can diagnose Torsades de Pointes on an electrocardiogram (EKG or ECG) 30, 47. Patients with Torsades de Pointes usually present with recurrent episodes of palpitations, dizziness, and syncope (fainting) that correspond to arrhythmia episodes; however, sudden cardiac death can occur with the first episode 48. Nausea, cold sweats, shortness of breath, and chest pain also may occur but are nonspecific and can be produced by any form of tachyarrhythmia 48.
In a young patient with Torsades de Pointes, a diagnosis of congenital long QT syndrome should be considered, especially if a family history of sudden cardiac death or sudden infant death syndrome is present 48. In these patients, episodes of Torsades de Pointes are triggered by adrenergic stimulation such as stress, fear, or physical exertion, but other predisposing factors also should be considered 49.
A family history of congenital deafness may also be suggestive, although a prolonged QT is found in only 0.25-0.3% of deaf-mute children 48. Patients with Jervell and Lange-Nielsen syndrome commonly have congenital sensorineural deafness representing an autosomal dominant pattern of inheritance for cardiac abnormalities, whereas deafness usually is autosomal recessive 48.
Another form of familial or congenital long QT syndrome is Romano-Ward syndrome, in which hearing is normal and an autosomal dominant pattern of inheritance is observed 48.
Patients with acquired long QT syndrome usually develop Torsades de Pointes during periods of bradycardia. The most common causes of acquired long QT syndrome are medications and electrolyte disorders (eg, hypokalemia, hypomagnesemia, hypocalcemia). Drug-associated Torsades de Pointes is relatively rare, but is becoming increasingly common; its incidence is as high as 2-3% with certain drugs 44, 6. Hence, asking your patient about all current medications is important.
Tests to diagnose Torsades de Pointes include:
- Electrocardiogram (EKG or ECG). An ECG is the most common test used to diagnose Torsades de Pointes and long QT syndrome (LQTS). It records the electrical signals in the heart and shows how fast or how slow the heart is beating. Sticky patches called electrodes attach to the chest and sometimes the arms and legs. Wires connect the electrodes to a computer, which prints or displays the test results. The heart’s signals are shown as waves on the test results.
- Blood tests to check electrolyte levels.
- Echocardiogram.
- A heart monitor you wear at home.
- Holter monitor. This small, portable ECG device records the heart’s activity. It’s worn for a day or two while you do your regular activities.
- Event recorder. This device is like a Holter monitor, but it records only at certain times for a few minutes at a time. It’s typically worn for about 30 days. You usually push a button when you feel symptoms. Some devices automatically record when an irregular heart rhythm is detected.
- Genetic testing. A genetic test is available to confirm long QT syndrome (LQTS). The test checks for gene changes that can cause long QT syndrome (LQTS). If you have long QT syndrome, your heart doctor may suggest that other family members also get genetic testing to check for the disorder. Genetic tests for long QT syndrome can’t find all inherited cases of long QT syndrome. It’s recommended that families speak to a genetic counselor before and after testing.
Physical examination
The physical findings in Torsades de Pointes depend on the rate and duration of tachycardia and the degree of cerebral hypoperfusion 48. Findings include rapid pulse, low or normal blood pressure, or transient or prolonged loss of consciousness 48. This could be preceded by bradycardia or premature ventricular contractions (PVC) 48. An increase in premaure ventricular contractions in individuals with prolonged QTc may be useful in predicting imminent Torsades de Pointes 50.
Pallor and diaphoresis may be noted, especially with a sustained episode.
Other physical signs depend on the cause of Torsades de Pointes.
Electrocardiographic (ECG)
Torsades de Pointes is an electrocardiographic (ECG) diagnosis, and obtaining an ECG is essential.
Frequent ECG monitoring is indicated for patients who are at risk due to chronic conditions or drug therapy. When the patient is in sinus rhythm, examine the QT interval. Usually, a prolonged QT interval and pathological U waves are present, reflecting abnormal ventricular repolarization 30. The most consistent indicator of QT prolongation is a QT of 0.60 second or longer or a QTc (corrected for heart rate) of 0.45 second or longer 30.
Other electrocardiographic features helpful in diagnosing Torsades de Pointes include its typical mode of onset and its morphology, as follows 30:
- Patients have paroxysms of 5-20 beats at a rate faster than 200 bpm; sustained episodes occasionally can be seen
- Progressive change in polarity of QRS about the isoelectric line occurs
- Complete 180° twist of QRS complexes in 10-12 beats is present
- A short-long-short sequence between the R-R intervals occurs before the trigger response.
- Torsades de Pointes may revert spontaneously or convert to a nonpolymorphic ventricular tachycardia or ventricular fibrillation
- Occasionally, T-wave alternans may be seen before Torsades de Pointes.
Torsades de Pointes occurring in the setting of acquired long QT syndrome is preceded by pauses in almost all cases 30. In congenital long QT syndrome (adrenergic-dependent), pause dependence is found in most of the adult cases, whereas onset of Torsades de Pointes in children is not pause-dependent 30.
Failure to identify Torsades de Pointes may occur for various reasons. During very short runs of Torsades de Pointes, the typical oscillatory QRS complexes around the isoelectric line may not be apparent. Early events usually are short lived. In the case of a single-lead recording, the typical morphology of Torsades de Pointes may not be obvious.
The diagnosis of Torsades de Pointes should be considered in any patient with pause-dependent ventricular tachycardia, and ventricular bigeminy in a patient with long QT interval may be a sign of an impending Torsades de Pointes 30.
Findings from electrophysiological studies usually are negative in Torsades de Pointes 30.
Other Tests
Other tests, including lab and imaging studies, should be ordered based on the underlying causal factors 51:
- Laboratory studies
- Electrolytes: Check for hypoglycemia, hypokalemia, hypomagnesemia, and hypocalcemia.
- Cardiac enzymes: Rule out myocardial ischemia, especially in patients without QT prolongation.
- Imaging studies
- Chest radiographs and echocardiography should be performed to rule out structural heart disease, if any clinical suggestion is present.
Torsades de Pointes Differential Diagnosis
The differential diagnosis of Torsades de Pointes includes 52:
- Ventricular Tachycardia
- Atrial Tachycardia
- Syncope
- Dialysis Complications of Chronic Renal Failure
- Antidysrhythmic Toxicity
- Cough, Cold, and Allergy Preparation Toxicity. Because these medications are available over the counter (OTC) and are found in most households, they are frequently implicated in toxic ingestions, particularly in children. Antihistamines and cough and cold preparations, respectively, rank seventh and twenty-third on the list of substance categories most frequently involved in human exposures in the United States 53.
- Ventricular Fibrillation
- Sudden Cardiac Death – an unexpected death due to cardiac causes that occurs in a short time period (generally within 1 hour of symptom onset) in a person with known or unknown cardiac disease
Other considerations are the differentiation of acquired long QT syndrome from congenital long QT syndrome. In addition, Torsades de Pointes should be differentiated from polymorphic ventricular tachycardia or rarely, monomorphic ventricular tachycardia.
Supraventricular tachycardia (SVT) with aberrant conduction may be confused with Torsades de Pointes, especially when the degree of aberration is variable. One clue to supraventricular tachycardia (SVT) is that atrial fibrillation may be intermixed with narrower and typical QRS complexes.
Torsades de Pointes treatment
Treatment of Torsades de Pointes can be divided into short-term or immediate management and long-term management 24. The goal of short-term management or immediate management is to assess for, achieve, and maintain hemodynamic stability. Short-term management or immediate management of Torsades de Pointes is the same in both acquired and congenital long QT syndrome, except that beta1-adrenergic stimulation may be tried in the acquired form but is contraindicated in the congenital form 24.
In an otherwise stable patient, direct current (DC) cardioversion is kept as a last resort, because Torsades de Pointes is paroxysmal in nature and is characterized by its frequent recurrences following cardioversion 24. Although Torsades de Pointes frequently is self-terminating, it may degenerate into ventricular fibrillation, which requires direct current (DC) defibrillation 24.
Any offending agent should be withdrawn. Predisposing conditions such as hypokalemia, hypomagnesemia, and bradycardia should be identified and corrected 24.
Medical therapy
Magnesium is the drug of choice for suppressing early afterdepolarizations and terminating Torsades de Pointes arrhythmia 54, 24, 55, 56. Magnesium achieves this by decreasing the influx of calcium, thus lowering the amplitude of early afterdepolarizations 57. Magnesium can be given at 1 to 2 g IV initially in 30-60 seconds, which then can be repeated in 5-15 minutes 24. Alternatively, a continuous magnesium infusion can be started at a rate of 3-10 mg/minute 24. Magnesium is effective even in patients with normal magnesium levels. Because of the danger of hypermagnesemia (depression of neuromuscular function), the patient requires close monitoring 24.
Some authorities recommend supplemental potassium to increase the potassium concentration to high normal, which increases the efflux of potassium from myocardial cells, thus causing rapid repolarization 24.
Lidocaine usually has no effect in Torsades de Pointes. Occasionally, lidocaine can have an initial beneficial effect, but Torsades de Pointes recurs in all cases 24, 54.
Mexiletine also may be helpful in suppressing Torsades de Pointes 24. In one study, mexiletine was used in patients with HIV who had acquired long QT interval and Torsades de Pointes 58. Mexiletine effectively suppressed the torsade on a long-term basis 58. Nakashima et al 59 reported Mexiletine successfully preventing refractory Torsades de Pointes and ventricular fibrillation in a 28-year-old patient with congenital type 2 long QT syndrome.
Patients with congenital long QT syndromes are thought to have an abnormality of sympathetic balance or tone and are treated with beta-blockers 24. If the patient experiences breakthrough Torsades de Pointes, a short-acting beta-blocker, such as esmolol, can be tried 60.
Isoproterenol can be used in bradycardia-dependent Torsades de Pointes that usually is associated with acquired long QT syndrome (pause-dependent) 24. Isoproterenol should be administered as a continuous IV infusion to keep the heart rate above 90 bpm 24.
Isoproterenol accelerates AV conduction and decreases the QT interval by increasing the heart rate and reducing temporal dispersion of repolarization 24. Beta-adrenergic agonists such as isoproterenol are contraindicated in the congenital form of long QT syndrome (adrenergic-dependent) 24. Because of precautions, contraindications, and adverse effects associated with its use, Isoproterenol is used as an interim agent until overdrive pacing can be started 24.
Temporary transvenous pacing
Based on the fact that the QT interval shortens with a faster heart rate, pacing can be effective in terminating Torsades de Pointes 24. Pacing is effective in both forms of the long QT syndrome because it facilitates the repolarizing potassium currents and prevents long pauses, suppressing early afterdepolarizations and decreasing the QT interval.
Atrial pacing is the preferred mode because it preserves the atrial contribution to ventricular filling and also results in a narrower QRS complex and hence a shorter QT 24. In patients with AV block, ventricular pacing can be used to suppress Torsades de Pointes 24. This is dependent on intact atrial-to-ventricular conduction at the pacing rate found necessary.
Pacing should be instituted at a rate of 90-110 bpm until the QT interval is normalized. Overdrive pacing may be necessary at a rate of up to 140 bpm to control the rhythm 24.
Patient with Torsades de Pointes who is in extremis should be treated with electrical cardioversion or defibrillation 24. Anecdotal reports cite successful conversion with phenytoin (Dilantin) and lidocaine. A few cases of successful conversion using phenytoin and overdrive pacing have been reported 24.
If patient is unresponsive to cardioconversion with phenytoin and overdrive pacing, attempt electrical cardioversion 24.
Long-term treatment
Beta-adrenergic antagonists (beta blockers) at maximally tolerated doses are used as a first-line long-term therapy in congenital long QT syndrome 24. Propranolol is used most extensively, but other agents such as esmolol or nadolol also can be used 24. Beta-blockers should be avoided in those congenital cases in which bradycardia is a prominent feature 24. Beta-blockers are contraindicated in acquired long QT syndrome because bradycardia produced by these agents can precipitate Torsades de Pointes 24. One approach to assess the adequacy of beta-blockade is by exercise testing. One investigator recommends aiming for at least a 20% reduction in maximum heart rate compared to that of the baseline (pre-beta blocker therapy). Another approach is to check the blood levels of beta blockers (eg, propranolol) when possible 61.
Patients without syncope, ventricular tachyarrhythmia, or a family history of sudden cardiac death can be observed without starting any treatment 24.
Permanent pacing benefits patients who remain symptomatic despite receiving the maximally tolerated dose of beta-blockers and can be used adjunctively with beta-blockers 24. Permanent pacing decreases the QT interval by enhancing the repolarizing potassium currents and suppressing early afterdepolarizations 24.
High left thoracic sympathectomy or left cardiac sympathetic denervation (LCSD) surgery, another antiadrenergic therapy, is effective in patients who remain refractory to beta-blockade and pacing 24. This surgery may be done if you have long QT syndrome and continuing heart rhythm changes but beta blockers don’t work for you. It doesn’t cure long QT syndrome. Instead, the surgery helps lower the risk of sudden cardiac death. In this treatment, surgeons remove specific nerves along the left side of the spine. These nerves are part of the body’s sympathetic nervous system, which helps control the heart rhythm. Accidental ablation of ocular efferent sympathetic nerves may result in Horner syndrome 24.
Implantable cardioverter-defibrillators (ICDs) are useful in instances when Torsades de pointes recurs despite treatment with beta-blockers, pacing, and possibly left thoracic sympathectomy 24. Beta-blockers should be used along with implantable cardioverter-defibrillators (ICDs) because shock can further precipitate Torsades de pointes by adrenergic stimulation. In the United States, an implantable cardioverter-defibrillator (ICD) for refractory cases may often precede sympathectomy 24. The implantable cardioverter-defibrillator (ICD) device is placed under your skin near the collarbone. Placing an implantable cardioverter-defibrillator (ICD) requires surgery. It continuously checks the heart rhythm. If the device finds an irregular heartbeat, it sends out low- or high-energy shocks to reset the heart’s rhythm.
Most people with long QT syndrome don’t need an implantable cardioverter-defibrillator (ICD). But the device may be suggested for some athletes to help them return to competitive sports. The decision to place an implantable cardioverter-defibrillator (ICD), especially in children, needs to be carefully considered. Sometimes the device may send out shocks that aren’t needed. Talk with your heart specialist about the benefits and risks of an implantable cardioverter-defibrillator (ICD).
Long-term treatment in acquired long QT syndrome usually is not required because the QT interval returns to normal once the inciting factor or predisposing condition has been corrected 24. Pacemaker implantation is effective in cases that are associated with heart block or bradycardia 24. Implantable cardioverter-defibrillators (ICDs) are indicated in cases that cannot be managed by avoidance of the offending agent.
The boundary between acquired and congenital may not always be clear. Additive factors are often present, and individuals may show increased susceptibility to QT effects.
Exercise and physical activity warning
Competitive sports are prohibited in patients with congenital long QT syndrome 24.
Complications of Torsades de Pointes may include:
- Monomorphic ventricular tachycardia
- Ventricular fibrillation
- Sudden cardiac death.
Living with Torsades de Pointes
There are several things you should do to take care of yourself with Torsades de Pointes.
- If you inherited Long QT syndrome from a parent, you should talk to a heart rhythm specialist before your start exercising.
- Keep taking your medicines to keep Torsades de Pointes from happening again.
- Learn how to take your pulse and what’s normal.
- Eat a diet rich in magnesium and potassium.
- Drink plenty of fluids.
- If your heart doctor gave you a heart monitor to use, be sure to wear it.
- You should go to the emergency room (ER) if you have a very fast pulse rate or if you feel palpitations, dizziness or lightheadedness or you get fainting episodes.
Torsades de Pointes prognosis
Without treatment, Torsades de Pointes can keep coming back or may lead to ventricular fibrillation, which can be deadly. With treatment, your chances of survival are good, especially if you stop taking the medicine that caused Long QT interval.
In congenital long QT syndrome, the mortality rate for untreated patients is 50% in 5 years, which can be reduced to 3-4% with therapeutic intervention 62, 63. A systematic review of babies born with long QT syndrome from 83 studies comprising 265 newborns with postnatal confirmation of long QT syndrome found that a longer fetal QTc was more predictive of death than any other antenatal factor, and the mortality risk was significantly raised when the fetal QTc was longer than 600 ms 25. Other factors that were highly predictive of death included the combination of ventricular tachycardia/Torsade de Pointes or functional 2:1 heart block and lack of a family history of long QT syndrome 25. However, fetal heart rate and heart z-score did not predict death 25.
In acquired long QT syndrome, the prognosis is excellent once the inciting factor has been identified and discontinued 3.
- Dessertenne F. La tachycardie ventriculaire à deux foyers opposés variables [Ventricular tachycardia with 2 variable opposing foci]. Arch Mal Coeur Vaiss. 1966 Feb;59(2):263-72. French.[↩][↩]
- Kahlon SS, Sikandar R, Tejovath S, Nair S, Hassan D, K Patel K, Peddemul A, Mostafa JA. Diagnosing Torsades De Pointes Based on Correlation to QT Interval: A Systematic Review. Cureus. 2022 Aug 9;14(8):e27833. doi: 10.7759/cureus.27833[↩]
- Cohagan B, Brandis D. Torsade de Pointes. [Updated 2023 Aug 8]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK459388[↩][↩][↩][↩]
- Torsade de Pointes. https://emedicine.medscape.com/article/1950863-overview[↩]
- Viskin S. Long QT syndromes and torsade de pointes. Lancet. 1999 Nov 06;354(9190):1625–1633. doi: 10.1016/S0140-6736(99)02107-8.S0140-6736(99)02107-8[↩]
- Drew BJ, Ackerman MJ, Funk M, Gibler WB, Kligfield P, Menon V, Philippides GJ, Roden DM, Zareba W; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology; Council on Cardiovascular Nursing; American College of Cardiology Foundation. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. J Am Coll Cardiol. 2010 Mar 2;55(9):934-47. doi: 10.1016/j.jacc.2010.01.001[↩][↩][↩][↩][↩][↩]
- Niemeijer MN, van den Berg ME, Deckers JW, Franco OH, Hofman A, Kors JA, Stricker BH, Rijnbeek PR, Eijgelsheim M. Consistency of heart rate-QTc prolongation consistency and sudden cardiac death: the Rotterdam Study. Heart Rhythm. 2015 Oct;12(10):2078–2085. doi: 10.1016/j.hrthm.2015.07.011.S1547-5271(15)00893-0[↩]
- Davies RA, Ladouceur VB, Green MS, Joza J, Juurlink DN, Krahn AD, McMurtry MS, Roberts JD, Roston TM, Sanatani S, Steinberg C, MacIntyre C. The 2023 Canadian Cardiovascular Society Clinical Practice Update on Management of the Patient With a Prolonged QT Interval. Can J Cardiol. 2023 Oct;39(10):1285-1301. doi: 10.1016/j.cjca.2023.06.011[↩]
- Groffen AJ, Bikker H, Christiaans I. Long QT Syndrome Overview. 2003 Feb 20 [Updated 2024 Mar 21]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1129[↩]
- Torsade de Pointes. https://emedicine.medscape.com/article/1950863-overview#a5[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Lee DH, Money DB, Deshpande A, Samuels B. A Case of Abiraterone-Related Hypokalemia Leading to Torsades de Pointes and Cardiac Arrest. Cureus. 2022 Mar 31;14(3):e23678. doi: 10.7759/cureus.23678[↩]
- Alipour A, Cruz R, Lott RS. Torsade de pointes after ziprasidone overdose with coingestants. J Clin Psychopharmacol. 2010 Feb;30(1):76-7. doi: 10.1097/JCP.0b013e3181c914d3[↩][↩]
- Schwartz PJ, Woosley RL. Predicting the Unpredictable: Drug-Induced QT Prolongation and Torsades de Pointes. J Am Coll Cardiol. 2016 Apr 5;67(13):1639-1650. doi: 10.1016/j.jacc.2015.12.063[↩][↩][↩]
- Nikolic G, Bishop RL, Singh JB. Sudden death recorded during Holter monitoring. Circulation. 1982 Jul;66(1):218-25. doi: 10.1161/01.cir.66.1.218[↩]
- Torsade de Pointes Epidemiology. https://emedicine.medscape.com/article/1950863-overview#a3[↩][↩][↩][↩]
- Lehmann MH, Timothy KW, Frankovich D, Fromm BS, Keating M, Locati EH, Taggart RT, Towbin JA, Moss AJ, Schwartz PJ, Vincent GM. Age-gender influence on the rate-corrected QT interval and the QT-heart rate relation in families with genotypically characterized long QT syndrome. J Am Coll Cardiol. 1997 Jan;29(1):93-9. doi: 10.1016/s0735-1097(96)00454-8[↩]
- Evaluation of drug-induced QT interval prolongation: implications for drug approval and labelling. Malik M, Camm AJ. Drug Saf. 2001;24:323–351. doi: 10.2165/00002018-200124050-00001[↩]
- Clinical practice. Long-QT syndrome. Roden DM. N Engl J Med. 2008;358:169–176. doi: 10.1056/NEJMcp0706513[↩]
- The long QT syndrome. Prospective longitudinal study of 328 families. Moss AJ, Schwartz PJ, Crampton RS, et al. Circulation. 1991;84:1136–1144. doi: 10.1161/01.cir.84.3.1136[↩]
- Krahn AD, Laksman Z, Sy RW, Postema PG, Ackerman MJ, Wilde AAM, Han HC. Congenital Long QT Syndrome. JACC Clin Electrophysiol. 2022 May;8(5):687-706. doi: 10.1016/j.jacep.2022.02.017[↩]
- Lankaputhra M, Voskoboinik A. Congenital long QT syndrome: a clinician’s guide. Intern Med J. 2021 Dec;51(12):1999-2011. doi: 10.1111/imj.15437[↩]
- Long QT Syndrome. https://rarediseases.org/rare-diseases/romano-ward-syndrome/[↩]
- Long QT syndrome. https://www.mayoclinic.org/diseases-conditions/long-qt-syndrome/symptoms-causes/syc-20352518[↩][↩]
- Treatment of Torsades de Pointes. https://emedicine.medscape.com/article/1950863-overview#a9[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Chivers S, Ovadia C, Regan W, Zidere V, Vigneswaran T, Sharland G, Rosenthal E, Seed PT, Simpson JM, Williamson C. Systematic review of long QT syndrome identified during fetal life. Heart Rhythm. 2023 Apr;20(4):596-606. doi: 10.1016/j.hrthm.2022.12.026[↩][↩][↩][↩][↩][↩]
- Jackobson G, Carmel NN, Lotan D, Kremer A, Justo D. Reckless administration of QT interval-prolonging agents in elderly patients with drug-induced torsade de pointes. Z Gerontol Geriatr. 2018 Jan;51(1):41-47. English. doi: 10.1007/s00391-016-1155-5[↩][↩]
- Salem M, Reichlin T, Fasel D, Leuppi-Taegtmeyer A. Torsade de pointes and systemic azole antifungal agents: Analysis of global spontaneous safety reports. Glob Cardiol Sci Pract. 2017 Jun 30;2017(2):11. doi: 10.21542/gcsp.2017.11[↩][↩]
- Porta-Sánchez A, Gilbert C, Spears D, Amir E, Chan J, Nanthakumar K, Thavendiranathan P. Incidence, Diagnosis, and Management of QT Prolongation Induced by Cancer Therapies: A Systematic Review. J Am Heart Assoc. 2017 Dec 7;6(12):e007724. doi: 10.1161/JAHA.117.007724[↩][↩]
- Heemskerk CPM, Pereboom M, van Stralen K, Berger FA, van den Bemt PMLA, Kuijper AFM, van der Hoeven RTM, Mantel-Teeuwisse AK, Becker ML. Risk factors for QTc interval prolongation. Eur J Clin Pharmacol. 2018 Feb;74(2):183-191. doi: 10.1007/s00228-017-2381-5[↩][↩]
- Torsade de Pointes Electrocardiography. https://emedicine.medscape.com/article/1950863-overview#a7[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- https://commons.wikimedia.org/wiki/File:Rhythm_torsade_(CardioNetworks_ECGpedia).png[↩]
- https://commons.wikimedia.org/wiki/File:Torsades_de_Pointes_TdP.png[↩]
- Electrocardiogram (EKG, ECG). https://cvphysiology.com/arrhythmias/a009[↩][↩]
- Al-Akchar M, Siddique MS. Long QT Syndrome. [Updated 2022 Dec 26]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441860[↩]
- Fukuyama M., Horie M., Aoki H., et al. School-based routine screenings of electrocardiograms for the diagnosis of long QT syndrome. Europace. 2022;24:1496–1503. doi: 10.1093/europace/euab320[↩]
- Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012 Aug 1;5(4):868-77. doi: 10.1161/CIRCEP.111.962019. Erratum in: Circ Arrhythm Electrophysiol. 2012 Dec;5(6):e119-20[↩]
- Schwartz PJ, Ackerman MJ. The long QT syndrome: a transatlantic clinical approach to diagnosis and therapy. Eur Heart J. 2013 Oct;34(40):3109-16. doi: 10.1093/eurheartj/eht089[↩]
- Pacha O, Kadikoy H, Amro M, Haque W, Abdellatif A. Torsades de pointes and prolonged QT syndrome in Takotsubo cardiomyopathy. J Cardiovasc Med (Hagerstown). 2012 Aug;13(8):536-40. doi: 10.2459/JCM.0b013e328336b4a6[↩]
- Komamura K, Fukui M, Iwasaku T, Hirotani S, Masuyama T. Takotsubo cardiomyopathy: Pathophysiology, diagnosis and treatment. World J Cardiol. 2014 Jul 26;6(7):602-9. doi: 10.4330/wjc.v6.i7.602[↩]
- Kim PY, Ewer MS. Chemotherapy and QT Prolongation: Overview With Clinical Perspective. Curr Treat Options Cardiovasc Med. 2014 May;16(5):303. doi: 10.1007/s11936-014-0303-8[↩]
- Dobesh PP, Trujillo TC. Ranolazine: a new option in the management of chronic stable angina. Pharmacotherapy. 2007 Dec;27(12):1659-76. doi: 10.1592/phco.27.12.1659[↩]
- De Vecchis R, Ariano C, Di Biase G, Noutsias M. Acquired drug-induced long QTc: new insights coming from a retrospective study. Eur J Clin Pharmacol. 2018 Dec;74(12):1645-1651. doi: 10.1007/s00228-018-2537-y[↩]
- Salem JE, Dureau P, Bachelot A, Germain M, Voiriot P, Lebourgeois B, Trégouët DA, Hulot JS, Funck-Brentano C. Association of Oral Contraceptives With Drug-Induced QT Interval Prolongation in Healthy Nonmenopausal Women. JAMA Cardiol. 2018 Sep 1;3(9):877-882. doi: 10.1001/jamacardio.2018.2251[↩]
- Yuan N, Oesterle A, Botting P, Chugh S, Albert C, Ebinger J, Ouyang D. High-Throughput Assessment of Real-World Medication Effects on QT Interval Prolongation: Observational Study. JMIR Cardio. 2023 Jan 20;7:e41055. doi: 10.2196/41055[↩][↩][↩][↩]
- Orozco BS, Lee SC, Fuchs RT, Fushianes GD, Cole JB. QT prolongation, torsades des pointes, and cardiac arrest after 4 mg of IV ondansetron. Am J Emerg Med. 2023 Jun;68:214.e3-214.e6. doi: 10.1016/j.ajem.2023.04.003[↩]
- Mann H, Kusayev J, Pandey S, Aryal B, Solaimanzadeh I. A Rare Presentation of Levetiracetam-Induced Torsades De Pointes. Cureus. 2023 Jun 23;15(6):e40866. doi: 10.7759/cureus.40866[↩][↩]
- Moskovitz JB, Hayes BD, Martinez JP, Mattu A, Brady WJ. Electrocardiographic implications of the prolonged QT interval. Am J Emerg Med. 2013 May;31(5):866-71. doi: 10.1016/j.ajem.2012.12.013[↩]
- Torsade de Pointes Presentation. https://emedicine.medscape.com/article/1950863-overview#a4[↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Cho Y. Management of Patients with Long QT Syndrome. Korean Circ J. 2016 Nov;46(6):747-752. doi: 10.4070/kcj.2016.46.6.747[↩]
- Marill KA, Lopez S, Hark D, Spahr J, Kapadia N, Liu SW. Increased ventricular ectopy precedes Torsades de Pointes in patients with prolonged QT. J Electrocardiol. 2023 Sep-Oct;80:17-23. doi: 10.1016/j.jelectrocard.2023.04.003[↩]
- Torsade de Pointes Other Tests. https://emedicine.medscape.com/article/1950863-overview#a8[↩]
- Torsade de Pointes Differential Diagnosis. https://emedicine.medscape.com/article/1950863-overview#a6[↩]
- Gummin DD, Mowry JB, Beuhler MC, Spyker DA, Bronstein AC, Rivers LJ, Pham NPT, Weber J. 2020 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 38th Annual Report. Clin Toxicol (Phila). 2021 Dec;59(12):1282-1501. doi: 10.1080/15563650.2021.1989785[↩]
- Thomas SH, Behr ER. Pharmacological treatment of acquired QT prolongation and torsades de pointes. Br J Clin Pharmacol. 2016;81(3):420-427. doi: 10.1111/bcp.12726[↩][↩]
- Jaiswal A, Goldbarg S. Dofetilide induced torsade de pointes: mechanism, risk factors and management strategies. Indian Heart J. 2014;66(6):640-648. doi: 10.1016/j.ihj.2013.12.021[↩]
- Khan IA. Long QT syndrome: diagnosis and management. Am Heart J. 2002 Jan;143(1):7-14. doi: 10.1067/mhj.2002.120295[↩]
- Baker WL. Treating arrhythmias with adjunctive magnesium: identifying future research directions. Eur Heart J Cardiovasc Pharmacother. 2017 Apr 1;3(2):108-117. doi: 10.1093/ehjcvp/pvw028[↩]
- Shimabukuro-Vornhagen A, Rybniker J, Zoghi S, Faetkenheuer G, Michels G, Erdmann E, von Bergwelt-Baildon M, Kochanek M. Acquired Long QT Syndrome and Torsade de Pointes Associated with HIV Infection. Case Rep Med. 2010;2010:278427. doi: 10.1155/2010/278427[↩][↩]
- Nakashima R, Takase S, Kai K, Sakamoto K, Tsutsui H. Mexiletine effectively prevented refractory Torsades de Pointes and ventricular fibrillation in a patient with congenital type 2 long QT syndrome. J Cardiovasc Electrophysiol. 2022 Jul;33(7):1592-1595. doi: 10.1111/jce.15517[↩]
- Kaye AD, Volpi-Abadie J, Bensler JM, Kaye AM, Diaz JH. QT interval abnormalities: risk factors and perioperative management in long QT syndromes and Torsades de Pointes. J Anesth. 2013 Aug;27(4):575-87. doi: 10.1007/s00540-013-1564-1[↩]
- Shah M, Carter C. Long QT syndrome: A therapeutic challenge. Ann Pediatr Cardiol. 2008 Jan;1(1):18-26. doi: 10.4103/0974-2069.41051[↩]
- Tse G, Gong M, Meng L, Wong CW, Bazoukis G, Chan MTV, Wong MCS, Letsas KP, Baranchuk A, Yan GX, Liu T, Wu WKK. Predictive Value of T peak – T end Indices for Adverse Outcomes in Acquired QT Prolongation: A Meta-Analysis. Front Physiol. 2018 Sep 3;9:1226. doi: 10.3389/fphys.2018.01226[↩]
- Ramalho D, Freitas J. Drug-induced life-threatening arrhythmias and sudden cardiac death: A clinical perspective of long QT, short QT and Brugada syndromes. Rev Port Cardiol (Engl Ed). 2018 May;37(5):435-446. English, Portuguese. doi: 10.1016/j.repc.2017.07.010[↩]


{kind=link}