Atypical hemolytic uremic syndrome
Atypical hemolytic uremic syndrome also called atypical HUS, is an extremely rare disease that causes abnormal blood clots (thrombi) to form in small blood vessels in the kidneys that primarily affects kidney function. These blood clots can cause serious medical problems if they restrict or block blood flow. Atypical hemolytic-uremic syndrome is characterized by three major features related to abnormal blood clotting:
- Low levels of circulating red blood cells due to their destruction also known as hemolytic anemia. Hemolytic anemia occurs when red blood cells break down (undergo hemolysis) prematurely. In atypical hemolytic-uremic syndrome, red blood cells can break apart as they squeeze past clots within small blood vessels. Anemia results if these cells are destroyed faster than the body can replace them. Anemia can lead to unusually pale skin (pallor), yellowing of the eyes and skin (jaundice), fatigue, shortness of breath, and a rapid heart rate.
- Thrombocytopenia is a reduced level of circulating platelets, which are cells that normally assist with blood clotting. In people with atypical hemolytic-uremic syndrome, fewer platelets are available in the bloodstream because a large number of platelets are used to make abnormal clots. Thrombocytopenia can cause easy bruising and abnormal bleeding.
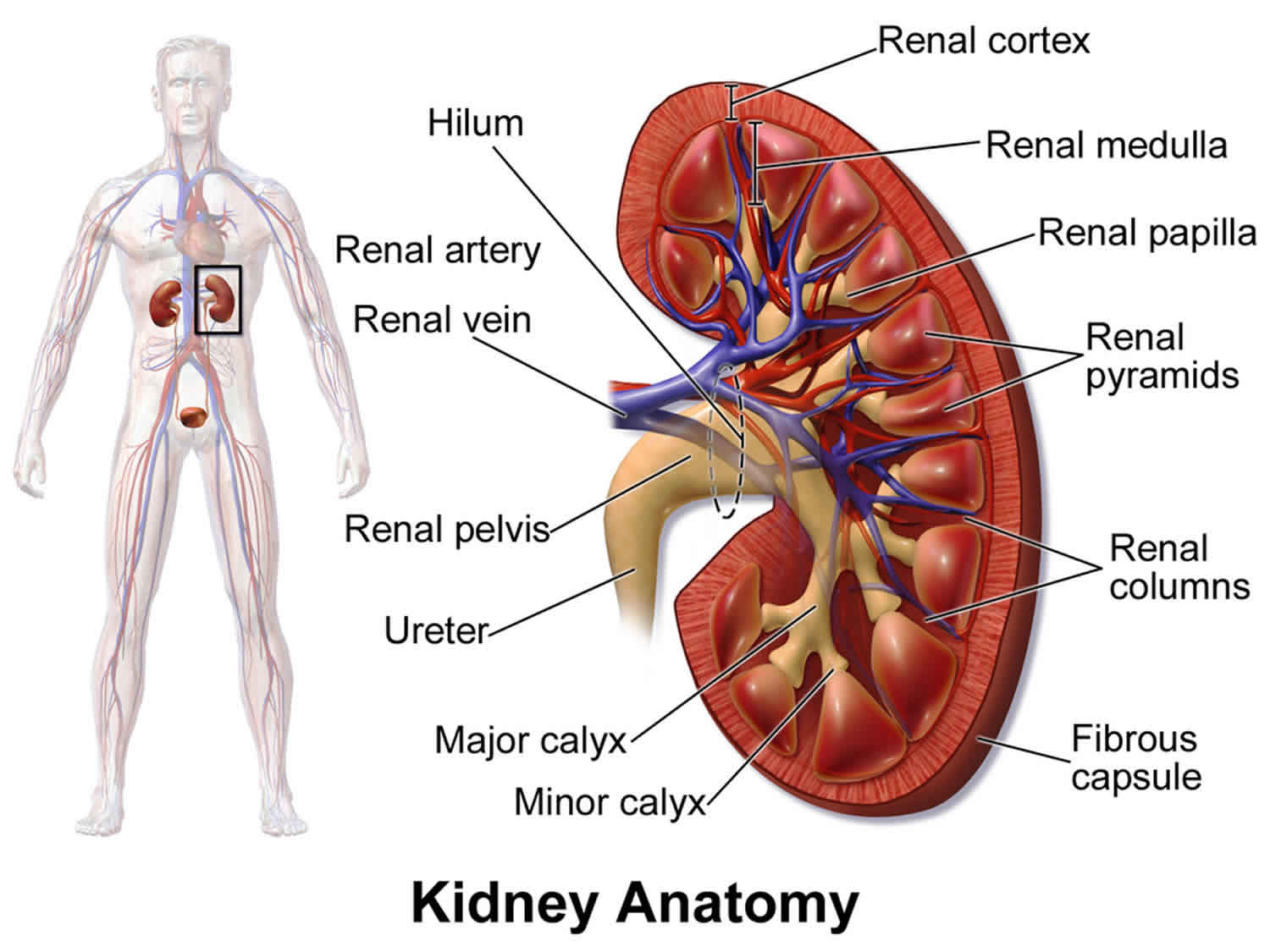
- As a result of clot formation in small blood vessels, people with atypical hemolytic-uremic syndrome experience kidney damage and acute kidney failure that lead to end-stage renal disease (ESRD) in about half of all cases. These life-threatening complications prevent the kidneys from filtering fluids and waste products from the body effectively, a condition known as uremia.
Atypical hemolytic-uremic syndrome is a complex disorder and multiple factors, including certain genetic, environmental and immunologic factors, all play a role in its development.
In childhood, atypical hemolytic uremic syndrome affects males and females in equal numbers. In adulthood, females are affected more often than males, most likely because pregnancy is a triggering event. The exact overall incidence and prevalence is unknown. One study placed the incidence in the United States at 2 individuals per 1 million in the general population. In Europe, the disorder is estimated to affect approximately 0.11 per 1 million individuals between the ages of 0-18. Atypical hemolytic uremic syndrome accounts for approximately 5-10% of all cases of hemolytic uremic syndrome.
Atypical hemolytic uremic syndrome can occur at any age and is often caused by a combination of environmental and genetic factors. Most cases of atypical hemolytic-uremic syndrome are sporadic, which means that they occur in people with no apparent history of the disorder in their family. Genetic factors involve genes that code for proteins that help control the complement system (part of your body’s immune system). Environmental factors include certain medications (such as anticancer drugs), chronic diseases (e.g., systemic sclerosis and malignant hypertension), viral or bacterial infections, cancers, organ transplantation, and pregnancy. In about 60% of atypical hemolytic-uremic syndrome, a gene mutation may be identified. The genes associated with genetic atypical hemolytic uremic syndrome include C3, CD46 (MCP), CFB, CFH, CFHR1, CFHR3, CFHR4, CFI, DGKE, and THBD. Mutations in these genes increase the likelihood (predisposition) to developing atypical hemolytic uremic syndrome, rather than directly causing the disease. In familial cases, predisposition to atypical HUS is inherited in an autosomal dominant or an autosomal recessive pattern of inheritance 1.
Atypical hemolytic-uremic syndrome should be distinguished from a more common condition called typical hemolytic-uremic syndrome. The two disorders have different causes and different signs and symptoms 2. The atypical hemolytic-uremic syndrome is probably about 10 times less common than the typical hemolytic-uremic syndrome 2. Unlike the atypical hemolytic uremic syndrome, the typical hemolytic uremic syndrome is caused by infection with certain strains of Escherichia coli (E.coli) bacteria that produce toxic substances called Shiga-like toxins (Stx HUS) and is generally foodborne. The typical hemolytic uremic syndrome is characterized by severe diarrhea and most often affects children younger than 10. The typical hemolytic uremic syndrome is less likely than the atypical form to involve recurrent attacks of kidney damage that lead to end-stage renal disease (ESRD) or kidney failure. Unlike individuals with typical HUS, who usually recover from the life-threatening initial episode and usually respond well to supportive treatment, individuals with atypical hemolytic-uremic syndrome are much more likely to develop chronic serious complications such as severe high blood pressure (hypertension) and kidney (renal) failure. The signs and symptoms of atypical hemolytic-uremic syndrome result from the formation of tiny blood clots (microthrombi) in various small blood vessels of the body. These clots reduce or prevent proper blood flow to various organs of the body, especially the kidneys.
Atypical hemolytic uremic syndrome causes
Atypical hemolytic-uremic syndrome often results from a combination of environmental and genetic factors. Most cases of atypical hemolytic uremic syndrome are associated with mutations amongst the multiple genes that produce (encode) proteins involved in the alternate pathway of complement, which is part of the complement system of the innate immune system. The complement system is a complex group of proteins that work together to fight infection in the body. Complement proteins respond to bacteria, viruses or other foreign substances in the body and ultimately produce a large multi-protein complex that directly attacks these foreign invaders. Other complement proteins regulate the formation of this attack complex in order to protect the body’s own cells from being damaged. Most individuals with atypical hemolytic uremic syndrome have a mutation in one or more of the genes that encode these regulatory proteins.
Some people with atypical hemolytic-uremic syndrome do not have any known genetic changes or environmental triggers for the disease. In these cases, the disorder is described as idiopathic (unknown reasons).
Mutations in at least seven genes appear to increase the risk of developing the disorder, but is not enough to cause atypical hemolytic uremic syndrome on its own. These genes most likely convey a genetic predisposition to developing atypical hemolytic uremic syndrome rather than causing the disorder outright. A genetic predisposition means that a person carries a gene (or genes) for a disorder, but it may not be expressed unless it is triggered or “activated” under certain circumstances such as because of particular environmental factors or because of an another illness.
In most individuals, there is a triggering occurrence or event such as an acute infection. Other triggers have included chicken pox (varicella) or H1N1 influenza. In women, pregnancy is a common trigger. In addition to an environmental trigger, affected individuals may require a mutation in a second complement gene or a second genetic variant such as single nucleotide polymorphisms (SNP) for the development the disorder. Single nucleotide polymorphisms are the most common genetic variation in humans and occur frequently in a person’s DNA. Most single nucleotide polymorphisms have no effect on a person’s health.
About 30% of the time, atypical hemolytic uremic syndrome is associated with malfunctions in the gene (CFH) responsible for the production of a blood protein known as factor H. This is the most common gene mutation associated with atypical hemolytic uremic syndrome. Factor H is one of the regulatory proteins of the complement system that protect blood vessels from injury. When factor H is deficient or inactive, there is the potential for damage to the small vessels in the kidneys with secondary injury to red blood cells and platelets.
Mutations in the other genes (e.g., C3, CD46 (MCP), CFB, CFHR1, CFHR3, CFHR4, CFI, DGKE, and THBD) have each been identified in a smaller percentage of cases. Loss-of-function mutations in genes encoding other complement regulatory proteins, membrane cofactor protein (MCP) and factor I, or gain-of-function mutations in genes encoding the key complement proteins complement factor B and C3 are associated with atypical hemolytic uremic syndrome. Finally, mutations in the gene encoding thrombomodulin (THBD), an endothelial anticoagulant glycoprotein with complement regulatory properties, have been found in 3-5% of individuals with atypical hemolytic uremic syndrome.
A seventh gene known as DGKE has been identified as being associated with atypical hemolytic uremic syndrome. Some researchers believe DGKE-associated atypical hemolytic uremic syndrome is a similar, but distinct disorder. The protein produced by the DGKE gene is not associated with the alternate pathway of complement and instead appears to be involved in the coagulation process.
The specific genetic mutation present may be more likely to be associated with specific symptoms or severity of the disorder. This is known as genotype-phenotype correlation. The response to treatment can also be influenced by the specific underlying genetic mutation. For example, MCP mutations have a lower risk of permanent kidney failure and a low risk of disease recurrence following a kidney transplant. Individuals with mutations in the CFH or THBD genes are more likely to present during childhood.
The genes associated with atypical hemolytic-uremic syndrome provide instructions for making proteins involved in a part of the body’s immune response known as the complement system. This system is a group of proteins that work together to destroy foreign invaders (such as bacteria and viruses), trigger inflammation, and remove debris from cells and tissues. The complement system must be carefully regulated so it targets only unwanted materials and does not attack the body’s healthy cells. The regulatory proteins associated with atypical hemolytic-uremic syndrome protect healthy cells by preventing activation of the complement system when it is not needed.
Mutations in the genes associated with atypical hemolytic-uremic syndrome lead to uncontrolled activation of the complement system. The overactive system attacks cells that line blood vessels in the kidneys, causing inflammation and the formation of abnormal clots. These abnormalities lead to kidney damage and, in many cases, kidney failure and end-stage kidney disease.
Some individuals develop atypical hemolytic uremic syndrome because of autoantibodies that target proteins encoded by complement genes. Antibodies are specialized proteins that react against foreign materials in the body, bringing about their destruction. When antibodies react against healthy tissue, they are known as autoantibodies. Anti-factor H autoantibodies have been reported in 6-10% of cases, mainly children. Less often, autoantibodies that target other complement proteins have been identified. The reason why these autoantibodies develop is unknown.
In approximately 30%-50% of individuals with atypical hemolytic uremic syndrome, no mutation in a complement gene and no autoantibodies can be detected. These individuals may be referred to as having idiopathic atypical hemolytic uremic syndrome. However, many researchers believe these individuals most likely have yet unidentified mutations in complement genes.
Although gene mutations increase the risk of atypical hemolytic-uremic syndrome, studies suggest that they are often not sufficient to cause the disease. In people with certain genetic changes, the signs and symptoms of the disorder may be triggered by factors including certain medications (such as anticancer drugs), chronic diseases, viral or bacterial infections, cancers, organ transplantation, or pregnancy.
Atypical hemolytic uremic syndrome inheritance pattern
Most cases of atypical hemolytic-uremic syndrome are sporadic, which means that they occur in people with no apparent history of the disorder in their family. Less than 20 percent of all cases have been reported to run in families. When the disorder is familial, these mutations are transmitted (inherited) as an autosomal dominant or an autosomal recessive pattern of inheritance. The dominant form affects adults more often than children.
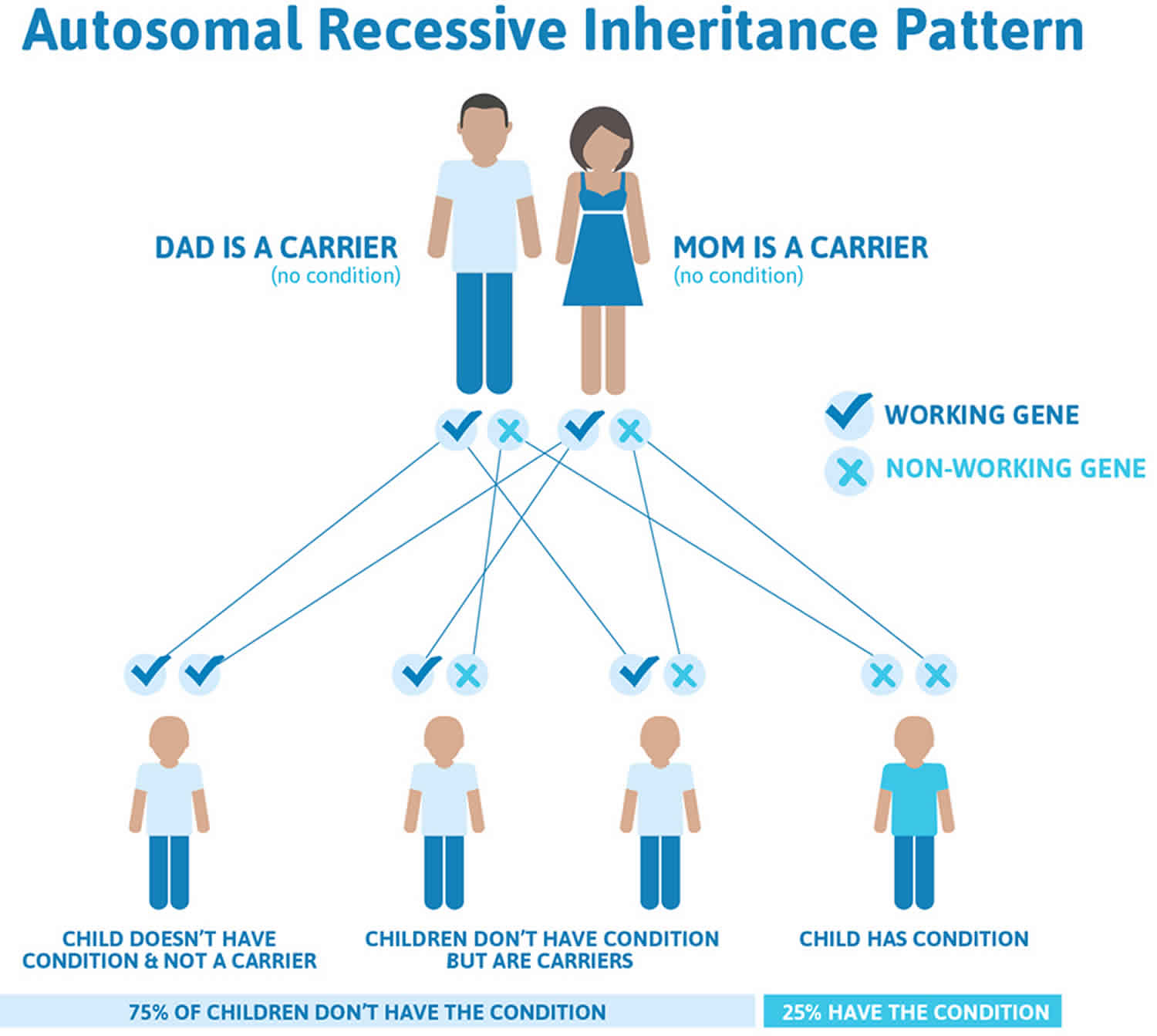
Autosomal dominant inheritance means one copy of an altered gene in each cell is sufficient to increase the risk of the disorder (see Figure 1). In some cases, an affected person inherits the mutation from one affected parent. The abnormal gene can be inherited from either parent or can be the result of a new mutation (gene change) in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females. However, most people with the autosomal dominant form of atypical hemolytic-uremic syndrome have no history of the disorder in their family. Because not everyone who inherits a gene mutation will develop the signs and symptoms of the disease, an affected individual may have unaffected relatives who carry a copy of the mutation.
Autosomal recessive inheritance means both copies of a gene in each cell have mutations (see Figure 2). The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition. If an individual inherits one normal gene and one gene for the disease, the person will be a carrier for the disease but usually will not show symptoms. The risk for two carrier parents to both pass the altered gene and have an affected child is 25% with each pregnancy. The risk to have a child who is a carrier like the parents is 50% with each pregnancy. The chance for a child to receive normal genes from both parents is 25%. The risk is the same for males and females.
To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Figure 1. Autosomal dominant inheritance pattern
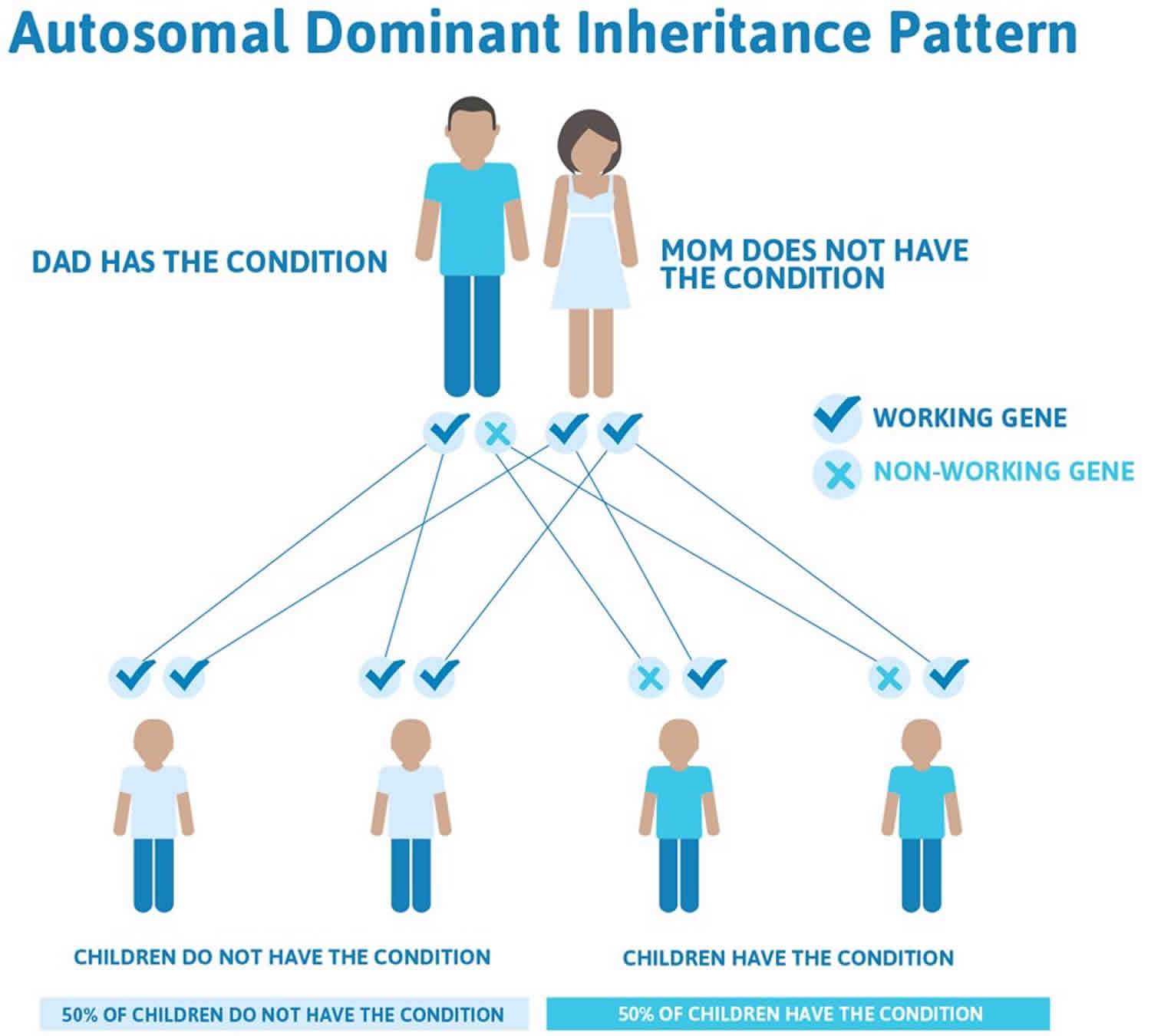
Figure 2. Autosomal recessive inheritance pattern
Atypical hemolytic uremic syndrome symptoms
The onset of atypical hemolytic uremic syndrome ranges from before birth (prenatally) to adulthood. In young children, the disorder often develops suddenly and usually follows an infection, particularly an upper respiratory infection or gastroenteritis. When it follows an episode of gastroenteritis it can more easily be confused with Shiga-like toxins hemolytic uremic syndrome which almost always is preceded by diarrhea. The disease has different causes and can be unpredictable in how it will progress in one individual as opposed to another.
Many affected individuals present with vague feelings of illness, fatigue, irritability, and lethargy that can potentially lead to hospitalization. The early phases may be difficult to diagnose, and the condition tends to be progressive. Because complications and relapse are common, it is critical that atypical hemolytic uremic syndrome be recognized at this stage.
The three main findings of atypical hemolytic uremic syndrome are hemolytic anemia, thrombocytopenia, and acute kidney failure. Although most affected individuals develop these three conditions, some individuals will not. Hemolytic anemia is a condition in which there is a premature destruction (hemolysis) of red blood cells. Thrombocytopenia is a condition in which there are low levels of platelets, a blood cell that is involved in clotting.
Kidney disease can be mild or severe. Kidney damage tends to worsen with each subsequent episode. Blood and protein in the urine (hematuria and proteinuria), frequent indicators of kidney disease, are common, especially during acute episodes. Kidney disease is progressive and can potentially progress to cause end stage renal failure, necessitating chronic dialysis or a kidney transplant.
High blood pressure (hypertension) is common and can result from kidney disease or because of lack of blood flow (ischemia) due to the formation small blood clots (microthrombi). Hypertension can be severe and may be associated with headaches and seizures.
Because small blood clots can potentially form in blood vessels serving other organs of the body, organ damage and failure can occur elsewhere besides the kidneys (which is the organ that is most commonly affected). The brain, gastrointestinal tract, liver, lungs, and heart can also be affected. Specific symptoms can vary based upon the specific organ system involved. Cardiovascular complications can include disease of the heart muscle (cardiomyopathy) or heart attack (myocardial infarction). Neurological complications can include headaches, double vision (diplopia), irritability, drowsiness, facial paralysis, seizures, transient ischemic attacks, stroke, and coma. Gastrointestinal bleeding may also occur. The lungs can be affected and bleeding or fluid accumulation in the lungs (pulmonary edema) can occur.
Atypical hemolytic uremic syndrome diagnosis
Diagnosing atypical hemolytic uremic syndrome is complicated by the fact that it is more difficult to establish without a family history of the disorder. The diagnostic criteria associated with atypical hemolytic uremic syndrome are hemolytic anemia (anemia in the presence of broken red blood cells), low platelet count (thrombocytopenia) and kidney dysfunction. Atypical hemolytic uremic syndrome is considered genetic when two or more members of the same family are affected by the disease at least six months apart and exposure to a common triggering infectious agent has been excluded, or when a disease-causing mutation(s) is identified in one of the genes known to be associated with atypical hemolytic uremic syndrome, irrespective of familial history.
Individuals with atypical hemolytic uremic syndrome do not present with the aggressive and bloody diarrhea that characterize the onset of Shiga-like toxins HUS, although 30-50% of children with atypical hemolytic uremic syndrome may have diarrhea. The absence of bloody diarrhea, negative stool cultures for Shiga toxin producing-E. coli (most frequently E. coli 0157:H7) associated with hemolytic uremic syndrome, a progressive course, and prior manifestations of nephrotic syndrome, such as swelling from the accumulation of fluid (edema), presence of blood in the urine (hematuria), excessive protein in the urine (proteinuria), and reduced albumin in the serum (hypoalbuminemia), with marked elevation in blood pressure are features that alert pediatricians and kidney specialists (nephrologists) to the possible diagnosis of atypical hemolytic uremic syndrome.
AHUS genetic testing
GeneTests (https://www.ncbi.nlm.nih.gov/gtr) lists the names of laboratories that are performing genetic testing for atypical hemolytic-uremic syndrome. To view the contact information for the clinical laboratories conducting testing and follow the “testing” link pertaining to each gene.
Please note: Most of the laboratories listed through GeneTests do not accept direct contact from patients and their families; therefore, if you are interested in learning more, you will need to work with a health care provider or a genetics professional. In the Genetic Services section of this letter we provide a list of online resources that can assist you in locating a genetics professional near you.
To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Atypical hemolytic uremic syndrome treatment
Treatment by a medical team familiar with the unique challenges of atypical hemolytic uremic syndrome is recommended and can include pediatricians or general internists, kidney specialists (nephrologists), intensive care physicians, nurses, nutritionists and social workers.
Initially, affected individuals may receive supportive care including maintaining proper nutrition and electrolyte and fluid balance through intravenous feeding (parenteral) when and if necessary. Blood transfusions are administered when the hemoglobin level is below 7 g/dl. Platelet transfusions are avoided if at all possible. Drugs that expand the blood vessels (vasodilators) are used to control blood pressure (hypertension). In some instances, individuals are diagnosed with atypical hemolytic uremic syndrome when they already have kidney damage and may initially require supportive measures such as peritoneal dialysis or hemodialysis.
In 2011, the U.S Food and Drug Administration (FDA) approved the use of the humanized anti-C5 monoclonal antibody eculizumab (Soliris®, Alexion Pharmaceuticals, Inc., Boston, MA, USA) as a treatment for acute hemolytic uremic syndrome. This drug is used to block excessive complement activation in individuals with atypical hemolytic uremic syndrome. Eculizumab has led to improvement with the blood abnormalities (reduced hemolysis and stabilized platelet counts) and reversed acute kidney injury. Eculizumab is now recommended as first-line therapy in both children and adults with a confirmed or strongly suspected diagnosis of atypical hemolytic uremic syndrome.
In 2019, the FDA approved Ultomiris (ravulizumab-cwvz), a long-acting C5 complement inhibitor, for the treatment of adults and pediatric patients one month of age and older with atypical hemolytic uremic syndrome to inhibit thrombotic microangiopathy.
For years, plasma therapy (plasma exchange and plasma infusion) was the standard treatment for individuals with atypical hemolytic uremic syndrome. Both infusions of fresh frozen plasma (plasma infusion) as well as plasma exchange (plasmapheresis) were utilized. Fresh frozen plasma is a blood derivative that is obtained from donors. Plasma exchange is a method for removing potentially harmful substances (e.g. toxins, metabolic substances, and plasma parts) from the blood. Blood is removed from the affected individual and blood cells are separated from the plasma. The plasma is then replaced with other human plasma and the blood is transfused into the affected individuals. Plasma exchange can also remove mutant factors and autoantibodies.
Plasma therapy has led to a remission in a subset of individuals. However, many of these individuals experience relapses if they don’t receive long term maintenance therapy. Other individuals, while seeing an improvement in the blood complications (e.g. hemolysis and thrombocytopenia), still experience progressive kidney damage, ultimately progressing to end stage renal disease. Plasma therapy has not been studied as a treatment for atypical hemolytic uremic syndrome in a controlled fashion.
Individuals who fail to recover kidney function after treatment may require a kidney (renal) transplant. Renal transplantation had been a controversial option for atypical hemolytic uremic syndrome because an estimated 50% of affected individuals who underwent this procedure had a recurrence of the disease in the newly grafted organ. Molecular genetic tests could help to define graft prognosis; thus, all patients should undergo such testing prior to transplantation. Molecular genetic testing should be particularly recommended before live related donation to avoid the risk of triggering disease in the donors. Eculizumab has been shown to be effective in preventing and treating post-transplant atypical hemolytic uremic syndrome recurrences.
Plasmapheresis in conjunction with drugs that suppress the immune system (immunosuppressive therapy) can be used to treat individuals with atypical hemolytic uremic syndrome due to autoantibodies to factor H.
The optimal treatment strategy for individuals with atypical hemolytic uremic syndrome due to mutations in the DGKE gene has not been established. The effectiveness of eculizumab for these individuals has not been established presumably because the underlying defect does not involve complement proteins. Several affected individuals received a kidney transplant with no reported recurrence of the disorder.
Atypical hemolytic uremic syndrome prognosis
Patients with atypical hemolytic uremic syndrome have an extremely poor prognosis. Among those with the most commonly identified atypical hemolytic uremic syndrome genetic mutation, the proportion of patients experiencing negative outcomes (e.g., need for dialysis, permanent kidney damage, death) within the first year rises to 70% 3. However, sudden morbidity and mortality can occur regardless of mutational status. Atypical hemolytic uremic syndrome can arise at any age, with more than 40% of cases first reported after 18 years of age 4. The oldest presentation in one study was at age 83 4. Kidney transplantation for atypical hemolytic uremic syndrome patients with end-stage renal disease was rarely considered because of a high incidence of graft loss due to thrombotic microangiopathy (TMA) recurrence in the transplanted organ in up to 90% of patients 5. Consequently, most atypical hemolytic uremic syndrome patients with end-stage renal disease undergo chronic dialysis, which is associated with significant morbidities and worsened prognosis 5. Combined liver-kidney transplantation has been attempted in patients with atypical hemolytic uremic syndrome, although this high-risk procedure has a mortality rate approaching 50% 6.
Quality of life is very poor for patients with atypical hemolytic uremic syndrome, who are burdened with fatigue, renal complications, hypertension, neurological impairment, gastrointestinal distress, clotting at the site of venous access, and ultimately, death 7. Plasma exchange or plasma infusion is also reported to be associated with significant safety risks and is highly disruptive to patients’ lives due to the requirements for extensive vascular access and frequent administration 8.
Since the approval of eculizumab (Soliris) the prognosis for atypical hemolytic uremic syndrome patients has improved greatly 9. Pediatric age at disease onset and presence of genetic or autoimmune complement abnormalities are risk factors for thrombotic microangiopathy events off treatment. Overall, patients who discontinue eculizumab may be at risk for additional thrombotic microangiopathy manifestations and renal function decreases. Discontinuation of eculizumab, with careful monitoring, is an option in select patients with consideration of patient preference, organ function normalization, and risk factors for relapse, including mutational analysis, age of onset, and history of multiple thrombotic microangiopathy episodes 9.
- Alpers CE. The Kidney. In: Kumar ed. Robbins and Cotran Pathologic Basis of Disease, Professional Edition , 8th ed. Philadelphia, PA: Saunders; 2009[↩]
- Atypical hemolytic-uremic syndrome. https://ghr.nlm.nih.gov/condition/atypical-hemolytic-uremic-syndrome[↩][↩]
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-1279. doi:10.1182/blood-2005-10-007252 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1895874[↩]
- Noris M, Caprioli J, Bresin E, et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin J Am Soc Nephrol. 2010;5(10):1844-1859. doi:10.2215/CJN.02210310 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2974386[↩][↩]
- Zuber J, Le Quintrec M, Sberro-Soussan R, Loirat C, Frémeaux-Bacchi V, Legendre C. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. 2011;7(1):23-35. doi:10.1038/nrneph.2010.155[↩][↩]
- Bresin E, Daina E, Noris M, et al. Outcome of renal transplantation in patients with non-Shiga toxin-associated hemolytic uremic syndrome: prognostic significance of genetic background. Clin J Am Soc Nephrol. 2006;1(1):88-99. doi:10.2215/CJN.00050505[↩]
- Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267-1279. doi:10.1182/blood-2005-10-007252[↩]
- Ariceta G, Besbas N, Johnson S, et al. Guideline for the investigation and initial therapy of diarrhea-negative hemolytic uremic syndrome. Pediatr Nephrol. 2009;24(4):687-696. doi:10.1007/s00467-008-0964-1[↩]
- Menne J, Delmas Y, Fakhouri F, et al. Outcomes in patients with atypical hemolytic uremic syndrome treated with eculizumab in a long-term observational study. BMC Nephrol. 2019;20(1):125. Published 2019 Apr 10. doi:10.1186/s12882-019-1314-1 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6456946[↩][↩]




{kind=link}