Autoimmune polyendocrine syndrome
Autoimmune polyendocrine syndrome also called autoimmune polyglandular syndromes, are clusters of endocrine abnormalities that occur in discreet patterns in subjects with immune dysregulation and that permit treatment and anticipation of associated systemic or other hormonal deficiencies 1. Three major entities recognized are: autoimmune polyendocrine syndrome type 1 (APS1), autoimmune polyendocrine syndrome type 2 (APS2) and autoimmune polyendocrine syndrome type 3 (APS3). Recently, a new category has emerged. It is iatrogenic polyendocrinopathy due to use of immunoregulatory agents in patients with cancer, by which the tumor’s blockade of immune regulatory checkpoints is inhibited, so that tumor antigens that have evaded recognition can now be attacked, but at the expense of activating autoimmunity 2.
The term “polyendocrine” itself is a misnomer, in that not all patients have multiple endocrine disorders, and many have nonendocrine autoimmune diseases 3. Nevertheless, the recognition that patients in whom multiple autoimmune disorders are diagnosed may have a specific genetic syndrome, may be at increased risk for multiple autoimmune disorders, and may have relatives who have an increased risk should spur clinicians toward early diagnosis and treatment 4.
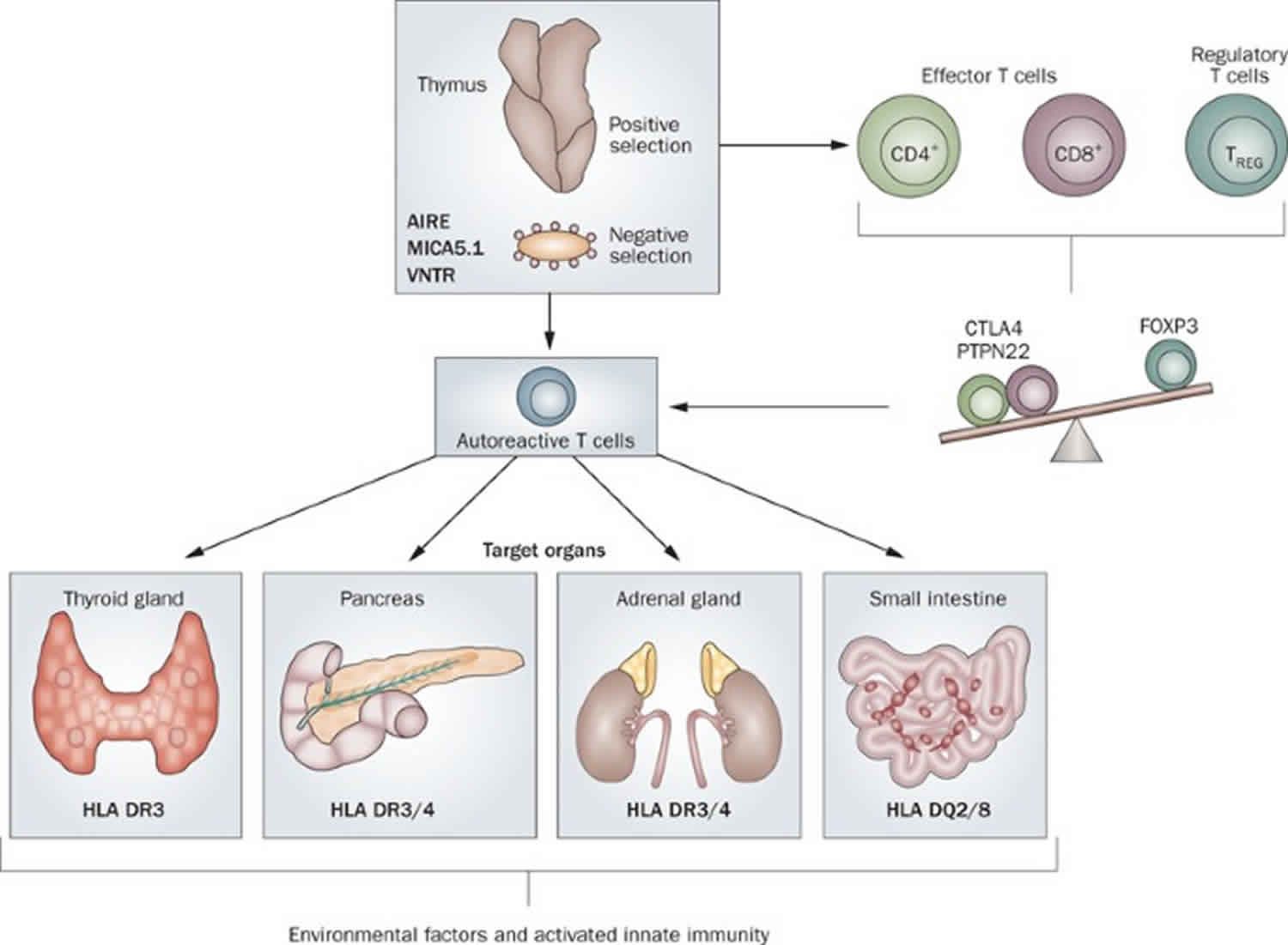
Autoimmune polyendocrine syndrome type 1 (APS1) is a syndrome characterized by chronic mucocutaneous candidiasis, hypoparathyroidism, and primary adrenal insufficiency (Addison’s disease), as well as ectodermal dystrophy and a host of other endocrine and non-endocrine tissue involvement in autoimmune destructive processes. The underlying cause is homozygous inactivating mutation in the autoimmune regulator gene (AIRE) which permits the intra-thymic expression of ectopic antigens normally expressed only in specific peripheral tissues (e.g. insulin), so that T-cells as they mature within the thymus and acquire a receptor for the self-antigen are eliminated (negative selection), thereby avoiding autoimmunity 1. Recent studies demonstrate that in addition to the classical homozygous mutations, single gene dominant mutations in AIRE (autoimmune regulatory gene) play an important role in autoimmune regulation and its disorders. Autoimmune polyendocrine syndrome type 2 (APS2) and autoimmune polyendocrine syndrome type 3 (APS3) are both due to mutations in the HLA DQ/DR regions which regulate antigen presentation to T-cell receptors; autoimmune polyendocrine syndrome type 2 (APS2) is characterized by type 1 diabetes mellitus, Addison disease, and hypothyroidism, whereas autoimmune polyendocrine syndrome type 3 (APS3) is similar but without Addison disease. In keeping with other autoimmune disorders, these entities are more frequent in females, whereas autoimmune polyendocrine syndrome type 1 (APS1) has no sexual predominance. The recent emergence of autoimmune endocrinopathies in patients treated with immunoregulatory agents for cancer adds a new dimension to considerations of autoimmune polyendocrinopathy syndromes.
Autoimmune polyendocrine syndrome type 1
Autoimmune polyendocrine syndrome type 1 also called autoimmune polyglandular syndrome type 1 or autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), is an inherited autoimmune disease which is characterized by a triad of hypoparathyroidism, Addison disease, and chronic mucocutaneous candidiasis 5. In most cases, the signs and symptoms of autoimmune polyendocrine syndrome type 1 begin in childhood or adolescence. This condition commonly involves three characteristic features: chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal gland insufficiency. Affected individuals typically have at least two of these features, and many have all three.
Chronic mucocutaneous candidiasis is a tendency to develop fungal infections of the skin, the nails, and the moist lining of body cavities (mucous membranes) caused by a type of fungus called Candida. These infections, which are commonly known as yeast infections, are chronic, which means they recur and can last a long time. Chronic mucocutaneous candidiasis is usually the first of the three characteristic features of autoimmune polyendocrine syndrome type 1 to become apparent in people with this disorder. Almost all affected individuals develop infections of the oral cavity (known as thrush). Infections of the tube that carries food from the mouth to the stomach (the esophagus) are also common, while the skin and nails are affected less often. In women, vaginal infections frequently occur.
Other features of autoimmune polyendocrine syndrome type 1 result from the body’s immune system attacking the network of hormone-producing glands (the endocrine system). The second characteristic feature of the disorder is hypoparathyroidism, which is a malfunction of the parathyroid glands. These glands secrete a hormone that regulates the body’s use of calcium and phosphorus. Damage to the parathyroid glands leads to reduced parathyroid hormone production (hypoparathyroidism). Hypoparathyroidism can cause a tingling sensation in the lips, fingers, and toes; muscle pain and cramping; weakness; and fatigue. Serious effects of hypoparathyroidism, such spasms of the voicebox (larynx) leading to breathing problems and seizures, can be life-threatening.
Damage to the small hormone-producing glands on top of each kidney (adrenal glands) results in a third major feature of autoimmune polyendocrine syndrome type 1, adrenal gland insufficiency (autoimmune Addison disease). Reduced hormone production by the adrenal glands leads to signs and symptoms that can include fatigue, muscle weakness, loss of appetite, weight loss, low blood pressure, and changes in skin coloring. Other endocrine problems that can occur in autoimmune polyendocrine syndrome type 1 include type 1 diabetes resulting from impaired production of the hormone insulin; a shortage of growth hormone leading to short stature; problems affecting the internal reproductive organs (ovaries or testes) that can cause inability to conceive children (infertility); and dysfunction of the thyroid gland (a butterfly-shaped tissue in the lower neck), which can result in many symptoms including weight gain and fatigue.
Besides the above triad, a host of other autoimmune diseases may present with autoimmune polyendocrine syndrome type 1. Autoimmune problems affecting non-endocrine tissues can lead to a variety of additional signs and symptoms in people with autoimmune polyendocrine syndrome type 1. These features occur more often in North American populations than in European populations. Rashes that resemble hives (urticarial eruptions) are common and often occur in infancy and early childhood. Other early signs and symptoms may include thin enamel on the teeth (enamel hypoplasia) and chronic diarrhea or constipation associated with difficulty in absorbing nutrients from food. Additional features that occur in people with autoimmune polyendocrine syndrome type 1, many of which can lead to permanent organ and tissue damage if left untreated, include stomach irritation (gastritis), liver inflammation (hepatitis), lung irritation (pneumonitis), dry mouth and dry eyes (Sjogren-like syndrome), inflammation of the eyes (keratitis), kidney problems (nephritis), vitamin B12 deficiency, hair loss (alopecia), loss of skin color in blotches (vitiligo), high blood pressure (hypertension), or a small (atrophic) or absent spleen (asplenia).
Autoimmune polyendocrine syndrome type 1 occurs in about 1 in 90,000 to 200,000 people in most populations studied, which have been mainly in Europe. The prevalence in Norway is 1 per 90,000 and in Ireland is 1 per 130,000, but it is more common in certain populations, affecting about 1 in 9,000 to 25,000 people among Iranian Jews (1 per 9000), Sardinians (1 per 14000) and Finns 6. There is no discernable gender preponderance. The age of the first manifestation also differs widely ranging from 0.2 to 18 years 7.
Autoimmune polyendocrine syndrome type 1 is caused by mutations in the autoimmune regulatory gene (AIRE); so far about 60 different mutations have been described; this is a potentially underdiagnosed condition due to the rarity and enormous variability in its presentation. HLA-DR/DQ genes also play a role in predisposing to which of the component autoimmune disease the patient actually develops 8.
Autoimmune polyendocrine syndrome type 1 results from a failure to eliminate T-cells that have acquired receptors to auto-antigens, as these T-cells mature and traverse the thymic epithelium during their development 1. Normally, such T-cells are prevented from entering the periphery because of the ectopic expression of multiple antigens within the thymus that usually are expressed only in discrete tissues, e.g. insulin in pancreatic β-cells. A developing T-cell that acquires and expresses a receptor for insulin will be bound to the ectopically expressed insulin antigen within the thymus, undergo apoptosis and be excluded from entering the periphery to initiate auto-immunity. This ectopic expression of antigens within the thymus is mediated by the Auto-Immune REgulator gene (AIRE) located on chromosome 21.
Diagnosis is made by finding antibodies against interferon-omega or interferon- alpha or by detecting disease-causing mutations in the AIRE gene. Management of autoimmune polyendocrine syndrome type 1 requires collaboration among multiple specialties because of the multiplicity of organs affected 7.
Autoimmune polyglandular syndrome type 1 causes
Mutations in the autoimmune regulatory (AIRE) gene located in the short arm of chromosome 21 cause autoimmune polyendocrine syndrome type 1 5. AIRE is expressed in the thymic medullary cells and the AIRE gene provides instructions for making a protein called the autoimmune regulator. As its name suggests, this protein plays a critical role in regulating certain aspects of immune system function. Specifically, it helps the body distinguish its own proteins and cells from those of foreign invaders (such as bacteria, fungi, and viruses). This distinction is critical because to remain healthy, a person’s immune system must be able to identify and destroy potentially harmful invaders while sparing the body’s normal tissues.
There are over 60 recognized different mutations in the AIRE gene, which might explain the variability in presentation and natural course of the disease. Over 95% of patients with autoimmune polyendocrine syndrome type 1 have one of the two most common mutations, which are arginine substitution at position 257 and 13 base pair deletion in exon 8 9.
Mutations in the AIRE gene reduce or eliminate the function of the autoimmune regulator protein. Without enough of this protein function, the immune system’s ability to distinguish between the body’s proteins and foreign invaders is impaired, and it may attack the body’s own organs. This reaction, which is known as autoimmunity, results in inflammation and can damage otherwise healthy cells and tissues. Autoimmune damage to the adrenal glands, parathyroid glands, and other organs underlies many of the major features of autoimmune polyendocrine syndrome type 1.
Studies suggest that AIRE gene mutations also result in immune substances (antibodies) mistakenly attacking proteins involved in an immune process called the IL-17 pathway, which is important in the body’s defense against Candida. This pathway, which depends on specialized proteins called IL-17 cytokines for signaling, creates inflammation, sending additional cytokines and white blood cells to fight foreign invaders and promote tissue repair. In addition, the IL-17 pathway promotes the production of certain antimicrobial protein segments (peptides) that control growth of Candida on the surface of mucous membranes. By damaging IL-17 cytokines, AIRE gene mutations are thought to impair the IL-17 pathway’s function, resulting in chronic mucocutaneous candidiasis in people with autoimmune polyendocrine syndrome type 1.
Researchers believe that differences in the effects of specific AIRE gene mutations as well as variations in other genes that have not been identified may help explain why the signs and symptoms of autoimmune polyendocrine syndrome type 1 can vary among affected individuals and populations.
Autoimmune polyglandular syndrome type 1 inheritance pattern
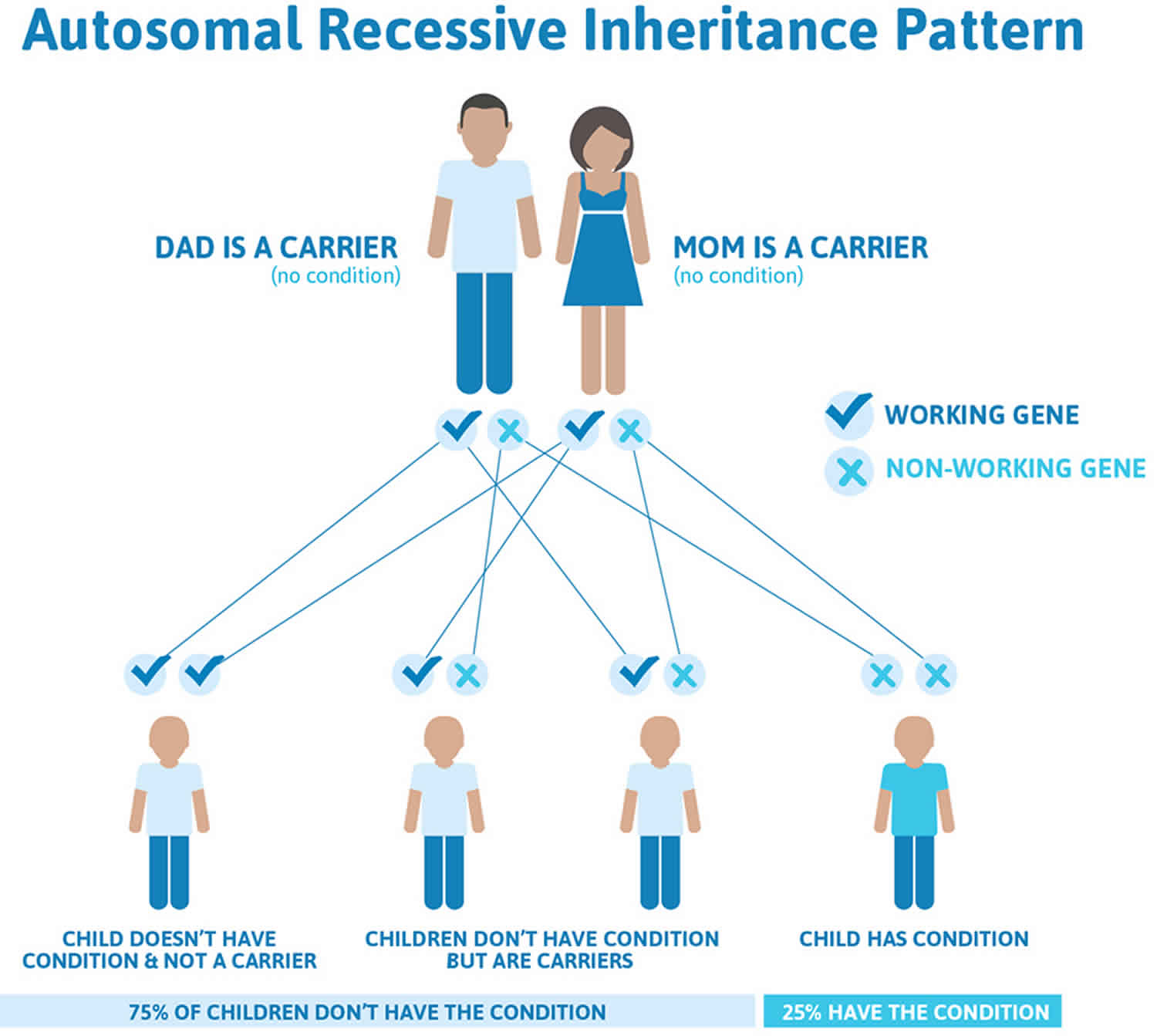
Autoimmune polyglandular syndrome type 1 is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
In rare cases, people with one copy of certain AIRE gene mutations in each cell have some features of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED), such as chronic mucocutaneous candidiasis, hypoparathyroidism, or vitamin B12 deficiency, but do not have the full pattern of signs and symptoms that typically characterize the disorder. These individuals usually have one similarly-affected parent.
Figure 1. Autoimmune polyglandular syndrome type 1 autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Autoimmune polyglandular syndrome type 1 signs and symptoms
The clinical signs and symptoms of autoimmune polyglandular syndrome type 1 are variable, even between siblings who have the same mutation. The most common initial manifestation is chronic mucocutaneous candidiasis presenting at a median age of 1.7 to 3 years. Hypoparathyroidism is the second component of the triad to appear; it appears earlier in females as compared to males. Addison’s disease (adrenocortical insufficiency) typically presents 5 years after hypoparathyroidism 10.
Patients with autoimmune polyendocrine syndrome type 1 have a defect of the immune system involving a particular subset of T-cells called Treg (T-regulatory) cells. It is suggested that this Treg-cell defect leads to the wide spread loss of immune tolerance, causing the autoimmunities in the disease. However, a specific defect in immunity to candidiasis indicates the presence of an immune effector defect also. Possibly, the invariable presence of auto-antibodies to the interferon family of immunological molecules called cytokines may prove to be the underlying reason.
Chronic mucocutaneous candidiasis (CMC), a condition of recurrent candidiasis infections that may involve the skin, nails, oral, anal and genital mucosa, is a hallmark of autoimmune polyendocrine syndrome type 1. It is often the first manifestation of autoimmune polyendocrine syndrome type 1, typically appearing and recurring frequently within the first two years of life. The chronic mucocutaneous candidiasis of autoimmune polyendocrine syndrome type 1 generally presents in babies as thrush (oral candidiasis), diaper rash, and/or nail involvement. Clinically, oral candidiasis presents as redness and ulceration at the angle of the mouth. It may also manifest as white or gray plaques and hyperkeratosis. Rarely, leukoplakic nodules or patches are present, which could be potentially precancerous. Esophageal candidiasis presents with chest pain and painful swallowing (odynophagia). Recurrent infections can lead to esophageal strictures which may need dilation. Rarely, genital and intestinal candidiasis can occur. There are also reports of squamous cell cancers of the oral cavity and esophagus in association with autoimmune polyglandular syndrome type 1 which could have been induced by chronic candidiasis 11.
Hypoparathyroidism (under functioning of the parathyroid glands) is the second most prevalent feature of autoimmune polyendocrine syndrome type 1 with about 33% presenting at age 5 years; it is more prevalent in females. More than 75% of patients develop hypoparathyroidism, usually before age 10-years. Dysfunction of the parathyroid glands leads to below-normal level of serum calcium together with elevated phosphorus levels. In turn, this can lead to a host of clinical findings, including muscle cramping and spasms, rigidity (tetany) and even seizures. Clinically, hypoparathyroidism presents as numbness or tingling in the perioral area and fingertips, muscle cramps and seizures. Chvostek and Trousseau signs are indicators of neuromuscular irritability and can be positive. Chvostek’s sign is elicited by gently tapping the angle of the jaw which would cause twitching of the lips. Trousseau sign is elicited by inflating a blood pressure cuff around the shoulder to a pressure higher than systolic blood pressure for 3 minutes which should lead to flexion of the wrist and metacarpophalangeal joints and extension of the interphalangeal joints 10.
Adrenal insufficiency also known as Addison’s disease is the last of the triad to appear and manifests between 5-15 years. Adrenocortical insufficiency is characterized by chronic and insufficient functioning of the cortex (outer layer) of the adrenal gland. This malfunction results in a deficiency of the glucocorticoid and salt retaining hormones cortisol and aldosterone respectively. Deficiencies of these hormones may presents as weakness, muscle cramps, faintness, fatigue, abdominal pain, diarrhea, nausea and vomiting, low blood pressure, dehydration, salt craving, dizziness, weight loss and hyperpigmentation. The physical exam may show hyperpigmentation of knuckles, joints, oral mucosa and scars. In females, there may be an absence of axillary and pubic hair because of the lack of production of adrenal androgens 12. These effects can become pronounced and life-threatening if not correctly identified and treated. However, steroid replacement therapy can precipitate or worsen hypocalcemia when hypoparathyroidism has not been already identified.
Other manifestations of autoimmune polyendocrine syndrome type 1 include:
Alopecia is the most common dermatological manifestation and typically appears around 40 years of age. The presentation varies widely from patchy alopecia to alopecia totalis. Alopecia correlates with the levels of anti-tyrosine hydroxylase antibodies 13.
Keratitis presents in about a quarter of the patients with autoimmune polyendocrine syndrome type 1. It appears around the age of 20 years. Its characteristics include decreased lacrimation, photophobia, and blepharospasm. Treatment is with topical glucocorticoids and cyclosporine. Topical vitamin A may prevent corneal ulcerations 14.
Gastritis and pernicious anemia may also present in association with autoimmune polyendocrine syndrome type 1. Anti-intrinsic factor antibodies are present which hinder the absorption of vitamin B12 leading to megaloblastic anemia. Management involves supplementation of vitamin B12.
There are reports of asplenia or absence of a spleen in patients with 20% of adults and 10% of children with autoimmune polyendocrine syndrome type 1. Howell-Jolly bodies are visible in the erythrocytes. Abdominal CT should be done to confirm the diagnosis of asplenia if Howell-Jolly bodies are detected. Asplenia makes the patient susceptible to infections by encapsulated organisms; therefore vaccinations against pneumococcus, meningococcus, and Hemophilus influenza are necessary 14.
Type 1 diabetes mellitus may present in patients aged 30 to 50 years. Diagnosis is by detecting fasting blood sugars greater than 126, or hemoglobin A1c of >6.5, or a random blood sugar of greater than 200 with symptoms of polyuria, polydipsia, or weight loss. Insulin antibodies or IA-2 antibodies may be detected 15.
Primary hypogonadism may be seen late in the disease in patients with anti-side-chain cleavage enzyme antibodies. The clinical features include fatigue, muscle loss, and decreased libido. Diagnosis is by low measured testosterone in conjunction with a high luteinizing hormone (LH). Treatment includes testosterone supplementations. Occasionally, isolated azoospermia may also accompany the hypogonadism 15.
Pituitary failure may be seen early in the course of the disease secondary to pituitary antibodies against tudor domain protein. Deficiencies of growth hormone, gonadotropin, or ACTH deficiency 10.
Autoimmune hepatitis is present in 20% of patients with autoimmune polyendocrine syndrome type 1. It does not appear after the age of 20 years. It characteristically presents with elevated levels of AST and ALT. Treatment involves immunosuppression with the use of glucocorticoids 16.
Autoimmune hypothyroidism or Hashimoto hypothyroidism, characterized by the presence of anti-thyroid peroxidase (TPO) antibodies anti-thyroglobulin antibodies may also be present. Clinical features include weight gain, loss of appetite, cold intolerance, and constipation. Diagnosis is by measuring thyroid-stimulating hormone (TSH) and thyroxine (T4) levels.
Enamel dysplasia is observable in 75% of Finnish patients. In milder forms, alternate bands of intact enamel and dysplastic enamel with observable pits and fissures present, while in more severe forms the entire enamel is involved. Prompt evaluation by a dentist is warranted, and in most instances extensive dental treatment is necessary. Due to the wearing of teeth and resulting malocclusion, temporomandibular joint pain can occur 17.
Vitiligo may be the initial manifestation and is a clinical feature in 33% of patients with autoimmune polyendocrine syndrome type 1. The degree of skin involvement is variable, and it can manifest as isolated spots or involving the whole skin. It correlates with the antibodies against SOX9 and SOX10. Unfortunately, there is no effective therapy against vitiligo 18.
Autoimmune polyglandular syndrome type 1 diagnosis
The diagnosis of autoimmune polyglandular syndrome type 1 is made by diagnosing at least two of the three major components or having mutations in both the AIRE genes. However, it can be difficult because of the rarity and varied presentation of the disorder. However, it is essential to make the diagnosis as it helps anticipate or discover the presence of other associated disorders. Genetic testing may help families make informed decisions about family planning.
Diagnostic criteria for autoimmune polyglandular syndrome type 1 5:
Definite diagnosis:
- Presence of two of the triad of chronic mucocutaneous candidiasis, hypoparathyroidism, or Addison disease.
- One of the above triad plus a sibling with proven autoimmune polyglandular syndrome type 1
- Known disease-causing mutations in the AIRE gene.
Probable diagnosis:
- Presence of one of the triad (before 30 years) along with one of the following components chronic diarrhea, keratitis, periodic rash with fever, severe constipation, autoimmune hepatitis, vitiligo, alopecia, and enamel hypoplasia.
- Any one of the above triad and anti-interferon antibodies
- Any one of the above triad and antibodies against NALP5, aromatic L-amino acid decarboxylase (AADC), tryptophan hydroxylase (TPH), or tyrosine hydroxylase (TH) 10.
Autoimmune polyglandular syndrome type 1 treatment
Treatment with autoimmune polyendocrine syndrome type 1 is multi-faceted and needs collaboration among different specialties. Referral to a center with experience in the treatment of autoimmune polyendocrine syndrome type 1 is recommended.
Treatment of autoimmune polyendocrine syndrome type 1 is currently directed toward the specific diseases that are apparent in each patient. In general, replacement therapy of the endocrine hormones that may be lacking, and patient education about the signs and symptoms of these deficiencies, are integral to treatment success. The educational aspect is of extreme importance, as this allows the patient to self-monitor, hopefully avoiding a life-threatening situation.
- Mucocutaneous candidiasis: Oral candidiasis treatment is with two topical polyene antifungals for 4-6 weeks. Recurrent oral candidiasis treatment is with weekly pulse prophylaxis with a polyene antifungal given every 3 weeks. Therapy for infection of the angle of the mouth is with natamycin or chlorhexidine gel. Vaginal candidiasis should have treatment with fluconazole. Nail infection can be challenging to treat and may require a referral to a podiatrist.
- Hypoparathyroidism: The objective of treatment is to maintain normocalcemia. Acute hypocalcemia warrants intravenous supplementation of calcium gluconate or calcium chloride. In nonacute circumstances, oral calcium and activated forms (1, 25 dihydroxy) of vitamin D such as Calctriol or Rocaltrol may suffice to maintain normocalcemia. The goal is to maintain calcium in the low normal range. Recombinant parathyroid hormone may be an option in cases where conventional treatment fails to normalize the calcium levels.
- Adrenal insufficiency or Addison’s disease: The mainstay of treatment is glucocorticoid supplementation with hydrocortisone. The recommended dose is 10 mg in the morning and 5 mg around 3:00 to 4:00 PM. In cases of malabsorption, higher doses of hydrocortisone may be necessary. Fludrocortisone (0.05 to 2 mg) may be used to replace mineralocorticoids; however, sensitivity varies among individuals. Dosing will require titration to maintain normokalemia and prevent pedal edema. All patients with adrenal insufficiency should wear an alert bracelet 10.
Autoimmune polyendocrine syndrome type 2
Autoimmune polyendocrine syndrome type 2 also known as Schmidt syndrome, is a rare autoimmune disorder in which there is a steep drop in production of several essential hormones by the glands that secrete these hormones. When first described, this disorder was thought to involve only adrenal insufficiency (Addison’s disease) and thyroid insufficiency (Hashimoto’s thyroiditis). However, over time, as more patients were studied, the scope of the disorder was expanded to include disorders of other underperforming endocrine glands. These include the gonads, which secrete sex hormones; the pancreas which secretes insulin and is intimately tied up with diabetes mellitus; and sometimes the parathyroid glands. Failure of the endocrine glands to function is usually accompanied by signs of malnutrition because the ability of the intestinal tract to absorb nutrients is reduced dramatically. Since the combination of affected glands differs from patient to patient, the signs of this disorder are diverse.
Autoimmune polyendocrine syndrome type 2 is characterized by the triad of type 1 diabetes mellitus, Addison disease (adrenal insufficiency) and thyroid autoimmunity with hypothyroidism, hyperthyroidism, or Hashimoto thyroiditis; type 1 diabetes mellitus and Addison disease are obligatory components, but thyroid autoimmunity is not and a host of other autoimmune entities can also be associated 1. These entities include celiac disease, vitiligo, alopecia, myasthenia gravis, pernicious anemia, IgA deficiency, hepatitis and hypogonadism. Peak prevalence is in the range of 20-40 years of age. In keeping with an autoimmune basis, autoimmune polyendocrine syndrome type 2 is more prevalent in females and associated with specific HLA DR3 and DR4 haplotypes and with the class II HLA alleles DQ2 and DQ8, also strongly linked to celiac disease 1. Autoantibodies to islet cell components (GAD65, IA2, ZnT8), thyroid (anti thyroglobulin TG, anti-thyroid peroxidase TPO), adrenal leading to Addison disease (anti-21-hydroxylase or anti 17-hydroxylase), and celiac disease (tissue transglutaminase and gliadin) are commonly present and should be periodically sought in those with one or more autoimmune endocrinopathies such as Addison disease and type 1 diabetes mellitus. Specific treatment for each entity should be continued in the hospital, with cortisol dosage adjusted for stress 19. A mechanism by which viral disease may trigger autoimmunity in the gut leading to celiac disease has recently been proposed and may have relevance to the other auto-immune diseases that form this entity 20.
Report suggest that the prevalence of autoimmune polyendocrine syndrome type 2 is about 14 to 20 cases per million of population 21 and that it affects females 3 to 4 times as often as it does males 22. Autoimmune polyendocrine syndrome type 2 usually strikes in the third or fourth decade of life 2.
Most cases of autoimmune polyendocrine syndrome type 2 are sporadic although some clinical researchers believe that there is a familial or hereditary trait associated with autoimmune polyendocrine syndrome type 2. If so, it may involve a complex interaction among many genes 23.
Autoimmune polyendocrine syndrome type 2 causes
The exact cause of autoimmune polyendocrine syndrome type 2 is not known, but it is thought to result from one or more abnormal immune responses 23. Autoimmune reactions occur when, for reasons not quite clear, the body mistakenly reacts to a normal antibody as if it were a foreign one.
Autoimmune polyendocrine syndrome type 2 is thought to be a polygenic disease, with significant heterogeneity due to multiple genetic loci and environmental factors responsible for the organ-specific damage 24. Major histocompatibility complex (MHC) genes located on chromosome 6 have been implicated in organ-specific damage in autoimmune polyendocrine syndrome type 2 21. It appears that HLA-DR3 and HLA-DR4 haplotypes and the class 2 HLA alleles DQ2 and DQ8 increase predisposition to autoimmune polyendocrine syndrome type 2 2.
Non-HLA genes can also predispose to autoimmune polyendocrine syndrome type 2 and include CD25-interleukin-2 receptor, cytotoxic T-lymphocyte protein 4 (CTLA-4), and protein tyrosine-protein phosphatase, non-receptor type 22 (PTPN22) 21.
Autoimmune polyendocrine syndrome type 2 signs and symptoms
Many conditions and symptoms are associated with autoimmune polyendocrine syndrome type 2. The symptoms may vary greatly among affected individuals.
Addison’s disease is a rare disorder characterized by chronic and insufficient functioning of the outer layer of the adrenal gland (adrenal cortex). Patients with Addison’s disease have a deficiency in the production of glucocorticoid hormones which are manufactured by the adrenal gland. These hormones (especially cortisol and aldosterone) are involved in carbohydrates, fat and protein metabolism, carbohydrate and blood sugar storage, and they fight inflammation and suppress the immune response. The deficiency in glucocorticoid causes an increased release of sodium and decreased release of potassium in the urine, sweat, saliva, stomach and intestines. These changes can cause low blood pressure and increased water excretion that can lead to severe dehydration.
Hypothyroidism (underactive thyroid) is a disorder that can be genetic or acquired and may occur alone or as a symptom of another illness. Major symptoms may include the development of an enlarged thyroid gland in the neck, a dull facial expression, puffiness and swelling around the eyes, drooping eyelids, thinning hair which is coarse and dry, and poor memory. Hypothyroidism can be caused by disorders of the hypothalamus or pituitary centers in the brain, disorders that affect control of the thyroid hormone, blockage in the metabolic process of transporting thyroid or iodine in the thyroid gland itself, or the result of a hereditary disorder called Hashimoto’s thyroiditis. Hashimoto’s thyroiditis is an autoimmune disorder in which the body’s natural defenses against invading organisms (i.e., antibodies, lymphocytes etc.) suddenly begin to attack healthy tissue.
Autoimmune polyendocrine syndrome type 2 patients may present with vague symptoms of weight loss, fatigue, nausea, vomiting, generalized weakness, anorexia, abdominal pain, diarrhea, polyuria, and polydipsia. Common signs in these patients may include mucosal and cutaneous hyperpigmentation low blood glucose levels and orthostatic hypotension if Addison disease is the diagnosis or polyuria and polydipsia with hyperglycemia if type 1 diabetes mellitus is present. Hypothyroidism can present with bradycardia, and delayed tendon reflexes.
Patients with Addison disease may present with shock-like features including hypotension, tachycardia, and altered mental status suggestive of adrenal crisis.
There is a 2.5-fold increase risk of adrenal crisis in patients who have Addison disease due to the autoimmune polyendocrine syndrome 25.
Some (but not all) of the following additional findings may be present in patients with autoimmune polyendocrine type 2:
- Type 1 diabetes mellitus: This type of diabetes generally starts during childhood or adolescence. The starches and sugars (carbohydrates) in the foods we eat are normally processed by digestive juices into glucose. The glucose circulates in the blood as a major energy source for body functions. A hormone produced by the pancreas (insulin) regulates the body’s use of glucose. In diabetes mellitus, the pancreas does not manufacture the correct amount of insulin needed to metabolize sugar. As a result, the patient needs daily injections of insulin to regulate blood sugar levels. Symptoms of this disorder may be frequent urination, extreme thirst, constant hunger, weight loss, itching of the skin, changes in vision, slow healing of cuts and bruises, and in children there is a failure to grow and develop normally.
- Hypoparathyroidism: This disorder causes lower than normal levels of calcium in the blood due to insufficient levels of parathyroid hormones. This condition can be inherited, associated with other disorders, or the result of a neck injury. Symptoms of hypoparathyroidism may be weakness, muscle cramps, abnormal sensations of the hands such as burning and numbness, excessive nervousness, loss of memory, headaches, cramping of wrists and feet, and spasms in facial muscles.
- Gonadal failure: This refers to the failure of the organ that produces sex cells (gonads-or testes in the male, and ovaries in the female) to function properly causing an absence of secondary sex characteristics.
- Pernicious anemia: This is a blood disorder resulting from an impaired absorption of vitamin B-12. This vitamin is used in the production of red blood cells. Healthy individuals absorb sufficient amounts of vitamin B-12 in their normal diet with the help of a substance secreted by the stomach called intrinsic factor. Patients with pernicious anemia generally lack intrinsic factor and can not absorb sufficient amounts of vitamin B-12. Symptoms of vitamin B-12 deficiency usually appear years after absorption of the vitamin ceases because B-12 is stored in large quantities in the liver. Symptoms of this disorder may be shortness of breath, fatigue, weakness, rapid heartbeat, angina, anorexia, abdominal pain, indigestion, and possibly intermittent constipation and diarrhea.
- Vitiligo: This is a skin condition in which there is an absence of pigment producing cells (melanocytes) causing decreased pigmentation of the skin. These “white spots” on the skin appear most often on the face, neck, hands, abdomen, and thighs although they may appear on all parts of the skin. Vitiligo is sometimes familial, but the exact mode of heredity is not yet understood.
- Celiac sprue: This chronic hereditary intestinal malabsorption disorder is caused by intolerance to gluten. The most common symptoms of this disorder are weight loss, chronic diarrhea, abdominal cramping and bloating, intestinal gas and abdominal distention and muscle wasting. Celiac sprue is a hereditary congenital disorder. Gluten is a protein that is present in wheat, oats, barley, rye and probably millet. Patients with celiac sprue cannot properly absorb a part of gluten called gliadin. This causes intestinal abnormalities as well as physiological deficiencies. Although the disorder begins in infancy, it is sometimes not diagnosed until the patient reaches adulthood.
- Myasthenia gravis: Sometimes this disorder can be associated with autoimmune polyendocrine syndrome type 2. Myasthenia gravis is a chronic neuromuscular disease characterized by weakness and abnormally rapid fatigue of the voluntary muscles, with improvement following rest. Any group of muscles may be affected, but those around the eyes and the muscles used for swallowing are the most commonly involved.
- Graves’ disease: This is a disorder that affects the thyroid gland. It is thought to occur as a result of an imbalance in the immune system. This disorder causes increased thyroid secretion (hyperthyroidism), enlargement of the thyroid gland and protrusion of the eyeballs. The exact cause of this disorder is not known. It is thought to be inherited as an autosomal recessive trait.
Manuela Dittmar et al. 26 followed 151 out of 360 autoimmune polyendocrine syndrome type 2 patients for 13 years and found that autoimmune thyroid disease was most prevalent in 99 patients (65.6%), and out of these, 50 patients (33.1%) were found to have Graves’ disease, and 49 patients (32.5%) had Hashimoto’s thyroiditis. Type 1 diabetes mellitus was found in 92 patients (60.9%), and Addison disease was found in 28 patients (18.5%). Type 1 diabetes mellitus occurred early in life with a mean age 27.5 years while other diseases manifested around age 36.5 to 40 years. The coexistence of type 1 diabetes mellitus and thyroid disease was most common while the coexistence of Addison and thyroid disease was less common 26.
Autoimmune polyendocrine syndrome type 2 diagnosis
Autoimmune polyendocrine syndrome type 2 is diagnosed by occurrence in the same patient of at least 2 out of 3 manifestations including primary adrenal insufficiency (Addison disease), autoimmune thyroid disease-causing Graves disease or hypothyroidism and type 1 diabetes mellitus. Other endocrine and non-endocrine manifestations of autoimmune polyendocrine syndrome type 2 are primary hypogonadism, myasthenia gravis and celiac disease, alopecia, vitiligo, pernicious anemia, idiopathic heart block, Stiff-man syndrome, Parkinson disease, IgA deficiency, serositis, dermatitis herpetiformis, idiopathic thrombocytopenia, and hypophysitis 21.
Diagnosis of Addison disease or primary adrenal insufficiency is based on a morning serum cortisol level less than 6.0 mcg/dl or a serum cortisol less than 18 mcg/dl at 60 minutes after ACTH stimulation test using 250-mcg intravenous or intramuscular bolus of cosyntropin 26. The presence of 21-hydroxylase or 17-hydroxylase autoantibodies can confirm autoimmune adrenalitis 27. Patients with positive 21-hydroxylase or 17-hydroxylase antibodies should have annual monitoring of morning cortisol and ACTH and cosyntropin stimulation test if suspicion is high.
Diagnosis of hypothyroidism due to Hashimoto’s thyroiditis or hyperthyroidism due to Graves’ disease can be made by evaluation of TSH and T4 for the former and TSH, T4, and T3 for the latter. In euthyroid patients, the presence of anti-thyroglobulin antibodies, thyroid microsomal antibodies and thyrotropin receptor antibodies (Graves’ disease) can detect patients at risk of thyroid disease in the future.
Diagnosis of type 1 diabetes mellitus can be made with classic symptoms of polyuria, polydipsia, and polyphagia associated with elevated serum glucose level (fasting greater than 125 mg/dl and random over 200 mg/dl and or elevated HbA1c, greater than 6.4%). Standard guidelines should be used for the diagnosis of individual organ dysfunction. These patients can be tested for anti-glutamic acid decarboxylase antibodies (GAD), anti-islet cell antigen 2 and anti-Zn transporter 8 antibodies. Also following a challenge with glucagon (1 mg) the plasma C-Peptide is less than 0.6 ng/ml.
A timely diagnosis of autoimmune polyendocrine syndrome type 2 requires knowledge of the complete spectrum of this disease. A complete history and thorough physical exam may give important clues. In many cases, the diagnosis of autoimmune polyendocrine syndrome type 2 may be delayed due to the heterogeneous presentation. It is uncommon for these patients to have dysfunction of all 3 major endocrine organs simultaneously and there is usually a latent phase between the endocrinopathies.
Patients with autoimmune polyendocrine syndrome type 2 and their family members should be monitored long-term due to the risk of development of organ-specific dysfunction over time. Family members who are not affected with autoimmune polyendocrine syndrome type 2 should watch for symptoms related to adrenal, thyroid and endocrine pancreatic dysfunction. Asymptomatic carriers should be followed on an annual basis 21.
Organ-specific antibodies like 21-hydroxylase antibody for Addison disease, an antibody against GAD 65 for type 1 diabetes, thyrotropin receptor antibody, and TPO antibody for autoimmune thyroid disease can be assayed for making a diagnosis. Transglutaminase antibodies are useful for the diagnosis of Celiac disease. However, the presence of autoantibodies to thyroid, adrenal, and islets does not predict glandular failure.
Autoimmune polyendocrine syndrome type 2 can be differentiated from Autoimmune polyendocrine syndrome type 1 due to late-onset on clinical manifestation mostly after age 20, different patterns of disease combination with no mucocutaneous candidiasis, and polygenic inheritance versus monogenic.
Delay in diagnosis can cause significant morbidity and mortality in these patients due to the risk of severe hypothyroidism, adrenal crisis, and diabetic ketoacidosis.
Thyroid ultrasound is an excellent noninvasive tool to evaluate thyroid disease. The diffuse or multifocal hypoechoic pattern is commonly seen in autoimmune thyroid disease.
CT scan and MRI of the adrenal gland is often normal, but sometimes there is a decrease in the volume of the gland suggestive of atrophy.
Unfortunately, there is no reliable imaging technique which can indicate endocrine pancreatic disease.
Autoimmune polyendocrine syndrome type 2 treatment
Each disorder in a case of autoimmune polyendocrine type 2 is treated separately. Replacement with appropriate hormones is the key. These patients should be followed up by an interprofessional team lead by an endocrinologist. Patients should be followed at least every 6 months with appropriate blood work to avoid over and under-treatment. These patients are at risk of adrenal crisis, hypoglycemia, diabetic ketoacidosis, among others. The treating physician should be proactive to diagnose these conditions and possible manifestations expected to occur over time without delay to avoid complications.
Care should be taken to treat patients with thyroxine as this can precipitate life-threatening Addisonian crisis if the patient has undiagnosed adrenal insufficiency. In these patients, testing for adrenal insufficiency should be done before treating hypothyroidism with levothyroxine 28. Hydrocortisone replacement should precede thyroxine therapy by about a week.
Family members at risk for developing autoimmune polyendocrine type 2 can be identified by checking organ-specific antibodies.
Due to the autoimmune nature of this disease, multiple immunosuppressants and immune-modulators have been tested, but none of these agents are being used on a regular basis due to the potential risk of side effects.
Autoimmune polyendocrine syndrome type 3
Autoimmune polyglandular syndrome type 3 sometimes referred to as Carpenter’s syndrome, is an autoimmune condition that affects the body’s endocrine glands 29. Autoimmune polyglandular syndrome type 3 has the same array of endocrine tissue autoimmune abnormalities as autoimmune polyendocrine type 2, but without Addison disease 1. Almost 20% of patients with type 1 diabetes mellitus have thyroglobulin (TG) and thyroid peroxidase (TPO) antibodies, but only a minority progress to clinical or biochemical hypothyroidism, so autoimmune polyglandular syndrome type 3 could be considered as a relatively common disorder 30.
Autoimmune polyglandular syndrome type 3, which typically affects women during middle age, results from failure of the glands to produce their hormones. This condition is characterized by autoimmune thyroiditis along with another organ-specific autoimmune disease 31. The other autoimmune diseases may include diabetes mellitus, pernicious anemia, vitiligo, alopecia, myasthenia gravis, and Sjogren’s syndrome 32. The adrenal cortex (the outer layer of the adrenal gland) is not involved 31. There are three types of autoimmune polyglandular syndrome type 3 31:
- APS3A – Autoimmune thyroiditis with immune-mediated diabetes mellitus (IMDM)
- APS3B – Autoimmune thyroiditis with pernicious anemia
- APS3C – Autoimmune thyroiditis with vitiligo and/or alopecia and/or other organ-specific autoimmune disease
The cause is still unknown, but it is believed that it may be an autoimmune disease, where environmental factors (such as viral infections) and genetic factors (such as variations in the HLA II genes) are also involved in the disease. In many cases more than one member of the same family is affected with autoimmune polyglandular syndrome type 3, suggesting that its inheritance could be autosomal dominant, and therefore, familiar screening is recommended. It is very important that people with autoimmune polyglandular syndrome type 3 are monitored closely by their doctors for early detection of any glandular problems. Treatment includes lifelong hormone replacement therapy for any established glandular failure 31.
Autoimmune polyglandular syndrome type 3 causes
Autoimmunity, environmental factors, and genetic factors are the 3 major factors that should be considered in the pathophysiology of autoimmune polyendocrine syndrome type 3 33.
Autoimmunity
Autoimmune disease affecting a single endocrine gland is frequently followed by impairment of other glands, resulting in multiple endocrine failure. The autoimmune pathogenesis of these disorders began to emerge in the mid-20th century. In 1956, Roitt and colleagues discovered circulating precipitating autoantibodies to thyroglobulin in patients with Hashimoto thyroiditis.
The identification of circulating organ-specific autoantibodies provided the earliest and strongest evidence for the autoimmune pathogenesis of polyglandular failure syndromes. Endocrine autoimmunities are associated with autoantibodies that react to specific antigens, whereas patients with collagen diseases synthesize immunoglobulins that recognize nonorgan-specific cellular targets, such as nucleoproteins and nucleic acids.
Cellular autoimmunity is also important in the pathogenesis of polyglandular failure syndromes. Histologic examination of the affected glands (eg, thyroid, parathyroid, ovaries, pancreatic islets, gastric mucosa) has demonstrated similar results, that is, mononuclear infiltrate composed mainly of lymphocytes, macrophages, natural killer (NK) cells, and plasma cells. The striking feature is the sparing of adjacent nontarget tissue. As the disease progresses, atrophy and fibrosis predominate.
Experimental animal models of autoimmune polyendocrine syndrome type 3 have been described. In BioBreeding/Worcester (BB/W) rats, the frequency of chronic lymphocytic thyroiditis was remarkably increased in diabetic insulin-treated BB/W rats 34.
Animal models have provided many of the insights into endocrine immunities. Polyglandular immunity, including gastritis, oophoritis, orchitis, and thyroiditis, could be induced in genetically susceptible mice by depleting T lymphocytes permanently or transiently. By using the model of neonatal thymectomy, it has been demonstrated that early interactions between the lymphoid system and target organs are important in the pathogenesis of autoimmunity. Furthermore, it also was demonstrated that CD4+ splenocytes from adult (but not neonatal mice) contain regulatory populations that can prevent the transfer of autoimmune endocrinopathies.
An autoimmune attack of a target organ often begins in individuals who have a genetic predisposition after an unknown precipitating event. The early process manifests by provoking autoantibody production, and it may arrest at this stage. Progressive disease is associated with secondary responses against antigens released by damaged tissue. Disease initially is detectable by observing minimal biochemical abnormalities such as elevation of trophic hormones. Organ function loss may plateau before the threshold of critical organ mass is reached, or it may progress to clinically overt disease. Early hormone replacement therapy may decelerate the destruction of surviving tissue; but, at the late stage, complete organ atrophy is inevitable.
Environmental factors
Some authorities postulate that environmental precipitators of autoimmunity might play a role in polyglandular autoimmunity. Viral infection may exaggerate the ongoing immune response and precipitate glandular failure, although no human epidemiological studies show infection triggering polyglandular autoimmunity.
The links between congenital rubella infection, type 1 diabetes mellitus, and hypothyroidism are well known. Reovirus type 1 infection in susceptible mice causes type 1 diabetes mellitus and growth failure.
International comparisons show a positive correlation between type 1 diabetes mellitus prevalence and ingestion of cow milk. Circulating autoantibodies against a peptide with homology to bovine serum albumin and human islet cell surface protein have been observed in patients with immune-mediated diabetes mellitus.
Development of autoimmune polyendocrine syndrome type 3 after interferon-alpha therapy for hepatitis C has been described, raising the possibility of interferon-enhanced major histocompatibility complex expression, which in turn initiated the onset of organo-specific autoantibodies and the clinical manifestations of autoimmune diseases.
Genetic factors
Autoimmune polyendocrine syndrome type 3, as well as autoimmune polyendocrine syndrome type 2, is associated with HLA class 2 genes with apparently distinctive HLA alleles for each. The underlying non-HLA genes of autoimmune polyendocrine syndrome type 3 remain to be further defined genetically. autoimmune polyendocrine syndrome type 3 is often observed in individuals in the same family, suggesting that its inheritance could be an autosomal dominant trait with incomplete penetrance 35.
HLA-DRB1*04/DQA1*0301/DQB1*0302 is the predominant HLA haplotype associated with susceptibility in immune-mediated diabetes mellitus. Interestingly, the HLA-DQB1*0602 allele protects against immune-mediated diabetes mellitus, even if the HLA-DQB1*0301 or DQB1*0302 susceptibility gene is present. HLA-DQB1*0301 is the HLA haplotype frequently associated with autoimmune thyroiditis. HLA-DRB1*13 is associated with vitiligo. Alopecia areata is strongly associated with DQB1*03 and DRB1*1104, which appear to be markers of general susceptibility to alopecia areata. In addition, the frequency of HLA-DRB1*0401 and DQB1*0301 is remarkably increased among patients with alopecia totalis and those with alopecia universalis, the most extensive form of the condition.
Multigenetic involvement in the development of the individual components of autoimmune polyendocrine syndrome type 3 has been proved. For example, immune-mediated diabetes mellitus is linked to several loci in non-HLA genomic regions. Furthermore, autoimmune thyroiditis also is polygenic.
Family and population studies showed that the autoimmune polyendocrine syndrome type 3A (APS3A) has a strong genetic background. Several gene variations present in both autoimmune thyroiditis and immune-mediated diabetes mellitus have been identified by whole genome and candidate gene approaches. The most important susceptibility genes are human leucocyte antigen (chromosome 6), cytotoxic T-lymphocyte–associated antigen 4 (chromosome 2), protein tyrosine phosphatase nonreceptor type 22 (chromosome 1), forkhead box P3 (X chromosome), and the interleukin 2 receptor alpha/CD25 gene region (chromosome 10) 36.
Autoimmune polyglandular syndrome type 3 symptoms
The hallmark of polyglandular autoimmune syndrome type 3 is the absence of adrenal insufficiency. In fact, autoimmune polyendocrine syndrome type 3 is autoimmune polyendocrine syndrome type 2 without adrenocortical involvement 37. Once adrenocortical insufficiency develops, such patients are reclassified as having autoimmune polyendocrine syndrome type 2. The involvement of multiple glands may be apparent at the time of initial presentation, but, more commonly, individual glandular failure develops sequentially. No specific sequence exists by which the individual glandular failures develop.
The clinical symptoms of autoimmune polyendocrine syndrome type 3 are a constellation of manifestations of endocrine gland failures that comprise the syndrome.
Autoimmune thyroiditis
Autoimmune thyroiditis is the characteristic of all subcategories of autoimmune polyendocrine syndrome type 3. The presenting symptoms are goiter, those due to hypothyroidism, or both. Occasionally, destruction of the gland early in the process gives rise to the release of thyroid hormones, creating a transient hyperthyroid state (ie, Hashitoxicosis). When this process is complete, hypothyroidism becomes apparent.
Fatigue and depression are leading symptoms in many patients with autoimmune thyroiditis. Weight gain, cold intolerance, constipation, dry hair, sluggishness, somnolence, hoarseness, and menorrhagia also are major clinical symptoms.
Although some patients report a sensation of tightness in the neck, pain is usually not a prominent symptom. Patients may have a history of other autoimmune conditions such as inflammatory bowel disease, celiac disease, gonadal dysgenesis (Turner syndrome), and hepatitis C.
Immune-mediated diabetes
Classic symptoms of immune-mediated diabetes mellitus are polyuria, polydipsia, and polyphagia. Polyuria is secondary to osmotic diuresis caused by hyperglycemia. Polydipsia is secondary to hyperosmolality. Polyphagia is probably secondary to deficient glucose utilization in the cells of the hypothalamic ventromedial nuclei.
Weight loss despite polyphagia is characteristic.
Blurred vision is common and also is secondary to hyperosmolality.
Paresthesia in the extremities may be present at presentation, although it is usually reversible with better glycemic control. Paresthesia is thought to be secondary to transient impairment of peripheral sensory nerve function caused by hyperglycemia.
Rapid development of insulin deficiency, usually precipitated by infection or other forms of stress, could result in diabetic ketoacidosis (DKA) as the initial presentation of type 1 diabetes mellitus. Abdominal pain, nausea, and vomiting are common in diabetic ketoacidosis, along with above symptoms. Altered mental status and rapid breathing are symptoms associated with severe diabetic ketoacidosis.
Pernicious anemia
Usual presenting features include insidious onset of fatigue, weakness, lightheadedness, headache, vertigo, tinnitus, and palpitations secondary to anemia.
Vague gastrointestinal symptoms, such as anorexia or diarrhea, may be present. Sore tongue, numbness and tingling in the extremities, and difficulty with balancing may be present at onset or may develop later in the course.
Neuropsychiatric manifestations may not parallel symptoms of anemia. In addition to the above neurological symptoms, irritability, memory loss, depression, hallucinations, agitation, suicidal ideation, and sphincter disturbances are recognized manifestations.
Vitiligo
Vitiligo is associated with many autoimmune endocrinopathies. Patients with an early age of onset are less likely to have autoimmune polyendocrine syndrome type 2 or other endocrinopathies.
Loss of skin pigmentation in vitiligo has been linked to autoimmune destruction of melanocytes by antityrosinase and antimelanocyte antibodies. The leading symptom is loss of skin pigmentation, which is more noticeable around the mouth, eyes, nose, nipples, umbilicus, or anus. Trauma to the skin results in further loss of pigmentation (Koebner phenomenon).
Alopecia
Autoimmune alopecia (alopecia areata) ranges in severity from (1) small round patches of hair loss that regrow spontaneously to (2) persistent extensive patchy involvement to (3) the loss of all scalp hair (alopecia totalis) or all scalp and body hair including eyelashes, eyebrows, underarm hair, and pubic hair (alopecia universalis).
In the latter, absence of eyebrows results in perspiration trickling into the eyes; absence of eyelashes results in little protection from dust and glare. Absence of nasal hairs results in lack of protection in the nostrils or sinuses from foreign particles in the air. Spontaneous remission and recurrence are common.
Autoimmune polyglandular syndrome type 3 diagnosis
Physical examination
Autoimmune thyroiditis
Physical findings in autoimmune thyroiditis are goiter, hypothyroidism, or both. The thyroid gland is palpably enlarged in classic goitrous autoimmune thyroiditis (Hashimoto disease). The entire thyroid gland is diffusely enlarged and is firm. The surface of the thyroid gland often is bosselated, that is, characterized by numerous bosses or rounded protuberances.
Extrathyroidal signs of autoimmune thyroiditis and hypothyroidism include facial pallor; bradycardia; hypertension; delayed relaxation of deep-tendon reflexes; and nonpitting edema (myxedema) of the skin of the hands, feet, and eyelids.
Immune-mediated diabetes
Dry skin and mucous membranes may be observed secondary to fluid loss associated with osmotic diuresis.
Severe dehydration or severe diabetic ketoacidosis may lead to hypotension.
Pernicious anemia
The most striking physical sign of pernicious anemia is pallor.
Mild scleral icterus may be present secondary to indirect hyperbilirubinemia caused by intramedullary hemolysis.
The tongue usually is smooth, raw, and beefy.
Systolic flow murmur and tachycardia may be present secondary to anemia.
Neurological signs may vary from diminished vibration and joint position sense to gross motor, sensory, and cognitive deficits.
A marked discordance may be present between the severity of neurological signs and the degree of anemia (see History).
Vitiligo
Vitiligo is characterized by symmetric areas of complete pigment loss, particularly affecting the periorificial areas and bony prominences.
The hairs within the patches of vitiligo often remain pigmented. However, in older lesions, the hairs also become white. Wood-lamp examination reveals more apparent chalky-white areas.
Alopecia
Alopecia areata causes different patterns of hair loss.
These include a localized patch of hair loss, a netlike pattern of hair loss, a serpentine pattern of hair loss that covers the periphery of the scalp similar to a serpent forming a turban over the edges of the scalp, and a diffuse form that affects the whole scalp without distinct patches.
Laboratory Studies
Laboratory studies to diagnose polyglandular autoimmune syndrome type 3 include (1) serological tests for autoantibodies, (2) assessment of end-organ function, and (3) genetic tests.
Serological tests for autoantibodies
Some experts argue that measurement of levels of circulating antibodies may not be very useful, because many individuals have these antibodies without clinical manifestations. In a study by Hunger-Battefeld et al 38 involving 139 patients with immune-mediated diabetes mellitus, 63% of the patients were found to have 1 or more pathologically increased antibody titers associated with an autoimmune endocrine disease other than diabetes; however, only 31% of the patients presented with symptoms of this additional disease. In the study, thyropathy was the most prevalent autoimmune disease accompanying immune-mediated diabetes mellitus. The authors recommended that patients with immune-mediated diabetes mellitus be screened for other autoimmune endocrine diseases 38.
Serological tests may be helpful in the following circumstances:
- They can be used to verify the autoimmune nature of disease.
- They can be used to identify patients affected by an isolated glandular failure in which polyglandular failure is likely to develop.
- They can be used to screen asymptomatic family members of patients with autoimmune polyglandular syndrome type 3.
Some autoantibodies appear to predict the development of glandular failure. This may allow the initiation of immunomodulatory treatment before the development of overt disease.
The following list delineates autoantibodies detected in each glandular disease:
- For autoimmune thyroiditis, autoantibodies may include antithyroglobulin antibodies, antithyroid microsomal antibodies/antithyroid peroxidase antibodies (peroxidase is the microsomal antigen).
- For immune-mediated diabetes mellitus, they may include anti – islet cell antibodies and antibodies to glutamic acid decarboxylase.
- For pernicious anemia, antiparietal cell antibodies and anti – intrinsic factor antibodies may be present.
- In vitiligo, antimelanocyte antibodies are found.
Assessment of end-organ function
Autoimmune thyroiditis
- An elevated serum thyrotropin concentration is sufficient to confirm the diagnosis of hypothyroidism. Serum thyroxine is low in overt hypothyroidism and normal in subclinical hypothyroidism.
- Other laboratory abnormalities of hypothyroidism include elevated creatine kinase, cholesterol, triglyceride, and lactate dehydrogenase levels; a low serum sodium level; and anemia.
- Procedures: Perform fine-needle aspiration biopsy of the thyroid gland to exclude malignancy if a suggestive nodule (ie, dominant nodule) is present or if the goiter is growing rapidly.
Immune-mediated diabetes
- A fasting blood glucose level higher than 125 mg/dL, a random blood glucose level higher than 200 mg/dL in the presence of classic symptoms of diabetes mellitus, or a 2-hour postprandial blood glucose level higher than 200 mg/dL in an oral glucose tolerance test is diagnostic of diabetes mellitus.
- Glycosylated hemoglobin level is more useful for monitoring of the progress of disease than for diagnostic purposes.
Pernicious anemia
- Complete blood count (CBC) is one of the most important diagnostic tests. Macrocytic anemia (mean cell volume >100 fL) with marked anisopoikilocytosis and hypersegmented neutrophils observed on peripheral blood films is characteristic. Serum vitamin B-12 level is very low.
- Other laboratory findings include megaloblastic bone marrow and abnormal vitamin B-12 absorption test (Schilling test), corrected with the addition of intrinsic factor.
- Vitiligo: Skin biopsy from the affected area reveals an absence of melanocytes and mild inflammatory cell infiltrate.
Genetic testing
Mutations in the HLAD gene should be analyzed in patients with autoimmune polyglandular syndrome type 3 and in siblings with symptoms of component glandular diseases. HLA-DR oligotyping and HLA-DQ oligotyping by polymerase chain reaction are commercially available through Associated Regional and University Pathologists Laboratories. HLA-DR typing of loci 1 and loci 2 also is available through Specialty Laboratories.
Autoimmune polyglandular syndrome type 3 treatment
Medical care of patients with polyglandular autoimmune syndrome type 3 includes monitoring of glandular functions for early detection of glandular failure, lifelong hormone replacement therapy for established glandular failure or failures, and familial screening.
Autoimmune thyroiditis
Patients with hypothyroidism need lifelong thyroxine therapy. Thyrotropin levels should be monitored by using highly sensitive assays to maintain a euthyroid state.
Overreplacement with thyroxine may result in osteoporosis and increased risk of atrial fibrillation.
Immune-mediated diabetes
The mainstay of immune-mediated diabetes treatment is lifelong replacement therapy with exogenous insulin injections. Monitor the progress of disease by periodic retina examination, foot examination, and measurement of glycosylated hemoglobin level and the ratio of urine microalbumin to creatinine.
The late 20th-century development of recombinant human insulin was a major breakthrough in the treatment of immune-mediated diabetes mellitus. Intensive insulin therapy has improved the long-term outcome of the disease at the expense of frequent hypoglycemia.
Pancreatic transplantation is becoming an option but usually is reserved for patients with end-stage renal disease who already require renal transplantation.
Specific therapies aimed at suppressing the immune response of the pancreatic islet cells are being researched.
Pernicious anemia
The mainstay of pernicious anemia treatment is lifelong replacement therapy with parenteral hydroxocobalamin. Within 48-72 hours of the first injection, serum potassium levels drop precipitously because of rapid regeneration of red blood cells.
Hypokalemia may be severe enough to necessitate replacement therapy. Serum iron levels also drop precipitously for the same reason.
If the patient initially has a marginal iron reserve, this may halt the recovery of anemia. Some experts suggest giving a small dose of iron supplements concurrently to prevent this phenomenon.
Vitiligo
Treatment of vitiligo is currently unsatisfactory. Repigmentation treatment is most successful on face and trunk; hands, feet and areas with white hair respond poorly. Compared to longstanding patches, new ones are more likely to respond to medical therapy.
When successful repigmentation occurs, melanocyte stem cells in the bulb at the base of the hair follicle are activated and migrate to the skin surface. They appear as perifollicular brown macules.
General measures
- Minimize skin injury: wear protective clothing.
- A cut, a graze, a scratch may lead to a new patch of vitiligo.
- Cosmetic camouflage can disguise vitiligo. Options include:
- Make-up, dyes and stains
- Waterproof products
- Dihydroxyacetone-containing products “tan without the sun.”
- Micropigmentation or tattooing for stable vitiligo.
- Sun protection: stay indoors when sunlight is at its peak, cover up with sun protective clothing and apply SPF 50+ sunscreen to exposed skin.
- White skin can only burn on exposure to ultraviolet radiation (UVR); it cannot tan.
- Sunburn may cause vitiligo to spread.
- Tanning of normal skin makes vitiligo patches appear more visible.
Topical treatments
Topical treatments for vitiligo include:
- Corticosteroid creams. These can be used for vitiligo on trunk and limbs for up to 3 months. Potent steroids should be avoided on thin-skinned areas of the face (especially eyelids), neck, armpits and groin.
- Calcineurin inhibitors (pimecrolimus cream and tacrolimus ointment. These can be used for vitiligo affecting eyelids, face, neck, armpits and groin.
- Experimental treatment with topical ruxolitinib, a Janus kinase inhibitor, shows great promise for facial vitiligo.
Phototherapy
Phototherapy refers to treatment with ultraviolet (UV) radiation. Options include:
- Whole-body or localised broadband or narrowband (311 nm) UVB
- Excimer laser UVB (308 nm) or targeted UVB for small areas of vitiligo
- Oral, topical, or bathwater photochemotherapy (PUVA)
- In-office or home phototherapy.
Phototherapy probably works in vitiligo by two mechanisms.
- Immune suppression—preventing the destruction of the melanocytes
- Stimulation of cytokines (growth factors)
Treatment is usually given twice weekly for a trial period of 3–4 months. If repigmentation is observed, treatment is continued until repigmentation is complete or for a maximum of 1–2 years.
- Phototherapy is unsuitable for very fair-skinned people.
- The treatment intensity aims for the vitiligo skin to be a light “carnation” pink.
- If repigmentation is observed, treatment is continued until repigmentation is complete or for a maximum of 1–2 years.
- Treatment times are generally brief. The aim is to cause the treated skin to appear very slightly pink the following day.
- It is essential to avoid burning (red, blistered, peeling, itchy or painful skin), as this could cause the vitiligo to get worse.
A meta-analysis included 35 different studies reporting outcome after phototherapy for generalised vitiligo. A marked or clinically useful response was achieved in 36% after 12 months of narrowband UVB and in 62% after 12 months of PUVA. Face and neck responded better than the trunk, which responded better than extremities. It was not very effective on hands and feet.
Systemic therapy
Systemic treatments for vitiligo may include:
- Mini-pulses of oral steroids for 3 to 6 months, such as dexamethasone 2.5–4 mg, two consecutive days per week for 3–6 months
- Methotrexate
- Ciclosporin
- Mycophenolate mofetil
- Oral minocycline 100 mg/day, a tetracycline antibiotic with anti-inflammatory properties
- Subcutaneous afamelanotide.
None of these treatments is based on randomized controlled trial data in treating vitiligo. The aim is to stop the progression of the disease (stabilisation) and so immune-modulating treatments should be considered at an early stage, particularly for vitiligo affecting the face.
It is anticipated that monoclonal antibody biologic agents will be developed to treat vitiligo. To date, the results of Janus kinase (JAK) inhibitor trials in vitiligo have been encouraging, but there are high rates of relapse.
Surgical treatment of stable vitiligo
Surgical treatment for stable and segmental vitiligo requires removal of the top layer of vitiligo skin (by shaving, dermabrasion, sandpapering or laser) and replacement with pigmented skin removed from another site.
Techniques include:
- Non-cultured melanocyte-keratinocyte cell suspension transplantation.
- Punch grafting
- Blister grafts, formed by suction or cryotherapy
- Split skin grafting
- Cultured autografts of melanocytes grown in tissue culture.
Depigmentation therapy
Depigmentation therapy, using monobenzyl ether of hydroquinone, may be considered in severely affected, dark-skinned individuals.
Cryotherapy and laser treatment (eg, 755-nm Q-switched alexandrite or 694 nm Q-switched ruby) have also been used successfully to depigment small areas of vitiligo.
Alopecia
There is not yet any reliable cure for alopecia areata and other forms of autoimmune hair loss. Because spontaneous regrowth is common in alopecia areata, especially in the early stages of the disease, and research has often been of poor quality, the effectiveness of reported treatments is mostly unknown. Systemic therapy is reserved for patients with:
- More than 20% of scalp hair loss
- Rapid hair loss
- Chronic hair loss
- Severe distress.
Topical treatments
Several topical treatments used for alopecia areata are reported to result in temporary improvement in some people. Their role and efficacy are unknown. The hair may fall out when they are stopped. These include:
- Potent or ultrapotent topical steroids
- Minoxidil solution or foam
- Dithranol (anthralin) ointment
Intralesional corticosteroid injections
Injections of triamcinolone acetonide 2.5–10 mg/ml into patchy scalp, beard or eyebrow alopecia areata may speed up regrowth of hair. Its effect is temporary. If bald patches reappear, they can be reinjected.
Systemic corticosteroids
Oral and pulse intravenous steroids in high dose can lead to temporary regrowth of hair. Most physicians agree that long-term systemic steroid treatment is not justified because of potential and actual adverse effects.
Immunotherapy
The sensitisers diphenylcyclopropenone (diphencyprone) and dinitrochlorobenzene provoke contact allergic dermatitis in treated areas. These sensitisers can be reapplied once weekly to bald areas on the scalp. The resultant dermatitis is irritating and may be unsightly. It is often accompanied by a swollen lymph gland.
Other treatments
A combination of the lipid-lowering medications simvastatin and ezetimibe (which have immunomodulating effects) has been reported to be effective.
A single case is reported of substantial hair growth in a patient with longstanding alopecia totalis following the use of dupilumab for her concomitant severe atopic eczema.
There is no convincing data to support the use of methotrexate, sulfasalazine, azathioprine, ciclosporin or phototherapy.
JAK/STAT inhibitors
Several patients with severe alopecia areata have had improvement when treated with oral tofacitinib or oral ruxolitinib, which are Janus kinase (JAK) inhibitors. It is thought they may act by blocking interleukin (IL)-15 signalling and gamma interferon (IFNγ). Watch out for the results of clinical trials of these biologic medicines.
Autoimmune polyglandular syndrome type 3 prognosis
Prognosis of autoimmune polyendocrine syndrome type 3 depends on the individual glandular failures involved.
No systematic studies of long-term prognosis of patients with autoimmune polyendocrine syndrome type 3 have been conducted.
Autoimmune polyendocrine syndrome associated with cancer immunotherapy
An increasingly important and frequent cause of endocrine-autoimmune syndromes is their appearance in association with the increasing use of immunotherapy as a front line or back-up therapy in various cancers 2. The variety and severity of endocrine autoimmune syndromes associated with the use of inhibitors of CTLA4 (cytotoxic T-lymphocyte-associated protein 4) such as ipilimumab, and immune checkpoint blockade of programmed death 1 (PD-1) and its ligands PDL1 and PDL2, has recently been termed “the Achilles heel of cancer immunotherapy” 39. The range of autoimmune endocrine manifestations includes hypophysitis with disturbances in anterior pituitary hormones, hypo-and hyperthyroidism, adrenal insufficiency and type 1 diabetes mellitus 39. Key checkpoints by which autoimmunity is regulated in normal individuals are also exploited by tumors to evade recognition and elimination via the immune system; employing immuno-regulatory agents that block these checkpoints facilitates the recognition of tumor antigens as foreign and activates their destruction, but at the same time stimulates autoimmune responses to self-antigens. Clinicians should be aware of these autoimmune manifestations and screen for involvement of endocrine tissues or their clinical manifestations. Notably, some of these endocrine autoimmune manifestations may appear months to years after initiation of immune therapy for cancer 40.
- Sperling M, Yau M. Autoimmune Polyglandular Syndromes. [Updated 2017 Oct 29]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279152[↩][↩][↩][↩][↩][↩]
- Sperling M, Yau M. Autoimmune Polyglandular Syndromes. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. MDText.com, Inc.; South Dartmouth (MA): Oct 29, 2017.[↩][↩][↩][↩]
- Autoimmune Polyendocrine Syndromes. N Engl J Med 2004; 350:2068-2079 DOI: 10.1056/NEJMra030158[↩]
- Eisenbarth GS, Gottlieb PA. The immunoendocrinopathy syndromes. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, eds. Williams textbook of endocrinology. 10th ed. Philadelphia: Saunders, 2003:1763-76.[↩]
- Bello MO, Garla VV. Polyglandular Autoimmune Syndrome Type I. [Updated 2020 Aug 15]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537211[↩][↩][↩]
- Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. https://ghr.nlm.nih.gov/condition/autoimmune-polyendocrinopathy-candidiasis-ectodermal-dystrophy[↩]
- Meloni A, Willcox N, Meager A, Atzeni M, Wolff AS, Husebye ES, Furcas M, Rosatelli MC, Cao A, Congia M. Autoimmune polyendocrine syndrome type 1: an extensive longitudinal study in Sardinian patients. J. Clin. Endocrinol. Metab. 2012 Apr;97(4):1114-24.[↩][↩]
- Autoimmune Polyglandular Syndrome Type 1. https://rarediseases.org/rare-diseases/autoimmune-polyglandular-syndrome-type-1/[↩]
- Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D. Projection of an immunological self shadow within the thymus by the aire protein. Science. 2002 Nov 15;298(5597):1395-401.[↩]
- Husebye ES, Perheentupa J, Rautemaa R, Kämpe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J. Intern. Med. 2009 May;265(5):514-29.[↩][↩][↩][↩][↩]
- Rautemaa R, Hietanen J, Niissalo S, Pirinen S, Perheentupa J. Oral and oesophageal squamous cell carcinoma–a complication or component of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED, APS-I). Oral Oncol. 2007 Jul;43(6):607-13.[↩]
- Winqvist O, Rorsman F, Kämpe O. Autoimmune adrenal insufficiency: recognition and management. BioDrugs. 2000 Feb;13(2):107-14.[↩]
- Hedstrand H, Ekwall O, Haavik J, Landgren E, Betterle C, Perheentupa J, Gustafsson J, Husebye E, Rorsman F, Kämpe O. Identification of tyrosine hydroxylase as an autoantigen in autoimmune polyendocrine syndrome type I. Biochem. Biophys. Res. Commun. 2000 Jan 07;267(1):456-61.[↩]
- Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 2006 Aug;91(8):2843-50[↩][↩]
- Söderbergh A, Myhre AG, Ekwall O, Gebre-Medhin G, Hedstrand H, Landgren E, Miettinen A, Eskelin P, Halonen M, Tuomi T, Gustafsson J, Husebye ES, Perheentupa J, Gylling M, Manns MP, Rorsman F, Kämpe O, Nilsson T. Prevalence and clinical associations of 10 defined autoantibodies in autoimmune polyendocrine syndrome type I. J. Clin. Endocrinol. Metab. 2004 Feb;89(2):557-62.[↩][↩]
- Obermayer-Straub P, Perheentupa J, Braun S, Kayser A, Barut A, Loges S, Harms A, Dalekos G, Strassburg CP, Manns MP. Hepatic autoantigens in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Gastroenterology. 2001 Sep;121(3):668-77.[↩]
- Ahonen P, Myllärniemi S, Sipilä I, Perheentupa J. Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N. Engl. J. Med. 1990 Jun 28;322(26):1829-36.[↩]
- Perheentupa J. Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Clin. Endocrinol. Metab. 2006 Aug;91(8):2843-50.[↩]
- Kakleas, K., et al., Associated autoimmune diseases in children and adolescents with type 1 diabetes mellitus (T1DM). Autoimmun Rev, 2015. 14(9): p. 781-97.[↩]
- Verdu, E.F. and A. Caminero, How infection can incite sensitivity to food. Science, 2017. 356(6333): p. 29-30.[↩]
- Husebye ES, Anderson MS, Kämpe O. Autoimmune Polyendocrine Syndromes. N. Engl. J. Med. 2018 Mar 22;378(12):1132-1141.[↩][↩][↩][↩][↩]
- Förster G, Krummenauer F, Kühn I, Beyer J, Kahaly G. [Polyglandular autoimmune syndrome type II: epidemiology and forms of manifestation]. Dtsch. Med. Wochenschr. 1999 Dec 10;124(49):1476-81.[↩]
- Autoimmune Polyendocrine Syndrome Type II. https://rarediseases.org/rare-diseases/autoimmune-polyendocrine-syndrome-type-ii[↩][↩]
- Singh G, Jialal I. Polyglandular Autoimmune Syndrome, Type II (Carpenters, Schmidt) [Updated 2020 Aug 13]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK525992[↩]
- Meyer G, Badenhoop K, Linder R. Addison’s disease with polyglandular autoimmunity carries a more than 2·5-fold risk for adrenal crises: German Health insurance data 2010-2013. Clin. Endocrinol. (Oxf). 2016 Sep;85(3):347-53.[↩]
- Dittmar M, Kahaly GJ. Polyglandular autoimmune syndromes: immunogenetics and long-term follow-up. J. Clin. Endocrinol. Metab. 2003 Jul;88(7):2983-92.[↩][↩][↩]
- Brandão Neto RA, de Carvalho JF. Diagnosis and classification of Addison’s disease (autoimmune adrenalitis). Autoimmun Rev. 2014 Apr-May;13(4-5):408-11[↩]
- Wang X, Ping F, Qi C, Xiao X. Delayed diagnosis with autoimmune polyglandular syndrome type 2 causing acute adrenal crisis: A case report. Medicine (Baltimore). 2016 Oct;95(42):e5062[↩]
- Autoimmune polyglandular syndrome type 3. https://rarediseases.info.nih.gov/diseases/10980/autoimmune-polyglandular-syndrome-type-3[↩]
- Gimenez-Barcons, M., et al., Autoimmune predisposition in Down syndrome may result from a partial central tolerance failure due to insufficient intrathymic expression of AIRE and peripheral antigens. J Immunol, 2014. 193(8): p. 3872-9.[↩]
- Type III Polyglandular Autoimmune Syndrome. https://emedicine.medscape.com/article/124398-overview[↩][↩][↩][↩]
- Polyglandular Autoimmune Syndrome Type 3 (PAS-3). http://labs.pathology.jhu.edu/cihakova/about/about/endocrine-diseases/polyglandular-autoimmune-syndrome-type-3-pas-3[↩]
- Type III Polyglandular Autoimmune Syndrome Pathophysiology. https://emedicine.medscape.com/article/124398-overview#a5[↩]
- Sternthal E, Like AA, Sarantis K, Braverman LE. Lymphocytic thyroiditis and diabetes in the BB/W rat. A new model of autoimmune endocrinopathy. Diabetes. 1981 Dec. 30(12):1058-61.[↩]
- Hansen MP, Matheis N, Kahaly GJ. Type 1 diabetes and polyglandular autoimmune syndrome: A review. World J Diabetes. 2015 Feb 15. 6 (1):67-79.[↩]
- Dittmar M, Kahaly GJ. Genetics of the autoimmune polyglandular syndrome type 3 variant. Thyroid. 2010 Jul. 20(7):737-43.[↩]
- Neufeld M, Blizzard RM. Polyglandular autoimmune disease. Pinchera A, Doniach D, Fenzi DF, Baschieri L, eds. Autoimmune aspects of endocrine disorders. London, UK: Academic Press; 1980. 357-65.[↩]
- Hunger-Battefeld W, Fath K, Mandecka A, et al. [Prevalence of polyglandular autoimmune syndrome in patients with diabetes mellitus type 1]. Med Klin (Munich). 2009 Mar 15. 104(3):183-91.[↩][↩]
- June, C.H., J.T. Warshauer, and J.A. Bluestone, Is autoimmunity the Achilles’ heel of cancer immunotherapy? Nat Med, 2017. 23(5): p. 540-547.[↩][↩]
- Ryder, M., et al., Endocrine-related adverse events following ipilimumab in patients with advanced melanoma: a comprehensive retrospective review from a single institution. Endocr Relat Cancer, 2014. 21(2): p. 371-81.[↩]

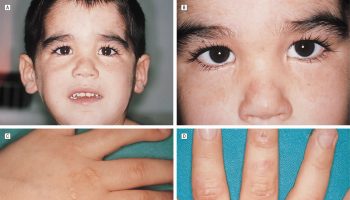

{kind=link}