
Werdnig Hoffmann disease
Werdnig Hoffmann disease is now called spinal muscular atrophy type 1 or severe infantile spinal muscular atrophy, is a genetic neuromuscular disorder that affects the nerve cells that control voluntary muscles (motor neurons) 1. Werdnig Hoffmann disease or spinal muscular atrophy type 1 is the most severe form of spinal muscular atrophy. It accounts for between 50 – 70% of cases of childhood onset spinal muscular atrophy. Without treatment, symptoms of Werdnig Hoffmann disease become apparent before 6 months of age and include worsening muscle weakness and poor muscle tone (hypotonia) due to loss of the lower motor neurons in the spinal cord and brain stem. Feeding and breathing problems are also present 2. Werdnig Hoffmann disease is caused by changes (pathogenic variants also called mutations) in the SMN1 gene and is typically inherited in an autosomal recessive manner 3.
Each baby with Werdnig Hoffmann disease or spinal muscular atrophy type 1 is different. The symptoms of spinal muscular atrophy type 1 usually appear within the first few months of life. In some cases spinal muscular atrophy can affect babies even before birth and mothers may remember that their baby had become less active towards the end of their pregnancy. Generally, the earlier the onset of symptoms, the more severe the condition. The most severely affected babies may die before, during, or very shortly after birth. This is sometimes referred to as spinal muscular atrophy Type 0.
Babies with spinal muscular atrophy type 1 or Werdnig Hoffmann disease are often described as ‘floppy’ babies, due to their low muscle tone (hypotonia) and severe muscle weakness. The muscle weakness affects movement, swallowing and breathing. Babies with Werdnig Hoffmann disease or spinal muscular atrophy type 1 are unable to lift their heads, have difficulty rolling over and are unable to sit unsupported. They may also have a weak cry.
In spinal muscular atrophy, muscle weakness is usually symmetrical. The muscles close to the centre of the body (proximal muscles) are usually affected more severely than the muscles situated away from the center of the body (distal muscles) and the legs are generally weaker than the arms. This results in babies having difficulty lifting their arms and legs, while still being able to use their hands and fingers.
The brain is unaffected and babies affected by spinal muscular atrophy are often described as bright, alert and responsive. The muscles of the face are not usually affected so babies can smile and frown.
Weakness of the respiratory muscles can cause severe difficulties with breathing and coughing. It can also increase susceptibility to respiratory viruses and infections which can be life-threatening.
The muscles used for sucking and swallowing are also affected and this may cause difficulty with feeding and weight gain. Difficulty with swallowing can increase the risk of fluids, or food, passing into the lungs (aspiration) which can cause choking and, in some cases,pneumonia.
Werdnig-Hoffmann disease is a rare disorder that affects males and females in equal numbers. The prevalence of all types of spinal muscular atrophy has been estimated to be 4-7.8 per 100,000 live births. Approximately 80% of spinal muscular atrophy patients have the Werdnig-Hoffmann form.
Diagnosis of Werdnig Hoffmann disease or spinal muscular atrophy type 1 is suspected by symptoms and confirmed by genetic testing. Spinal muscular atrophy has been added to the list of recommended newborn screening tests in the United States, so that it can be detected prior to symptoms developing. This occurred because treatments are being developed that are changing the course of the disease. In December 2016, nusinersen (Spinraza) became the first FDA approved treatment for Werdnig Hoffmann disease. Continued treatment with nusinersen (Spinraza) is allowing many babies with Werdnig Hoffmann disease to reach and maintain age appropriate developmental milestones, including sitting, crawling, and walking. On average, breathing problems, nutrition problems, and hospital admissions have also decreased. However, response to treatment does vary. Some babies with Werdnig Hoffmann disease may not respond to the nusinersen at all or may have medical complications that prevent use of the treatment 4. Other treatments remain supportive 5.
Is there a cure for Werdnig Hoffmann disease?
There is no cure for Werdnig Hoffmann disease or spinal muscular atrophy. At present there is one approved treatment for spinal muscular atrophy, nusinersen (Spinraza). Its availability varies from country to country. There is also a range of options aimed at managing symptoms, reducing complications of weakness and maintaining the best quality of life for as long as possible.
A baby with Werdnig Hoffmann disease or spinal muscular atrophy type 1 should receive care from a multidisciplinary team which may include specialists in neuromuscular conditions, palliative care, respiratory medicine, physiotherapy, occupational therapy, speech and language therapy, dietetics and a hospital or community consultant paediatrician. Where available, a keyworker should assist in the co-ordination of services for the family.
The impact of a diagnosis of spinal muscular atrophy type 1 on families is enormous. Parents should be offered ongoing support with emotional, practical and financial issues following the diagnosis of their baby.
Parents should also be offered a referral for genetic counseling to help understand how spinal muscular atrophy is passed on and the chances of other children being affected. Genetic counseling also provides the opportunity to discuss choices for future pregnancies.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Werdnig Hoffmann disease causes
The specific underlying cause of Werdnig-Hoffmann disease is unknown. In spinal muscular atrophy, it appears that the SMN1 (survival motor neuron 1) and SMN2 (survival motor neuron 2) genes produce (encode) a protein, which is essential for the proper function of motor neurons. Mutation of the SMN1 causes the gene to produce a defective protein that cannot perform its intended function. It is believed that SMN2 gene produces a partially-effective protein required by motor neurons to function. This is why individuals with more copies of SMN2 have a milder form of spinal muscular atrophy.
Parents of several individuals with Werdnig-Hoffmann disease have been closely related by blood (consanguineous). All individuals carry 4-5 abnormal genes. Parents who are consanguineous have a higher chance than unrelated parents to both carry the same abnormal gene, which increases the risk to have children with a recessive genetic disorder.
All forms of spinal muscular atrophy are caused by mutations in the SMN1 (survival motor neuron 1) gene at chromosomal locus 5q11-q13. A second gene, known as the SMN2 (survival motor neuron 2) gene, plays a role in the development of spinal muscular atrophy. The SMN2 gene is next to (adjacent) to the SMN1 gene on chromosome 5. While the mutations of the SMN1 cause spinal muscular atrophy, evidence has been developed that SMN2 influences disease severity; individuals with more copies of the SMN2 gene tend to have a milder form of spinal muscular atrophy.
Chromosomes, which are present in the nucleus of human cells, carry the genetic information for each individual. Human body cells normally have 46 chromosomes. Pairs of human chromosomes are numbered from 1 through 22 and the sex chromosomes are designated X and Y. Males have one X and one Y chromosome and females have two X chromosomes. Each chromosome has a short arm designated “p” and a long arm designated “q”. Chromosomes are further sub-divided into many bands that are numbered. For example, “chromosomal locus 5q11-q13” refers to bands 11-13 on the long arm of chromosome 5. The numbered bands specify the location of the thousands of genes that are present on each chromosome.
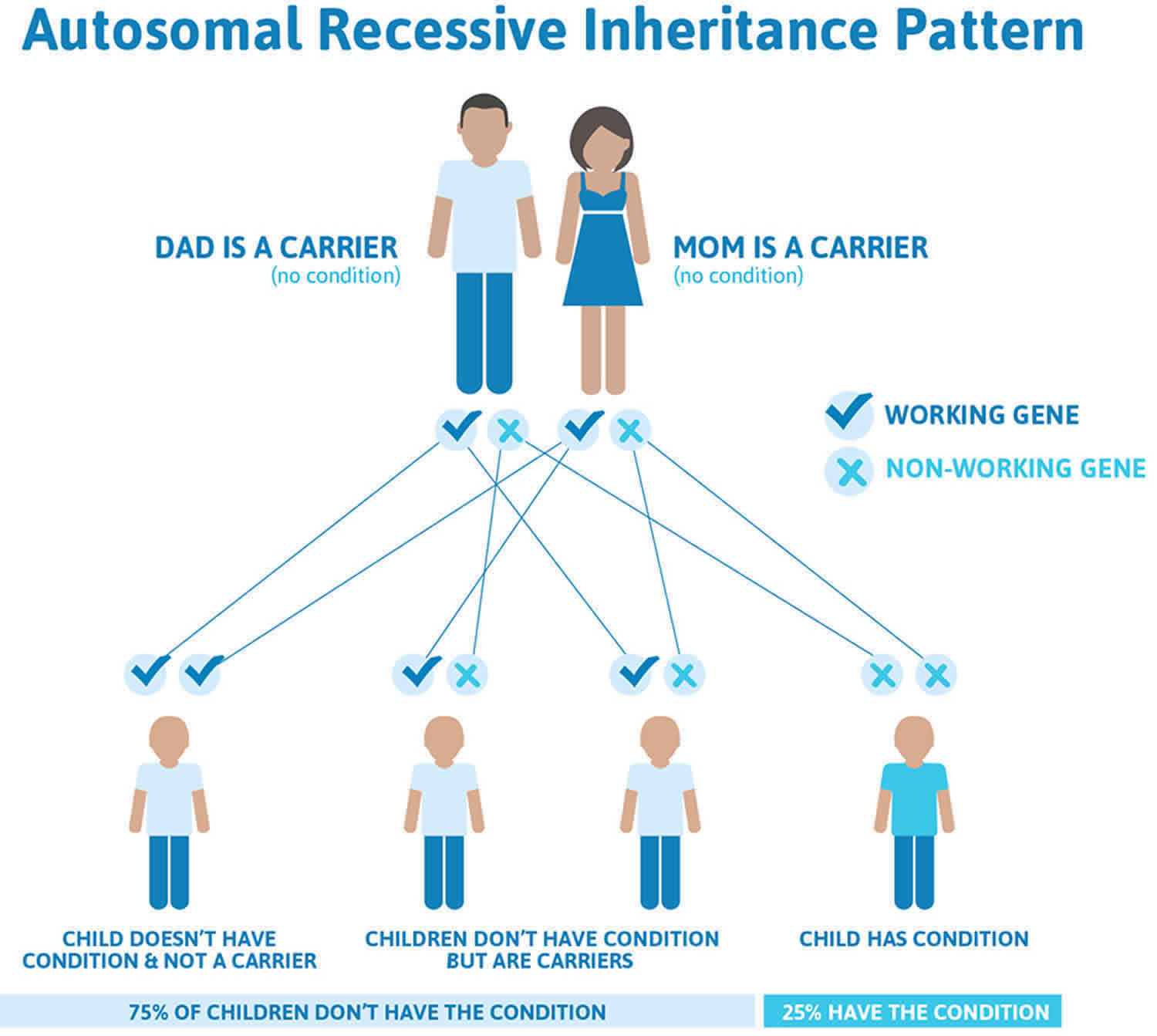
Genetic diseases are determined by the combination of genes for a particular trait that are on the chromosomes received from the father and the mother. All spinal muscular atrophys are inherited in an autosomal recessive manner. Recessive genetic disorders occur when an individual inherits the same abnormal gene for the same trait from each parent. If an individual receives one normal gene and one gene for the disease, the person will be a carrier for the disease, but usually will not show symptoms. The risk for two carrier parents to both pass the defective gene and, therefore, have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents and be genetically normal for that particular trait is 25%. The risk is the same for males and females.
Additional genes may influence the development of spinal muscular atrophy. Deletion of the NAIP (neuronal apoptosis inhibitory protein) gene that is close to the SMN gene may also be associated with spinal muscular atrophy. More patients with Werdnig-Hoffmann disease (spinal muscular atrophy type 1) than other types of spinal muscular atrophy have NAIP deletions. Some researchers suggest that loss of the NAIP gene and/or different mutations of the SMN gene may play a role in affecting the disorder’s severity. Some investigators also indicate that other genetic factors may contribute to the variable clinical expression of the disorder.
Werdnig Hoffmann disease inheritance pattern
Werdnig Hoffmann disease or spinal muscular atrophy type 1 is inherited in an autosomal recessive manner. This means that to be affected, a person must have a change (mutation) in both copies of the responsible gene in each cell. The parents of an affected person usually each carry one mutated copy of the gene and are referred to as carriers. Carriers are typically unaffected and do not have any signs or symptoms of the condition.
When two carriers of an autosomal recessive condition have children, each child has a:
- 25% chance to have the condition
- 50% chance to be an unaffected carrier like each of the parents
- 25% chance to not have the condition and not be a carrier.
An unaffected sibling of a person with spinal muscular atrophy1 has a 2/3 chance to be a carrier.
Approximately 2% of cases of spinal muscular atrophy1 are not inherited from both parents. In these cases, the affected person inherits one mutated copy of the gene from one carrier parent, and has a new mutation that occurs for the first time in the other copy of the gene 6.
Figure 1. Werdnig Hoffmann disease autosomal recessive inheritance pattern
Werdnig Hoffmann disease symptoms
The symptoms and progression of Werdnig Hoffmann disease or spinal muscular atrophy type 1 varies among affected individuals. Infants with Werdnig Hoffmann disease or spinal muscular atrophy type 1 experience severe weakness before 6 months of age. Muscle weakness, lack of motor development and poor muscle tone (hypotonia) are the major features of Werdnig Hoffmann disease 7. Infants with the poorest outlook have problems with breathing and feeding (sucking and/or swallowing) 8. Some children develop scoliosis (curvature of the spine) or other skeletal abnormalities. Intellectual development is usually normal 8. Affected children are not able to sit up or stand, and the vast majority do not survive past 2 years of age due to respiratory failure 9.
The early signs include a generalized muscle weakness, diminished muscle tone (hypotonia) resulting in “floppiness,” abnormal flexibility (hypermobility) of the joints, absent tendon reflexes, twitching (fasciculation) of the tongue, a frog-like position with the hips moved apart (abducted) and knees bent or flexed, and an alert appearance. Muscles of the face are not affected initially. Mental development is usually normal. Typically, the child does not gain head control, cannot turn over and is unable to sit or stand. In addition, children with spinal muscular atrophy may develop difficulties sucking, swallowing, and breathing; have an increased susceptibility to respiratory infections, or develop other complications that may lead to potentially life-threatening abnormalities within the first months or years of life.
For infants who appear to have normal development for several months prior to the onset of muscle weakness, the disorder may tend to have a more slowly progressive course. Muscles of the lower extremities appear to be disproportionately affected. With disease progression, diminished muscle tone and weakness may gradually spread to affect almost all voluntary muscles, with the exception of certain muscles controlling movements of the eyes.
The rate of progression of Werdnig-Hoffmann disease varies. Within a few months, breathing (respiratory) and bowel (constipation) difficulties may develop. The infant may be unable to swallow. Respiratory failure may occur or food inhaled into the lungs (aspiration) may cause choking. Most affected children die before 2 years of age but survival may be dependent on the degree of respiratory function.
Werdnig Hoffmann disease diagnosis
If a doctor suspects spinal muscular atrophy after taking a medical history and physically examining a baby, the diagnosis is confirmed by taking a blood sample for DNA testing. The blood sample is tested for a deletion in the survival motor neurone 1 (SMN1) gene on chromosome 5. The result of the test is usually available within 2 – 4 weeks.
Further tests, such as an electromyogram (EMG) or muscle biopsy, may be considered if there is any uncertainty about the diagnosis, but are not usually needed to confirm spinal muscular atrophy.
Genetic testing for Werdnig Hoffmann disease or spinal muscular atrophy type 1 is available. Carrier testing for at-risk relatives and prenatal testing for pregnancies at increased risk are possible, if the disease-causing mutations in the family have been identified. Werdnig Hoffmann disease is caused by mutations in the SMN1 gene, and extra copies of the SMN2 gene affect the severity of the condition 2.
In some cases, interpreting results of carrier testing for spinal muscular atrophy is difficult. Approximately 6% of parents of a child with spinal muscular atrophy due to deletions in each copy of the gene have normal results of SMN1 dosage testing (carrier testing) for one of two reasons. Most people have one copy of SMN1 on each chromosome. However, about 4% of carriers have two copies of SMN1 on a single chromosome and a deletion on the other chromosome. These carriers with two copies of SMN1 on one chromosome are misdiagnosed as non-carriers by the SMN1 dosage test (they have a false negative test result). The second reason is that a new (de novo) deletion on one copy of the SMN1 gene occurs in 2% of people with SMA; in these cases, only one parent is a carrier 2.
The genetics of Werdnig Hoffmann disease is complex. People with specific questions about genetic risks and genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Werdnig Hoffmann disease treatment
No curative treatment exists for infants with Werdnig-Hoffmann disease. Treatment is aimed at the specific symptoms that are present in each individual. Treatment may require a team of specialists.
Feeding difficulties may cause nutritional concerns and may require a gastrostomy, a procedure in which a feeding tube is inserted directly into the stomach through a surgical opening.
Breathing (respiratory) function declines in individuals with Werdnig-Hoffmann disease. Treatment options range from offering no respiratory support to using noninvasive procedures or long-term invasive procedures such as a tracheotomy. A tracheotomy is a procedure in which a tube is inserted through a surgical opening in the windpipe (trachea). A non-invasive option is intermittent positive pressure ventilation (NIPPV), in which breathing is mechanically assisted without the creation of an artificial airway as is done with a tracheotomy. The specific treatment for the life-threatening respiratory complications of Werdnig-Hoffman disease is controversial. Any decisions for treatment should be made after consultation between the parents and the entire medical team and taken on an individual case basis.
Physical and occupational therapy is helpful to minimize contractures and help the patient/caretakers develop compensatory strategies; muscle strengthening is not a reason for therapy. Orthopedic devices (e.g., braces) and surgery to correct scoliosis may become necessary.
In 2017, Spinraza (nusinersen) was FDA approved as the first drug to treat children and adults with spinal muscular atrophy. Spinraza is manufactured by Biogen.
Genetic counseling may be of benefit for affected individuals and their families. Other treatment is symptomatic and supportive.
Support with breathing
Good respiratory management is important to provide comfort and reduce complications of muscle weakness. An overview of respiratory management in spinal muscular atrophy can be found in the booklet ‘Standards of Care for Spinal Muscular Atrophy – the family guide’ published by TREAT-NMD. This booklet can be obtained from here (https://treat-nmd.org/treat-nmd-diseases/spinal-muscular-atrophy).
There are a number of options for respiratory support. Not all options will be appropriate for all babies with Werdnig Hoffmann disease or spinal muscular atrophy type 1.
The options may include:
- Interventions to maintain comfort such as chest physiotherapy and suction to remove secretions, medications to reduce secretions, pain relief to reduce any distress caused by breathlessness
- Non-invasive ventilation with mechanical ventilation to increase comfort, in the management of acute infection or to correct night time hypoventilation. Non-invasive ventilation may not be appropriate for some babies e.g. babies with severe weakness of the muscles around the mouth and throat (bulbar muscles), and in babies aged less than 6 months, whilst for others it may help to relieve respiratory distress or facilitate discharge home from hospital
- Invasive ventilation – mechanical ventilation via an endotracheal tube or tracheostomy. Mechanical ventilation via an endotracheal tube may sometimes be used as a short term measure in a medical emergency. However, until there is an effective treatment to prevent the progression of muscle weakness, the use of tracheostomy with long term invasive ventilation remains an ethical dilemma
Choosing the most appropriate management involves very difficult decisions. Parents should have time and support to ask questions and discuss the different options, to help decide which is most appropriate for their baby. These discussions will be with the medical team who has knowledge of their baby’s condition and possible progression. Parents may also wish to seek emotional support from sources outside the medical team e.g. their family, community and spiritual leaders.
Support with feeding and swallowing
Babies with Werdnig Hoffmann disease or spinal muscular atrophy type 1 may encounter problems with feeding and swallowing due to muscle weakness. Feeding can become a tiring process for babies with spinal muscular atrophy type 1 and as a result they may lose weight. Babies with a weak swallow are also at risk of inhaling (aspirating) their feed which can cause chest infections.
Advice and support on feeding, swallowing and diet is available to parents from a number of healthcare professionals. These may include the health visitor at their local health centre, the child’s consultant, speech and language therapist, dietitian and community nurse. Occupational therapists and physiotherapists may also advise on positioning and seating which may assist feeding.
At this time there is no clear evidence that babies with Werdnig Hoffmann disease or spinal muscular atrophy type 1 need a therapeutic feed or one with an increase or decrease in particular nutrients.
If swallowing becomes unsafe, or if a baby is not gaining enough weight, alternative ways of feeding may be suggested. These may include feeding through a nasogastric (NG) tube, a nasojejunal (NJ) tube or a gastrostomy (G) tube.
Parents should have the opportunity to discuss the reasons for these suggestions and have time to ask questions, so they understand the possible benefits and risks to their baby. Whichever option is chosen, parents will be provided with training and support to enable them to feed their child safely at home.
Constipation is a common problem with babies with Werdnig Hoffmann disease or spinal muscular atrophy type 1. It can cause discomfort and respiratory distress. Some babies may also experience reflux. To reduce discomfort and prevent complications, the management of these symptoms should be discussed with the baby’s medical team.
Options for care and support
Parents should have the opportunity to discuss the range of care options in depth with their baby’s medical team to decide what support is most appropriate for their individual circumstances. These discussions are important for developing an anticipatory care plan, which records the treatment parents wish their baby to receive when his/her health deteriorates or in an emergency situation. This plan can be reviewed and parents can change their minds at any time.
Ideally the goal of care is to enable a child to enjoy a good quality of life at home with their family for as long as possible, with a minimum of hospital admissions.
In addition to medical interventions available for breathing and feeding, additional support is available to improve a baby’s health and wellbeing and to provide emotional support for the family. When a baby is at home this may be provided by their GP, community nursing or palliative care team. Children’s hospices also offer a wide range of services and support to children with life-limiting conditions and their families.
Physiotherapy can provide passive exercises to enable movement that babies are unable to make independently. Parents can be taught these techniques so they can enjoy doing these with their baby at home between appointments. Passive exercises are also good for babies’ circulation and help prevent stiffening of the joints (contractures).
Physiotherapists may suggest performing stretches and exercises when a baby is in the bath, swimming or hydrotherapy pool. Building these exercises into play can make them fun and babies enjoy the additional freedom of movement provided by warm water.
Chest physiotherapy is also very important to help clear the chest when babies have difficulty coughing.
Good posture management can improve comfort. An occupational therapist can advise on seating which will help to provide appropriate support for babies with low muscle tone. This will also make it easier for them to play.
Sleep systems may sometimes be provided to increase comfort and support a baby’s limbs in a good position at night time.
Werdnig Hoffmann disease prognosis
The outlook for children with Werdnig-Hoffmann disease is very poor:
- If no intervention is provided, children with Werdnig-Hoffmann disease generally do not survive beyond the first two years 10.
- Prognosticating for individual children is difficult, however – several studies have cited individual cases where children have had onset of symptoms before the age of 6 months and not developed respiratory failure for years. The genetic basis of this phenotypical variability is not understood 11.
- Non-invasive respiratory support and prompt treatment of respiratory complications also appear to prolong survival but not to alter the disease course 12.
- Werdnig G. Two early infantile hereditary cases of progressive muscular atrophy simulating dystrophy, but on a neural basis. 1891. Arch Neurol. 1971;25:276–8.[↩]
- Prior TW, Leach ME, Finanger E. Spinal Muscular Atrophy. 2000 Feb 24 [Updated 2019 Nov 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1352[↩][↩][↩]
- Spinal muscular atrophy. https://ghr.nlm.nih.gov/condition/spinal-muscular-atrophy[↩]
- Kariyawasam D, Carey KA, Jones KJ, Farrar MA. New and developing therapies in spinal muscular atrophy. Paediatr Respir Rev. April 5, 2018; pii: S1526-0542(18):30048-4. https://www.ncbi.nlm.nih.gov/pubmed/29703692[↩]
- Finkel RS, Mercuri E, Meyer OH, et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. March 2018; 28(3):197-207. https://doi.org/10.1016/j.nmd.2017.11.004[↩]
- Thomas W. Prior. Carrier screening for spinal muscular atrophy. Genetics in Medicine. November, 2008; 10(11):840-842.[↩]
- Spinal muscular atrophy. https://medlineplus.gov/ency/article/000996.htm[↩]
- Werdnig-Hoffmann Disease. https://rarediseases.org/rare-diseases/werdnig-hoffmann-disease[↩][↩]
- Spinal Muscular Atrophy Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Spinal-Muscular-Atrophy-Information-Page[↩]
- D’Amico A, Mercuri E, Tiziano FD, et al; Spinal muscular atrophy. Orphanet J Rare Dis. 2011 Nov 26:71. doi: 10.1186/1750-1172-6-71[↩]
- Hardart MK, Truog RD; Spinal muscular atrophy–type I. Arch Dis Child. 2003 Oct88(10):848-50.[↩]
- Iannaccone ST; Modern management of spinal muscular atrophy. J Child Neurol. 2007 Aug22(8):974-8.[↩]



{kind=link}