Asymmetric septal hypertrophy
Asymmetric septal hypertrophy is the most common type of hypertrophic cardiomyopathy in which the abnormal ventricular muscle thickening is confined to the interventricular septum, causing the walls of the lower heart chambers (typically the left ventricle) to become thick and stiff 1. The hypertrophy in this phenotype of hypertrophic cardiomyopathy is usually asymmetric and is typically most evident in the anteroseptal myocardium. Asymmetric septal hypertrophy accounts for an estimated 60%–70% of the cases of hypertrophic cardiomyopathy 2. Hypertrophic cardiomyopathy is defined as a diffuse or segmental left ventricular (LV) hypertrophy with a nondilated and hyperdynamic chamber, in the absence of another cardiac or systemic disease capable of producing the magnitude of hypertrophy that is evident 2. Hypertrophic cardiomyopathy may affect any portion of the left ventricle 3. Asymmetric involvement of the interventricular septum is the most common hypertrophic cardiomyopathy and other variants include apical, symmetric, midventricular, masslike, and noncontiguous hypertrophic cardiomyopathy (Figure 2) 4. The criteria for diagnosing asymmetric septal hypertrophy is septal thickness greater than or equal to 15 mm (normal wall thickness is 12 mm or less, measured during diastole) or ratio of septal thickness to the thickness of the left ventricular inferior wall at the mid-ventricular level is greater than 1.5 5. Asymmetric septal hypertrophy makes it difficult for the heart to relax and for a sufficient amount of blood to fill the heart chambers. While the heart squeezes normally, the limited filling prevents the heart from pumping enough blood, especially during physical activity. Children with asymmetric septal hypertrophy are not allowed to play competitive sports because of the possibility of a sudden collapse or increased heart failure. Hypertrophic cardiomyopathy is the leading cause of sudden death from arrhythmias in infants, teenagers, and young adults 6.
It is clinically important to distinguish between the obstructive and nonobstructive forms of hypertrophic cardiomyopathy, on the basis of the presence or absence of a gradient between the left ventricular outflow tract (LVOT) and the aorta with the patient at rest and/or with provocation 2. Approximately 20%–30% of the patients with asymmetric septal hypertrophic cardiomyopathy have a resting systolic pressure gradient of the LVOT caused by systolic anterior motion of the mitral valve leaflets and midsystolic contact with the interventricular septum 7. Concomitant mitral regurgitation is also frequently noted as a result of systolic anterior motion and incomplete leaflet apposition. Although the mechanism of systolic anterior motion remains unclear, a widely accepted explanation of this phenomenon is that raised flow velocities in an LVOT anatomically distorted by septal hypertrophy create a Venturi effect, pulling the mitral valve leaflets toward the septum and obstructing the outflow tract 8. Systolic anterior motion is not pathognomonic of hypertrophic cardiomyopathy because systolic anterior motion may occur in patients with a hypertensive heart, diabetes mellitus, acute myocardial infarction, or mitral valve repair or dysfunction 9.
Clinically, patients may be asymptomatic or may present with a wide variety of symptoms, including dyspnea (shortness of breath), chest pain, syncope, or sudden cardiac death 10. Dyspnea on exertion is the most common symptom because the key functional hallmark of hypertrophic cardiomyopathy is an impaired diastolic function with impaired left ventricular (LV) filling in the presence of preserved systolic function 11. Systolic dysfunction often develops with end-stage hypertrophic cardiomyopathy.
Figure 1. Hypertrophic cardiomyopathy
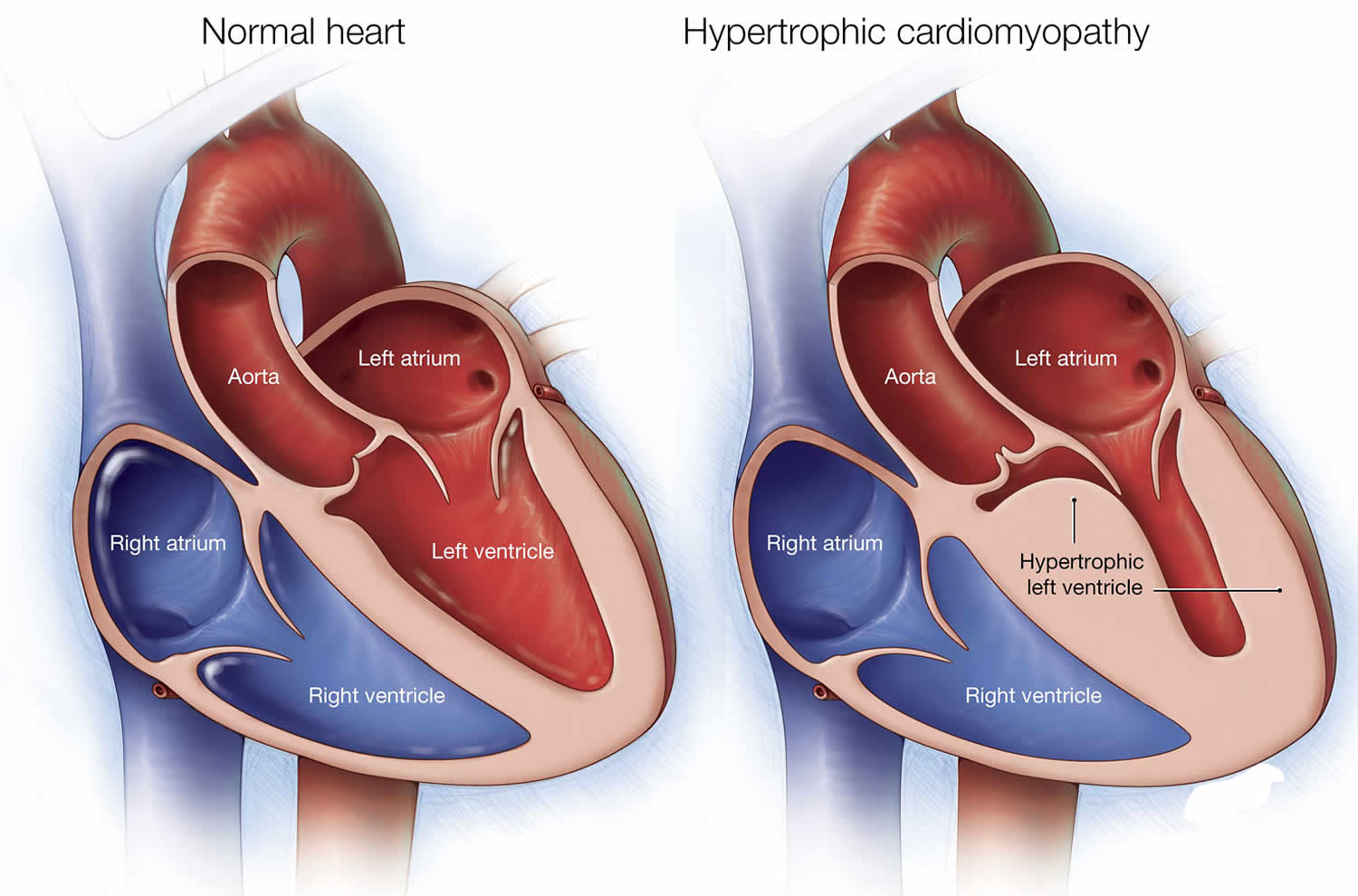
Footnote: Illustrations of a normal heart (left) and a heart with hypertrophic cardiomyopathy. Note that the heart walls (muscles) are much thicker (hypertrophied) in the heart with hypertrophic cardiomyopathy.
Figure 2. Types of hypertrophic cardiomyopathy
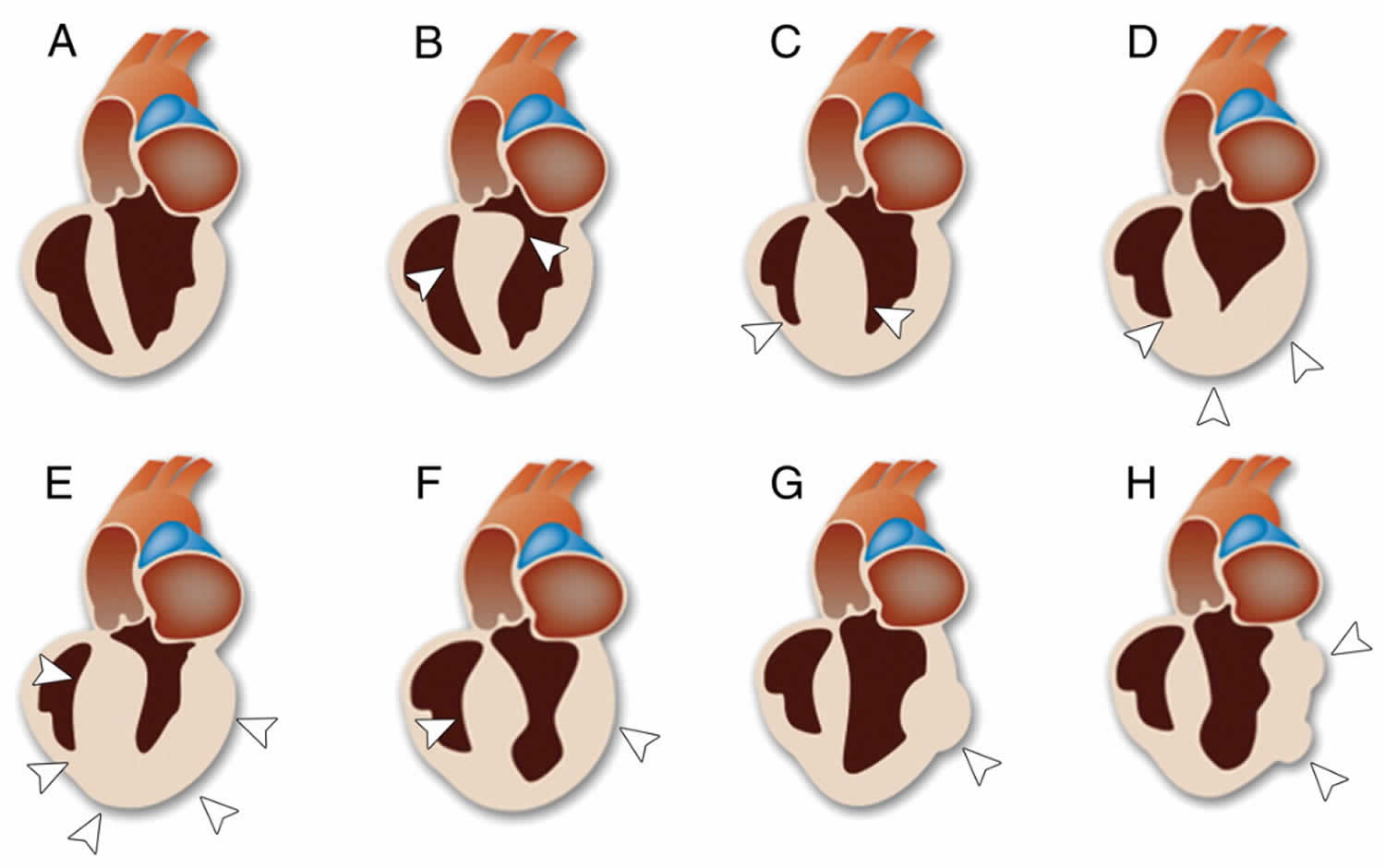
Footnote: Drawings of the normal heart and the heart in the various phenotypes of hypertrophic cardiomyopathy. The diagnostic criterion of hypertrophic cardiomyopathy is that the maximal LV wall thickness is greater than or equal to 15 mm in the end-diastolic phase. (A) Normal heart; (B) asymmetric septal hypertrophic cardiomyopathy with left ventricular outflow tract (LVOT) obstruction; (C) asymmetric septal hypertrophic cardiomyopathy without LVOT obstruction; (D) apical hypertrophic cardiomyopathy; (E) symmetric hypertrophic cardiomyopathy (concentric hypertrophic cardiomyopathy); (F) midventricular hypertrophic cardiomyopathy; (G) masslike hypertrophic cardiomyopathy; (H) noncontiguous hypertrophic cardiomyopathy. The drawings of the various phenotypes of hypertrophic cardiomyopathy show the areas of hypertrophy (arrowheads).
[Source 12 ]Figure 3. Asymmetric septal hypertrophy
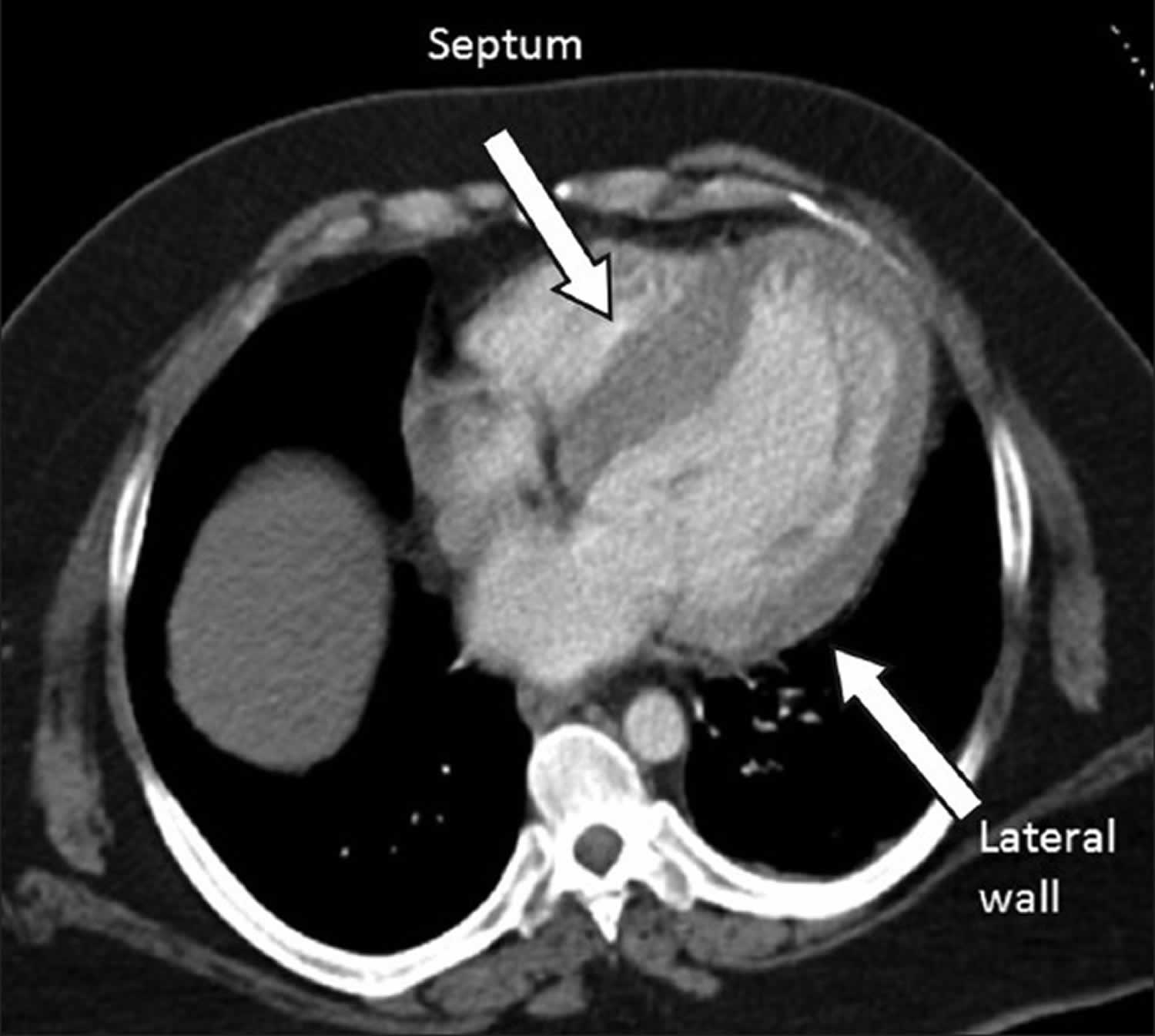
Footnote: Chest CT scan shows four-chamber of the heart. The intraventricular septum (IVS) and lateral wall thicknesses are 26 mm and 12.8 mm, respectively, with a resulting septal-to-posterior wall thickness ratio of 2.1.
[Source 13 ]Figure 4. Asymmetric septal hypertrophy echo
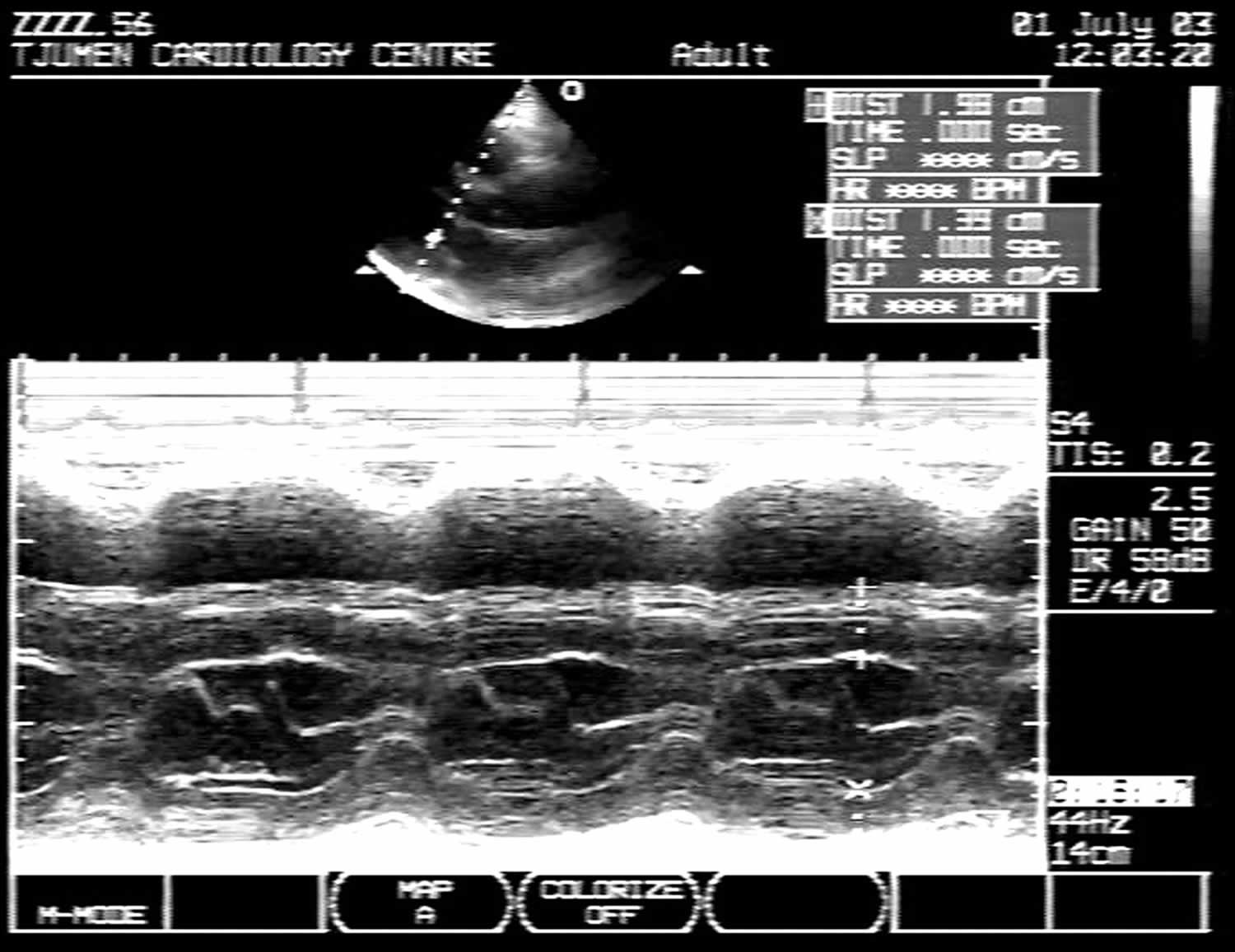
Footnote: M-mode echocardiogram of left ventricle in patients with asymmetrical septal hypertrophy. Thickness of intraventricular septum (IVS) = 20 mm; left ventricular posterior wall (LVPW) = 14 mm; ratio IVS/LVPW = 1.43.
[Source 14 ]A number of conditions can cause shortness of breath and heart palpitations. It’s important to get a prompt, accurate diagnosis and appropriate care. See your doctor if you have a family history of hypertrophic cardiomyopathy or any symptoms associated with hypertrophic cardiomyopathy.
Call your local emergency number if you have any of the following symptoms for more than a few minutes:
- Rapid or irregular heartbeat
- Difficulty breathing
- Chest pain
Asymmetric septal hypertrophy causes
Hypertrophic cardiomyopathy is most often passed down through families (inherited). Hypertrophic cardiomyopathy is usually caused by abnormal genes (gene mutations) that cause the heart muscle to grow abnormally thick. Individuals with hypertrophic cardiomyopathy who do not have acquired (secondary) hypertrophic cardiomyopathy or syndromic hypertrophic cardiomyopathy (see Table 2) have nonsyndromic hypertrophic cardiomyopathy. Genes causing hypertrophic cardiomyopathy without other systemic involvement will be referred to as hypertrophic cardiomyopathy genes. See Table 1 for a current list of known hypertrophic cardiomyopathy genes; the strength of the evidence associating each gene with hypertrophic cardiomyopathy varies significantly 15. The genes with the strongest clinical validity encode different components of the sarcomere 15. Pathogenic variants in one of the genes encoding a component of the sarcomere are found in approximately 50%-60% of probands (adults and children) with a family history of hypertrophic cardiomyopathy, and approximately 20%-30% of probands without a family history of hypertrophic cardiomyopathy 16. Approximately 3%-5% of affected individuals have more than one sarcomere gene variant (either biallelic variants in 1 gene or heterozygous variants in >1 gene) although fewer than 1% will have more than one pathogenic or likely pathogenic variant 17.
Acquired (secondary) left ventricular hypertrophy can be pathologic, occurring in response to pressure overload (e.g., systemic hypertension, aortic stenosis). This type of adverse remodeling can lead to diastolic abnormalities and heart failure. Physiologic hypertrophy (athlete’s heart) may result from rigorous athletic training. Such training may result in increased left ventricular wall thickness accompanied by increased left ventricular (LV) cavity size. This type of remodeling is thought to be adaptive and not associated with adverse consequences. Both pathologic and physiologic forms of secondary hypertrophy can regress if the underlying stimulus is removed (e.g., by adequate treatment of high blood pressure or a period of detraining for an athlete).
Younger people are likely to have a more severe form of hypertrophic cardiomyopathy. However, the condition is seen in people of all ages.
In most people with hypertrophic cardiomyopathy, the muscular wall (septum) between the two bottom chambers of the heart (ventricles) becomes thicker than normal. As a result, the thicker wall may block blood flow out of the heart. This is called obstructive hypertrophic cardiomyopathy (HOCM).
If there’s no significant blocking of blood flow, the condition is called nonobstructive hypertrophic cardiomyopathy. However, the heart’s main pumping chamber (left ventricle) may become stiff. This makes it hard for the heart to relax and reduces the amount of blood the ventricle can hold and send to the body with each heartbeat.
People with hypertrophic cardiomyopathy also have an abnormal arrangement of heart muscle cells (myofiber disarray). This can trigger arrhythmias in some people.
Table 1. Hypertrophic Cardiomyopathy Genes
| Gene | Mode of inheritance | % of Hypertrophic Cardiomyopathy Caused by Pathogenic Variants in This Gene 1 | ClinGen Gene Validity Classification 2 | Allelic Disorders 3 |
| MYBPC3 | AD | 50.00% | Definitive | DCM |
| MYH7 | AD | 33.00% | Definitive | Laing distal myopathy Myosin storage myopathy Left ventricular non-compaction Scapuloperoneal myopathy |
| TNNI3 | AD | 5.00% | Definitive | DCM Restrictive cardiomyopathy |
| TNNT2 | AD | 4.00% | Definitive | DCM Left ventricular non-compaction Familial restrictive cardiomyopathy |
| ACTC1 | AD | <3% | Definitive | DCM |
| MYL2 | AD | <3% | Definitive | |
| MYL3 | AD AR | <3% | Definitive | |
| TPM1 | AD | <3% | Definitive | DCM |
| PLN | AD | <3% | Definitive 5 | DCM |
| ALPK3 | AR | Rare | Strong | |
| ACTN2 | AD | <1% | Moderate 5 | DCM |
| CSRP3 | AD | <1% | Moderate | DCM |
| TNNC1 | AD | <1% | Moderate | DCM |
| JPH2 | AD | Rare | Moderate | |
| MYOZ2 | AD | <1% | Limited | |
| NEXN | AD | <1% | Limited | DCM |
| ANKRD1 | AD | Rare | Limited | |
| CALR3 | AD | Rare | Limited | |
| KLF10 | AD | Rare | Limited | |
| MYH6 | AD | Rare | Limited | DCM |
| MYLK2 | Digenic | Rare | Limited | |
| MYOM1 | AD | Rare | Limited | |
| MYPN | AD | Rare | Limited | DCM Nemaline myopathy |
| OBSCN | AD | Rare | Limited | |
| PDLIM3 | AD | Rare | Limited | |
| RYR2 | AD | Rare | Limited | ARVD2 CPVT1 |
| TCAP | AD | Rare | Limited | DCM LGMD2G |
| TRIM63 | AD | Rare | Limited | |
| TTN | AD | Rare | Limited | DCM Hereditary myopathy with early respiratory failure LGMD2J Salih myopathy Udd distal myopathy |
| VCL | AD | Rare | Limited | DCM |
Footnote:
1. Prevalence data list for genes included in Alfares et al 16. “Rare” denotes genes not included in this paper.
2. The ClinGen Gene Curation working group developed a framework to evaluate the clinical validity of a gene-disease relationship. The classification reflects the strength of evidence that a variant in that gene can cause a particular phenotype, in this case hypertrophic cardiomyopathy 15.
3. Allelic disorders = other phenotypes caused by pathogenic variants in the same gene
4. PLN and ACTN2 were curated for intrinsic cardiomyopathy given their association with a spectrum of cardiac phenotypes, including isolated left ventricular hypertrophy (LVH) and hypertrophic cardiomyopathy.
Abbreviations: AD = autosomal dominant; AR = autosomal recessive; DCM = dilated cardiomyopathy; LGMD2G = limb-girdle muscular dystrophy type 2G; LGMD2J = limb-girdle muscular dystrophy type 2J
[Source 18 ]Table 2. Syndromic hypertrophic cardiomyopathy (a selected list)
| Disorder | Gene(s) | Mode of inheritance | Additional Clinical Features |
|---|---|---|---|
| Danon disease | LAMP2 | XL |
|
| Fabry disease | GLA | XL |
|
| Friedreich ataxia | FXN | AR |
|
| Glycogen storage disease of the heart, lethal congenital | PRKAG2 | AD |
|
| Hereditary transthyretin amyloidosis | TTR | AD |
|
| Pompe disease | GAA | AR |
|
RASopathies 1 including:
| BRAF HRAS KRAS LZTR1 MAP2K1 MAP2K2 NRAS PTPN11 RAF1 RASA2 RRAS RIT1 SOS1 SOS2 | AD |
|
Footnote: 1) The RASopathies are a group of syndromes that have overlapping clinical features resulting from a common pathogenetic mechanism 19.
Abbreviations: AD = autosomal dominant; AR = autosomal recessive; XL = X-linked
[Source 18 ]People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Risk factors for asymmetric septal hypertrophy
Hypertrophic cardiomyopathy is usually passed down through families (inherited). If you have a parent with hypertrophic cardiomyopathy, you have a 50% chance of having the genetic mutation for the disease. Parents, children or siblings of a person with hypertrophic cardiomyopathy should ask their doctors about screening for the disease.
Asymmetric septal hypertrophy prevention
There is no known prevention for hypertrophic cardiomyopathy. But it’s important to identify the condition as early as possible to guide treatment and prevent complications.
If you have a first-degree relative — a parent, sibling or child — with hypertrophic cardiomyopathy, doctors may recommend genetic testing to screen for the condition. However, not everyone with hypertrophic cardiomyopathy has a currently detectable mutation. Also, some insurance companies may not cover genetic testing.
If genetic testing isn’t done, or if the results aren’t helpful, then your doctor may recommend echocardiograms on a regular basis if you have a family member with hypertrophic cardiomyopathy. Adolescents and competitive athletes should be screened once a year. Adults who don’t compete in athletics should be screened every five years.
Asymmetric septal hypertrophy symptoms
Some people with hypertrophic cardiomyopathy may have no symptoms. They may first find out they have the problem during a routine medical exam.
In many young adults, the first symptom of hypertrophic cardiomyopathy is sudden collapse and possible death. This can be caused by highly abnormal heart rhythms (arrhythmias). It may also be due to a blockage that prevents the outflow of blood from the heart to the rest of the body.
Signs and symptoms of hypertrophic cardiomyopathy may include one or more of the following:
- Chest pain, especially during exercise
- Fainting, especially during or just after exercise or exertion
- Dizziness
- Fatigue
- Lightheadedness, especially with or after activity or exercise
- Heart murmur, which a doctor might detect while listening to your heart
- Sensation of rapid, fluttering or pounding heartbeats (palpitations)
- Shortness of breath, especially during exercise or after lying down (or being asleep for a while).
Signs of hypertrophic cardiomyopathy may include:
- Abnormal heart sounds or a heart murmur. These sounds may change with different body positions.
- High blood pressure.
Asymmetric septal hypertrophy complications
Many people with hypertrophic cardiomyopathy don’t have significant health problems. But complications of hypertrophic cardiomyopathy can include:
- Atrial fibrillation. Thickened heart muscle, as well as the abnormal structure of heart cells, can cause changes in the heart’s electrical system, resulting in fast or irregular heartbeats. Atrial fibrillation can also increase your risk of developing blood clots, which can travel to your brain and cause a stroke.
- Blocked blood flow. In many people, the thickened heart muscle blocks the blood flow leaving the heart, causing shortness of breath with exertion, chest pain, dizziness and fainting spells.
- Mitral valve problems. If the thickened heart muscle blocks the blood flow leaving the heart, the valve between the left atrium and left ventricle (mitral valve) may not close properly. As a result, blood can leak backward into the left atrium (mitral valve regurgitation), possibly leading to worsening symptoms.
- Dilated cardiomyopathy. In a very small number of people with hypertrophic cardiomyopathy, the thickened heart muscle may become weak and ineffective. The ventricle becomes enlarged (dilated), and its pumping ability becomes less forceful.
- Heart failure. The thickened heart muscle can eventually become too stiff to effectively fill the heart with blood. As a result, your heart can’t pump enough blood to meet your body’s needs.
- Sudden cardiac death. Rarely, hypertrophic cardiomyopathy can cause heart-related sudden death in people of all ages. Because many people with hypertrophic cardiomyopathy don’t realize they have it, sudden cardiac death may be the first sign of a problem. It can happen in seemingly healthy young people, including high school athletes and other young, active adults.
Sudden cardiac death is the most devastating and unpredictable complication of hypertrophic cardiomyopathy and the overall annual mortality rate ranges from less than 1% in asymptomatic patients to 6% in patients with high-risk factors 6. Because sudden cardiac death may occur as the initial manifestation of hypertrophic cardiomyopathy without reliable warning signs or symptoms, particularly in young patients, the identification of individuals at a high risk for sudden cardiac death among the various manifestations of hypertrophic cardiomyopathy has become an important challenge. The American College of Cardiology and the European Society of Cardiology guidelines indicate the major risk factors for sudden cardiac death as follows 2:
- (a) cardiac arrest (ventricular fibrillation),
- (b) spontaneous sustained ventricular tachycardia,
- (c) a family history of premature sudden death,
- (d) unexplained syncope,
- (e) left ventricular (LV) wall thickness greater than or equal to 30 mm,
- (f) abnormal exercise blood pressure, and
- (g) nonsustained ventricular tachycardia (Holter).
Among such risk factors, those with potential applications in cardiac imaging for stratification of the risk of sudden cardiac death in patients with hypertrophic cardiomyopathy are as follows 20:
- (a) left ventricular (LV) maximal wall thickness of 30 mm or more,
- (b) left ventricular outflow tract (LVOT) gradient of 30 mm Hg or more at rest or 50 mm Hg or more with provocation,
- (c) left ventricular (LV) dilatation with depressed ejection fraction,
- (d) presence of fibrosis,
- (e) perfusion defect, and
- (f) reduced functional reserve flow.
Maximal left ventricular (LV) wall thickness, especially a thickness of 30 mm or more, has been reported as a strong predictor of the risk of sudden death in patients with hypertrophic cardiomyopathy 21. This relationship of extreme hypertrophy to sudden cardiac death is accentuated in younger patients; it reflects either preferential sudden cardiac death at a young age, structural remodeling with wall thinning, or both 2. With echocardiography, the magnitude of hypertrophy tends to be substantially underestimated in comparison with MR imaging (MRI) for the assessment of massive LV hypertrophy (wall thickness ≥30 mm) 22.
Historically, the left ventricular outflow tract (LVOT) gradient has been a prominent and quantifiable feature of hypertrophic cardiomyopathy. Maron et al 23 reported that patients with LVOT obstruction (defined as a basal gradient of ≥30mm Hg) have an increased risk—more than four times that among patients without obstruction—of sudden cardiac death from hypertrophic cardiomyopathy or progression to severe congestive symptoms. Kofflard et al 24, who investigated the clinical outcome and risk factors of hypertrophic cardiomyopathy in a community-based population (not a hospital-based population), also reported that patients with “significant” LVOT obstruction (≥50 mm Hg at rest or with provocation) are susceptible to clinical deterioration in their condition.
Although most patients with hypertrophic cardiomyopathy have diastolic dysfunction, a small distinctive subset of patients paradoxically have hypertrophic cardiomyopathy that evolves into a phase characterized by systolic dysfunction, LV dilatation, and wall thinning 6, which can be demonstrated with MR imaging and multidetector CT. This dilated-hypokinetic evolution of hypertrophic cardiomyopathy is often called the “end-stage” or “burned-out phase”. The clinical course is unfavorable once hypertrophic cardiomyopathy gets to this phase because it has usually progressed to a heart failure unresponsive to therapy with medications; and, ultimately, heart transplantation remains the only definitive treatment option 25. Therefore, vigilant follow-up and careful risk stratification should be required for timely detection of a transition to the burned-out phase and to permit early treatment before progression to heart failure 26. Such hypokinesia can occur after an acute myocardial infarction or it can develop gradually without a clinical infarction. Patients with midventricular or apical hypertrophic cardiomyopathy are at a higher risk of developing segmental or diffuse LV hypokinesia 27. MR imaging reveals a thin-walled apical aneurysm showing transmural enhancement that extends into substantial areas of the contiguous interventricular septum and LV free wall. Furthermore, thrombus, which is frequently associated with an apical aneurysm, is readily detected at delayed enhancement MRI (DE-MRI) because thrombus manifests as a low-signal-intensity mass with lack of enhancement.
Asymmetric septal hypertrophy diagnosis
Your doctor will examine you and ask questions about your signs, symptoms, medical and family history and will perform a physical exam and listen to your heart and lungs with a stethoscope.
The pulse in your arms and neck will also be checked. The provider may feel an abnormal heartbeat in the chest.
Asymmetric septal hypertrophy tests
Your doctor will likely order tests to diagnose hypertrophic cardiomyopathy or rule out other conditions that can cause similar symptoms.
Tests used to diagnose heart muscle thickness, problems with blood flow, or leaky heart valves (mitral valve regurgitation) may include:
- Echocardiogram. An echocardiogram is commonly used to diagnose hypertrophic cardiomyopathy. This test uses sound waves (ultrasound) to see if your heart’s muscle is abnormally thick. It also shows how well your heart’s chambers and valves are pumping blood. Sometimes, an echocardiogram is done while you exercise, usually on a treadmill. This is called an exercise stress test. Treadmill stress tests are commonly used to diagnose people with hypertrophic cardiomyopathy.
- Electrocardiogram (ECG or EKG). Sensors (electrodes) attached to adhesive pads are placed on your chest and sometimes legs. They measure electrical signals from your heart. An ECG can show abnormal heart rhythms and signs of heart thickening. In some cases, a portable ECG, called a Holter monitor, is needed. This device records your heart’s activity continuously over one to two days.
- Cardiac MRI. A cardiac MRI uses powerful magnets and radio waves to create images of your heart. It gives your doctor information about your heart muscle and shows how your heart and heart valves work. This test is often done with an echocardiogram.
- CT scan of the heart
- Transesophageal echocardiogram (TEE)
- 24-hour Holter monitor (heart rhythm monitor)
- Cardiac catheterization
- Chest x-ray
Blood tests may be done to rule out other diseases.
Close family members of people who have been diagnosed with hypertrophic cardiomyopathy may be screened for the condition.
Asymmetric septal hypertrophy treatment
The goal of hypertrophic cardiomyopathy treatment is to relieve symptoms and prevent sudden cardiac death in people at high risk. Your specific treatment depends on the severity of your symptoms. Together, you and your doctor will discuss the most appropriate treatment for your condition.
Medications
Medications can help reduce how strong the heart muscle squeezes and slow the heart rate so that the heart can pump blood better. Medications to treat hypertrophic cardiomyopathy and its symptoms may include:
- Beta blockers such as metoprolol (Lopressor, Toprol-XL), propranolol (Inderal, Innopran XL) or atenolol (Tenormin)
- Calcium channel blockers such as verapamil (Verelan, Calan SR,) or diltiazem (Cardizem, Tiazac)
- Heart rhythm drugs such as amiodarone (Pacerone) or disopyramide (Norpace)
- Blood thinners such as warfarin (Coumadin, Jantoven), dabigatran (Pradaxa), rivaroxaban (Xarelto) or apixaban (Eliquis) to prevent blood clots if you have atrial fibrillation
Surgeries and other procedures
Several different surgeries or procedures are available to treat cardiomyopathy or its symptoms. They range from open-heart surgery to implantation of a device to control your heart rhythm.
- Septal myectomy. This open-heart surgery may be recommended if medications do not improve your symptoms. It involves removing part of the thickened, overgrown wall (septum) between the heart chambers. Septal myectomy helps improve blood flow out of the heart and reduces backward flow of blood through the mitral valve (mitral regurgitation). The surgery may be done using different approaches, depending on the location of the thickened heart muscle. In one type, called apical myectomy, surgeons remove thickened heart muscle from near the tip of the heart. Sometimes the mitral valve is repaired at the same time.
- Septal ablation. This procedure destroys the thickened heart muscle with alcohol. The alcohol is injected through a long, thin tube (catheter) into the artery supplying blood to that area. Possible complications include disruption of the heart’s electrical system (heart block), which requires implantation of a pacemaker.
- Implantable cardioverter-defibrillator (ICD). An implantable cardioverter-defibrillator (ICD) is a small device that continuously monitors your heartbeat. It’s implanted in your chest like a pacemaker. If a life-threatening arrhythmia occurs, the ICD delivers precisely calibrated electrical shocks to restore a normal heart rhythm. ICD has been shown to help prevent sudden cardiac death, which occurs in a small number of people with hypertrophic cardiomyopathy.
Lifestyle and home remedies
Lifestyle changes can reduce your risk of complications related to hypertrophic cardiomyopathy. Your doctor may recommend lifestyle changes, including:
- Using caution when playing sports. Competitive sports are generally not recommended for people with hypertrophic cardiomyopathy, with the possible exception of some low-intensity sports. You may be able to participate in low- to moderate-intensity exercise as part of a healthy lifestyle. Discuss specific recommendations with your doctor.
- Eating a healthy diet. A healthy diet is an important part of maintaining your heart health.
- Maintaining a healthy weight. Maintaining a healthy weight will prevent excessive stress on your heart and reduce health risks associated with surgery or other procedures.
- Reducing alcohol use. If you have symptoms or a history of rhythms provoked by alcohol, ask your doctor for guidance about safe levels of alcohol use. Drinking too much alcohol can trigger irregular heart rhythms and can lead to increased blockage of blood flow in your heart.
- Taking your medications. Make sure to take your medications as prescribed.
- Having regular medical appointments. Your doctor may recommend regular follow-up appointments to evaluate your condition. Let your doctor know if you have any new or worse symptoms.
Pregnancy
Women who have hypertrophic cardiomyopathy can generally have normal pregnancies. However, if you have hypertrophic cardiomyopathy, your doctor may recommend that you see a doctor experienced in caring for women with high-risk conditions during your pregnancy.
Asymmetric septal hypertrophy prognosis
Some people with hypertrophic cardiomyopathy may not have symptoms and will have normal lifespan. Others may get worse slowly or quickly. In some cases, the condition may develop into dilated cardiomyopathy. Dilated cardiomyopathy is a condition in which the heart muscle becomes weakened and enlarged. As a result, the heart cannot pump enough blood to the rest of the body.
People with hypertrophic cardiomyopathy are at higher risk for sudden death than people without the condition. Sudden death can occur at a young age.
Compared to the U.S. general population, the mortality rate in individuals with hypertrophic cardiomyopathy is approximately threefold higher, but the mortality rate in younger individuals with hypertrophic cardiomyopathy, ages 20-29, is as much as fourfold higher than expected. Sudden death accounts for 16% of deaths 28.
There are different types of hypertrophic cardiomyopathy, which have different prognoses. The outlook may be better when the disease occurs in older people or when there is a particular pattern of thickness in the heart muscle.
Hypertrophic cardiomyopathy is a well-known cause of sudden death in athletes. Almost half of deaths due to this condition happen during or just after some type of physical activity.
- THE ECHOCARDIOGRAPHIC DIAGNOSIS OF ASYMMETRIC SEPTAL HYPERTROPHY: CAN THE USE OF CONTRAST AGENTS CHANGE THE DIAGNOSIS? Daniel Flannery, Diane Lilburn. J Am Coll Cardiol. 2012 Mar, 59 (13 Supplement) E1296. DOI: 10.1016/S0735-1097(12)61297-2[↩]
- Maron BJ, McKenna WJ, Danielson GK et al.. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol 2003;42(9):1687–1713.[↩][↩][↩][↩][↩]
- Hughes SE. The pathology of hypertrophic cardiomyopathy. Histopathology 2004;44(5):412–427.[↩]
- Hansen MW, Merchant N. MRI of hypertrophic cardiomyopathy. I. MRI appearances. AJR Am J Roentgenol 2007;189(6):1335–1343.[↩]
- Kansal S, Roitman D, Sheffield LT. Interventricular septal thickness and left ventricular hypertrophy: an echocardiographic study. Circulation 1979;60(5):1058–1065.[↩]
- Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002;287(10):1308–1320.[↩][↩][↩]
- Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet 2004;363(9424):1881–1891.[↩]
- Maron MS, Olivotto I, Zenovich AG et al.. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation 2006;114(21):2232–2239.[↩]
- Luckie M, Khattar RS. Systolic anterior motion of the mitral valve: beyond hypertrophic cardiomyopathy. Heart 2008;94(11):1383–1385.[↩]
- Hypertrophic Cardiomyopathy: Assessment with MR Imaging and Multidetector CT. Eun Ju Chun, Sang Il Choi, Kwang Nam Jin, Hyon Joo Kwag, Young Jin Kim, Byoung Wook Choi, Whal Lee, and Jae Hyung Park. RadioGraphics 2010 30:5, 1309-1328[↩]
- Spirito P, Autore C. Management of hypertrophic cardiomyopathy. BMJ 2006;332(7552):1251–1255.[↩]
- Hypertrophic Cardiomyopathy: Assessment with MR Imaging and Multidetector CT. Eun Ju Chun, Sang Il Choi, Kwang Nam Jin, Hyon Joo Kwag, Young Jin Kim, Byoung Wook Choi, Whal Lee, and Jae Hyung Park. RadioGraphics 2010 30:5, 1309-1328. https://doi.org/10.1148/rg.305095074[↩]
- Wong RC, Tan KB. Asymmetric left ventricular hypertrophy associated with morbid obesity mimicking familial hypertrophic cardiomyopathy. Singapore Med J. 2014;55(12):e201-e204. doi:10.11622/smedj.2014186 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4291995[↩]
- Vadim A. Kuznetsov, Elena I. Yaroslavskaya, Igor P. Zyrianov, Grigoriy V. Kolunin, Dmitriy V. Krinochkin, Marina I. Bessonova, Ivan S. Bessonov, Asymmetric septal hypertrophy in patients with coronary artery disease, European Journal of Echocardiography, Volume 11, Issue 8, September 2010, Pages 698–702, https://doi.org/10.1093/ejechocard/jeq046[↩]
- Ingles J, Goldstein J, Thaxton C, Caleshu C, Corty EW, Crowley SB, Dougherty K, Harrison SM, McGlaughon J, Milko LV, Morales A, Seifert BA, Strande N, Thomson K, Peter van Tintelen J, Wallace K, Walsh R, Wells Q, Whiffin N, Witkowski L, Semsarian C, Ware JS, Hershberger RE, Funke B. Evaluating the clinical validity of hypertrophic cardiomyopathy genes. Circ Genom Precis Med. 2019;12:e002460[↩][↩][↩]
- Alfares AA, Kelly MA, McDermott G, Funke BH, Lebo MS, Baxter SB, Shen J, McLaughlin HM, Clark EH, Babb LJ, Cox SW, DePalma SR, Ho CY, Seidman JG, Seidman CE, Rehm HL. Results of clinical genetic testing of 2,912 probands with hypertrophic cardiomyopathy: expanded panels offer limited additional sensitivity. Genet Med. 2015;17:880–8.[↩][↩]
- Burns C, Bagnall RD, Lam L, Semsarian C, Ingles J. Multiple gene variants in hypertrophic cardiomyopathy in the era of next-generation sequencing. Circ Cardiovasc Genet. 2017;10:e001666[↩]
- Cirino AL, Ho C. Hypertrophic Cardiomyopathy Overview. 2008 Aug 5 [Updated 2019 Jun 6]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1768[↩][↩]
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–6[↩]
- Nagueh SF, Mahmarian JJ. Noninvasive cardiac imaging in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 2006;48(12):2410–2422.[↩]
- Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med 2000;342(24):1778–1785.[↩]
- Rickers C, Wilke NM, Jerosch-Herold M et al.. Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation 2005;112(6):855–861.[↩]
- Maron MS, Olivotto I, Betocchi S et al.. Effect of left ventricular outflow tract obstruction on clinical outcome in hypertrophic cardiomyopathy. N Engl J Med 2003;348(4):295–303.[↩]
- Kofflard MJ, Ten Cate FJ, van der Lee C, van Domburg RT. Hypertrophic cardiomyopathy in a large community-based population: clinical outcome and identification of risk factors for sudden cardiac death and clinical deterioration. J Am Coll Cardiol 2003;41(6):987–993.[↩]
- Biagini E, Coccolo F, Ferlito M et al.. Dilated-hypokinetic evolution of hypertrophic cardiomyopathy: prevalence, incidence, risk factors, and prognostic implications in pediatric and adult patients. J Am Coll Cardiol 2005;46(8):1543–1550.[↩]
- Harris KM, Spirito P, Maron MS et al.. Prevalence, clinical profile, and significance of left ventricular remodeling in the end-stage phase of hypertrophic cardiomyopathy. Circulation 2006;114(3):216–225.[↩]
- Matsubara K, Nakamura T, Kuribayashi T, Azuma A, Nakagawa M. Sustained cavity obliteration and apical aneurysm formation in apical hypertrophic cardiomyopathy. J Am Coll Cardiol 2003;42(2):288–295.[↩]
- Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, Cirino AL, Fox JC, Lakdawala NK, Ware JS, Caleshu CA, Helms AS, Colan SD, Girolami F, Cecchi F, Seidman CE, Sajeev G, Signorovitch J, Green EM, Olivotto I, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy. Insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation. 2018;138:1387–98.[↩]





{kind=link}