Bohring Opitz syndrome
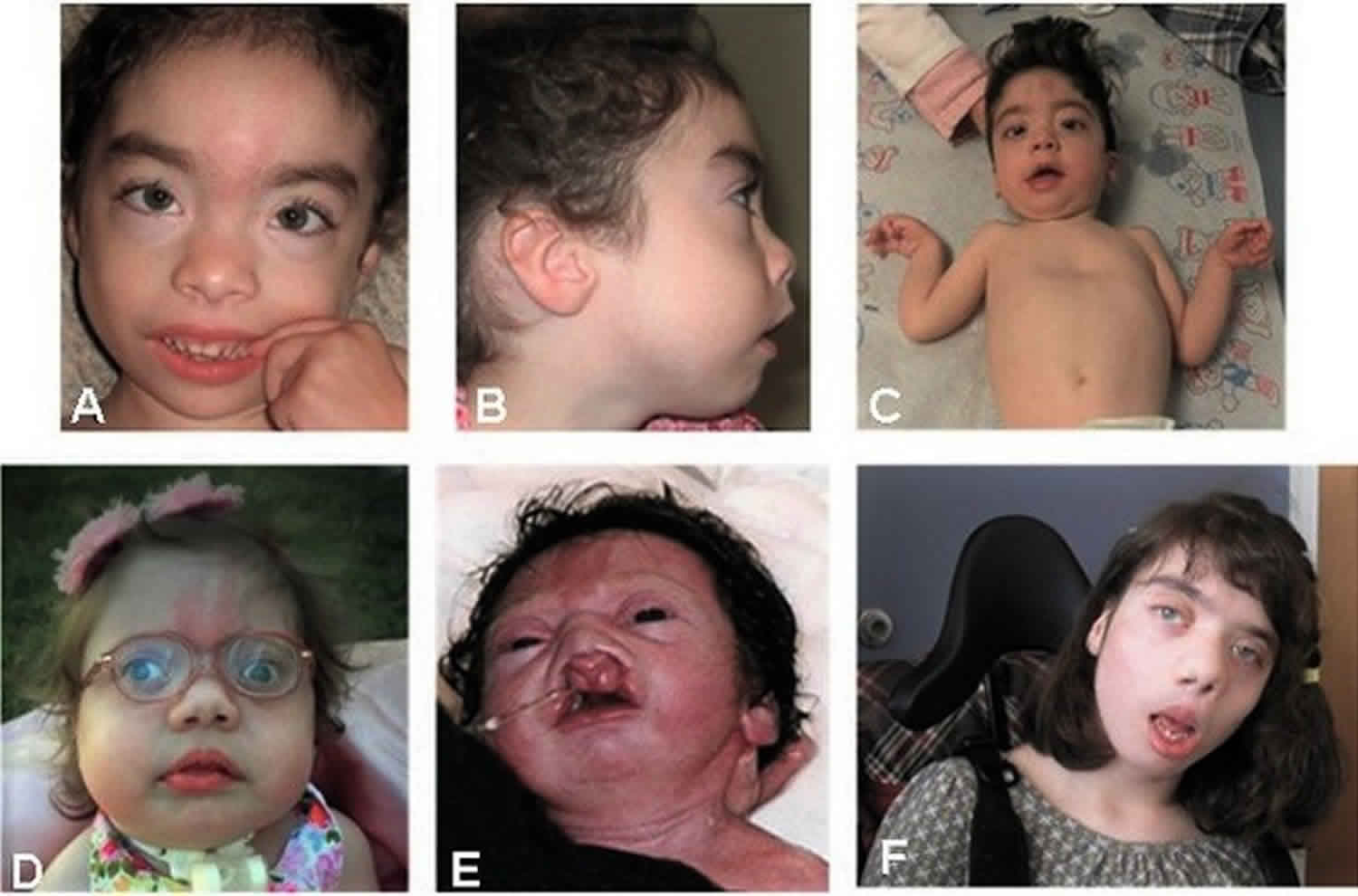
Bohring Opitz syndrome also called Opitz trigonocephaly-like syndrome, C-like syndrome or Oberklaid-Danks syndrome, is a ultra-rare genetic condition that affects the development of many parts of the body characterized by distinctive facial features, variable small head size (microcephaly), hypertrichosis, nevus flammeus, severe myopia, unusual posture (flexion at the elbows with ulnar deviation, and flexion of the wrists and metacarpophalangeal joints), severe intellectual disability, and feeding issues 1. Most individuals with Bohring-Opitz syndrome have profound to severe intellectual disability, developmental delay, and seizures. Most affected individuals have a normal head shape and size with no brain abnormalities; however, some have abnormal development of the head. Abnormal development can lead to a small head size (microcephaly) and a skull abnormality called trigonocephaly, which gives the forehead a pointed appearance. Structural brain abnormalities can occur with or without head abnormalities. For example, the fluid-filled spaces near the center of the brain (ventricles) may be usually large (ventriculomegaly) or the tissue that connects the left and right halves of the brain (the corpus callosum) can be abnormally thin.
Eye problems that can affect vision also occur in people with Bohring-Opitz syndrome. People with this disorder may have protruding eyes (exophthalmos), eyes that do not point in the same direction (strabismus), widely spaced eyes (hypertelorism), or outside corners of the eyes that point upward (upslanting palpebral fissures). Affected individuals may have severe nearsightedness (high myopia) or abnormalities in the light-sensitive tissue at the back of the eye (the retina) or the nerves that carry information from the eyes to the brain (optic nerves).
Additional facial differences associated with Bohring-Opitz syndrome can include a flat nasal bridge, nostrils that open to the front rather than downward (anteverted nares), a high arch or opening in the roof of the mouth (high arched or cleft palate), a split in the upper lip (cleft lip), a small lower jaw (micrognathia), low-set ears that are rotated backward, a red birthmark (nevus simplex) on the face (usually the forehead), a low frontal hairline often with eyebrows that grow together in the middle (synophrys), and excessive body and facial hair (hirsutism) that increases with age.
Some individuals with Bohring-Opitz syndrome have poor growth before birth (intrauterine growth retardation). During infancy, they grow and gain weight slowly and often have severe feeding difficulties with recurrent vomiting.
People with this condition often have characteristic body positioning, known as Bohring-Opitz syndrome posture. This posture consists of slouching shoulders, bent elbows and wrists, hands positioned with the wrists or all of the fingers angled outward toward the fifth finger (ulnar deviation), with the legs usually extended straight. Affected individuals usually stop exhibiting the Bohring-Opitz syndrome posture as they get older. Other abnormalities include joint deformities called contractures that are apparent at birth in the knees, hips, or other joints and abnormal muscle tone. Affected individuals can have recurrent infections and heart, kidney, or genital abnormalities. In rare cases, a childhood form of kidney cancer known as Wilms tumor can develop.
Some individuals with Bohring-Opitz syndrome do not survive past early childhood, while others live into adolescence or early adulthood. The most common causes of death are heart problems, abnormalities of the throat and airways that cause pauses in breathing (obstructive apnea), and lung infections.
All Bohring-Opitz syndrome individuals who have been reported to have a mutation in the ASXL1 gene appear to have developed it spontaneously (de novo mutations), and not inherited it from a parent. If Bohring Opitz syndrome were to be inherited, it is theorized that it would be in an autosomal dominant manner, meaning that the individual with Bohring Opitz syndrome would have a 50% chance of passing down the affected gene change to any offspring.
In some disorders which are dominant, individuals may have varying expression of certain signs and symptoms. Therefore, not all individuals with Bohring Opitz syndrome may be identical in regards to their medical issues, although many do share similar characteristics.
Bohring-Opitz syndrome is thought to be a rare condition, although its exact prevalence is unknown. Bohring Opitz syndrome is not thought to affect one population of individuals more than another, and does not show any higher prevalence in males or females. There are no areas of the world where Bohring Opitz syndrome is thought to be more common due to founder mutations or inbreeding (consanguinity). The current prevalence is unknown; there have been fewer than 250 cases worldwide, but there may be others who have either not had testing or who have not come to medical attention 2.
Figure 1. Bohring Opitz syndrome
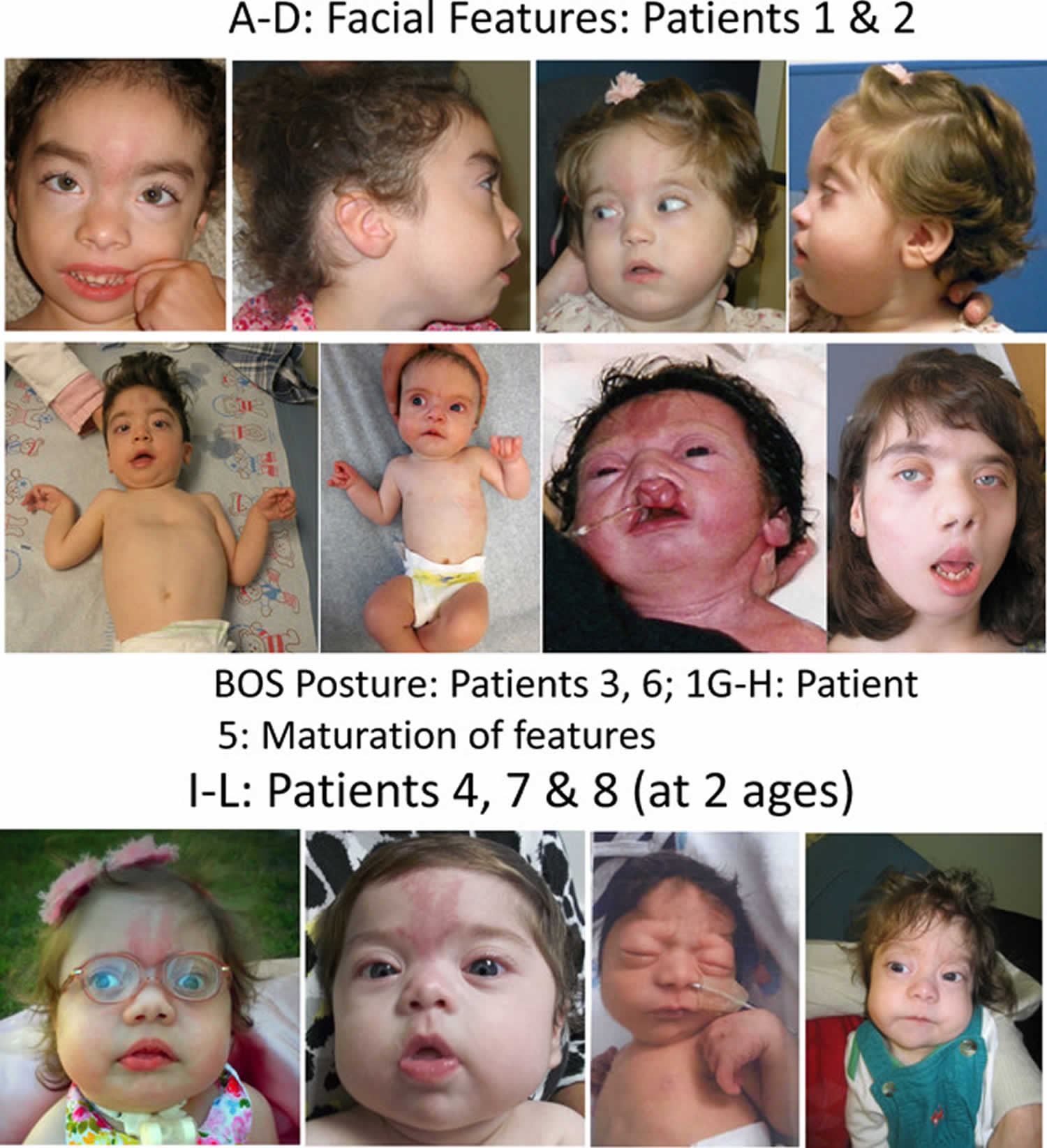
Footnote: Comparison of photographs of patients with Bohring–Opitz syndrome caused by ASXL1 mutations. Patient 1 at 9 years (panels A, B) and patient 2 at 2 years (panels C, D) with classic facial features. Patient 3 at 18 months (panel E) and patient 6 at 3 months (panel F) demonstrating the typical Bohring-Opitz syndrome posture. Patient 5 as an infant and at 12 years (panels G, H). Patient 4 at 2 years (panel I) and Patient 7 at 11 months (panels J) with prominent nevus flammeus. Patient 8 at birth (panel K) and 7 months (panel L) demonstrating progression of features over time. Facial features include nevus flammeus that fades with age, facial hypotonia, proptosis, widely spaced eyes, depressed and wide nasal bridge, anteverted nares, palatal anomalies including cleft lip and/or palate and prominent palatine ridges, micro and retrognathia, low-set ears with increased posterior angulation, a low posterior hairline, and hypertrichosis. The characteristic Bohring-Opitz syndrome posture can be seen in panels E and F with flexion at the elbows with ulnar deviation, and flexion of the wrists and metacarpophalangeal joints.
[Source 3 ]Bohring Opitz syndrome causes
Bohring-Opitz syndrome is caused by mutations in the ASXL1 gene 4. The ASXL1 gene provides instructions for making a protein that is involved in a process known as chromatin remodeling. Chromatin is the complex of DNA and proteins that packages DNA into chromosomes. The structure of chromatin can be changed (remodeled) to alter how tightly DNA is packaged. Through its role in chromatin remodeling, the ASXL1 gene regulates the activity (expression) of many genes, including a group of genes known as HOX genes, which play important roles in development before birth. The ASXL1 protein can turn on (activate) or turn off (repress) HOX genes depending on when they are needed.
ASXL1 gene mutations reduce the amount of functional ASXL1 protein available, which likely disrupts the regulation of the activity of HOX genes and other genes during development. Altered activity of these genes probably leads to the neurological and physical features of Bohring Opitz syndrome.
Bohring Opitz syndrome inheritance pattern
Most cases of Bohring-Opitz syndrome result from new (de novo) mutations in the gene that occur during the formation of reproductive cells (eggs or sperm) or in early embryonic development. These cases occur in people with no history of the disorder in their family. No one with Bohring-Opitz syndrome has been known to have children.
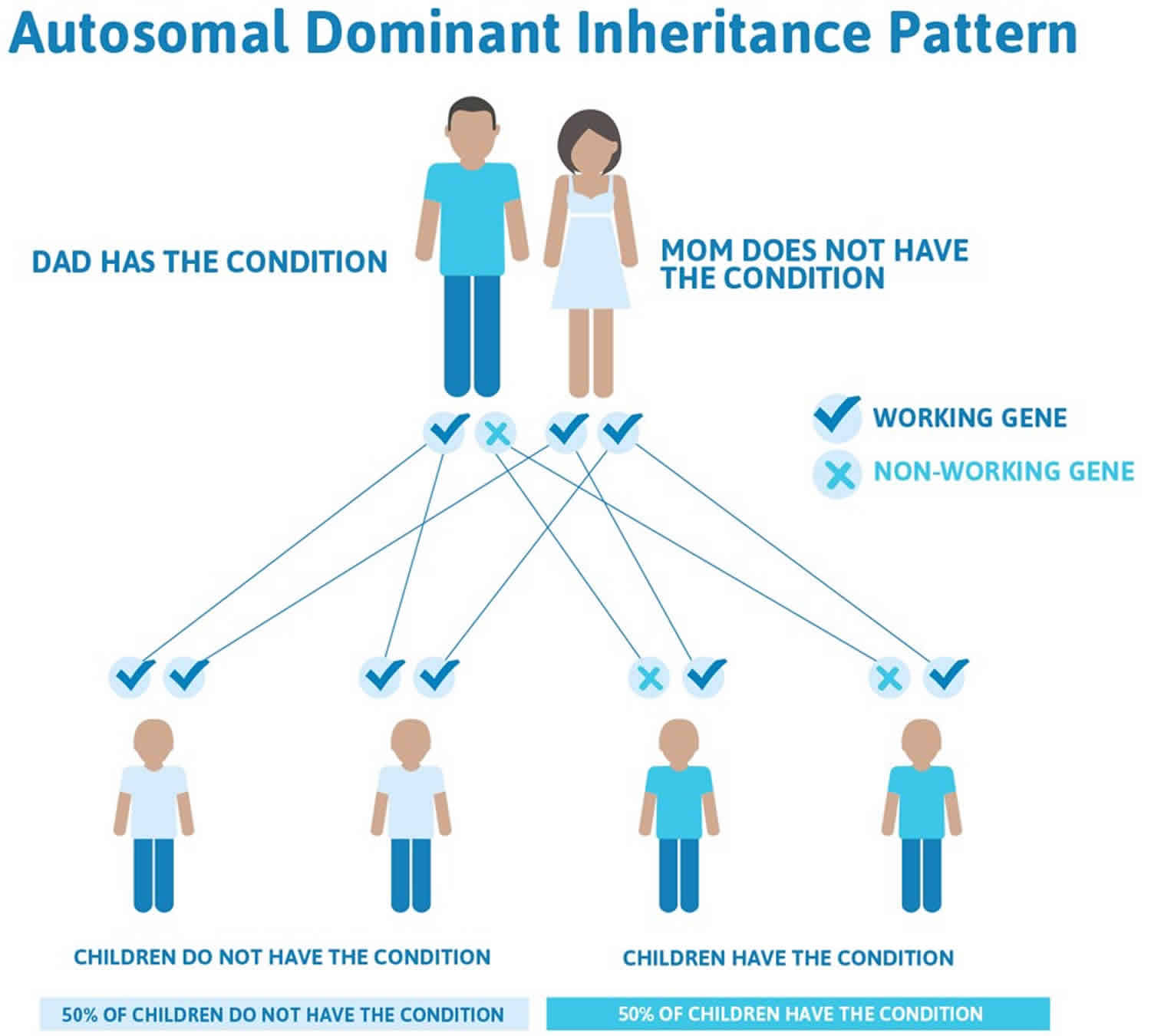
However, if Bohring Opitz syndrome were to be inherited, it is theorized that it would be inherited in an autosomal dominant manner, which means one copy of the altered gene in each cell is sufficient to cause the disorder. The individual with Bohring Opitz syndrome would have a 50% chance of passing down the affected gene change to any offspring. No one with Bohring-Opitz syndrome has been known to have children. Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
Very rarely, individuals with Bohring-Opitz syndrome inherit the altered gene from their unaffected mother, who has the mutation only in some cells, including egg cells, but not in others. This phenomenon is known as mosaicism.
Figure 2. Bohring Opitz syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Bohring Opitz syndrome signs and symptoms
Bohring Opitz syndrome presents typically at birth (congenital), but not all signs may be evident immediately. Infants with Bohring Opitz syndrome may display poor growth prenatally (intrauterine growth restriction), and may have brain abnormalities evident on prenatal ultrasound, including but not limited to agenesis of the corpus callosum, enlarged ventricles, or Dandy-Walker malformation.
Infants with Bohring Opitz syndrome may have feeding issues which may necessitate the use of a feeding tube (G-tube or gastrostomy tube), as well as cyclic vomiting, gastroesophageal reflux, or oral aversion. Feeding issues may improve or resolve as children get older. Some children with Bohring Opitz syndrome may have a cleft lip or palate which may further complicate feeding and swallowing. Individuals may have cardiac anomalies involving structure of the heart or heart rhythm abnormalities. Frequent infections may be common, and may be exacerbated by low tone and inability to clear secretions. Some children may need a breathing tube through the neck (tracheostomy) if they have significant airway issues or need help breathing. Although children with Bohring Opitz syndrome are more susceptible to common respiratory infections, there have been no documentations of immunodeficiency in Bohring Opitz syndrome. Some children may have sleep problems related to falling or staying asleep, and some may also have pauses in their breathing during sleep (sleep apnea). Children may also have eye abnormalities including difficulty seeing far away (myopia) or problems with eye muscles (strabismus).
All individuals with Bohring Opitz syndrome have learning and developmental differences, which are often severe. Most children do not attain speech, although many are able to use various adaptive communication devices. Although few individuals with Bohring Opitz syndrome walk independently, many also benefit from adaptive walkers, strollers, and leg bracing to assist with mobility. There are some individuals with Bohring Opitz syndrome who have been reported to have a type of childhood kidney cancer called Wilms tumor; while there may be an increased risk for children with Bohring Opitz syndrome to have Wilms tumor, and certain screening recommendations exist, not all children with Bohring Opitz syndrome develop cancer.
Bohring Opitz syndrome diagnosis
Making a diagnosis for a genetic or rare disease can often be challenging. Healthcare professionals typically look at a person’s medical history, symptoms, physical exam, and laboratory test results in order to make a diagnosis. The definitive diagnosis of Bohring Opitz syndrome is made after molecular genetic testing for mutations in the ASXL1 gene. Clinical suspicion for Bohring Opitz syndrome may be raised if an individual displays the characteristic Bohring Opitz syndrome posturing of the hands and elbows, is having delayed growth and developmental milestones, and has the characteristic facial features seen in Bohring Opitz syndrome, including the presence of characteristic birthmarks. While the aforementioned characteristics are considered classic, individuals may present more variably, so Bohring Opitz syndrome should not be entirely excluded necessarily on clinical presentation alone, unless obvious signs of another condition are present.
Clinical testing and work-up
Children with Bohring Opitz syndrome should receive regular abdominal ultrasounds every three months from birth (or at the time of diagnosis) until eight years to screen for Wilms tumor. They should also receive regular evaluations to focus on growth, feeding, nutrition, and management of other complications.
Bohring Opitz syndrome treatment
There are currently no definitive treatments, medications, or therapies that will reverse the symptoms of Bohring Opitz syndrome as there are no proven treatments to change an individual’s mutation in the ASXL1 gene. While some work is being done in general with gene therapy and other advanced technologies (CRISPR/CAS9), there have been no studies on children with Bohring Opitz syndrome.
Treatment is currently geared toward symptomatic treatment for an individual’s specific medical issues. Most children will benefit from a combination of various therapies, including physical, occupational, and speech. They may also benefit from an augmented communication device and other devices to assist with mobility, including standers, gait trainers, adaptive strollers, etc.
Children with feeding difficulties may benefit from G-tubes or GJ tubes; those who are at risk for recurrent aspiration and develop lung disease or who need supplemental oxygen may need a tracheostomy and/or ventilator support.
Individuals who obtain common respiratory infections should be aggressively treated with the assistance of clearance of secretions and managed appropriately to reduce complications. All individuals with Bohring Opitz syndrome, unless there is evidence of cellular immunodeficiency, should receive the standard schedule of childhood immunizations, including prophylaxis for respiratory syncytial virus (RSV), if appropriate, and influenza.
Bohring Opitz syndrome prognosis
The leading cause of death is respiratory infections. Children with Bohring Opitz syndrome can have feeding difficulties, recurring respiratory infections, sleep apnea, developmental delay, failure to thrive, abnormal hair density and length, Wilm’s Tumors, brain abnormalities, silent aspiration, and many more.
Reports of malignancies in patients with Bohring Opitz syndrome have been limited to a patient who died at 5 months and was found on autopsy to have bilateral nephroblastomatosis 5 and a patient who developed medulloblastoma at age 5 years 6. Neither had a mutation in ASXL1 7. ASXL1 mutations have been identified in patients with myelodysplastic syndromes 8. Leukemia cells with heterozygous ASXL1 mutations have reduced or absent ASXL1 protein expression, suggesting a role for ASXL1 as a tumor suppressor 9. Given the link between ASXL1 and myelodysplastic conditions 8, combined with the small number of reported patients with Bohring Opitz syndrome and high infant mortality, predisposition for neoplasms may be higher than reported.
- Russell, Bianca & Johnston, Jennifer & Biesecker, Leslie & Kramer, Nancy & Pickart, Angela & Rhead, William & Tan, Wen-Hann & Brownstein, Catherine & Clarkson, L. & Dobson, Amy & Rosenberg, Avi & Schrier Vergano, Samantha & Helm, Benjamin & Harrison, Rachel & Graham, John. (2015). Clinical Management of Patients with ASXL1 Mutations and Bohring-Opitz Syndrome, Emphasizing the Need for Wilms Tumor Surveillance. American journal of medical genetics. Part A 9999A:1–10. DOI 10.1002/ajmg.a.37131[↩]
- Bohring-Opitz Syndrome. http://bos-foundation.org/overview[↩]
- Russell, Bianca & Johnston, Jennifer & Biesecker, Leslie & Kramer, Nancy & Pickart, Angela & Rhead, William & Tan, Wen-Hann & Brownstein, Catherine & Clarkson, L. & Dobson, Amy & Rosenberg, Avi & Schrier Vergano, Samantha & Helm, Benjamin & Harrison, Rachel & Graham, John. (2015). Clinical Management of Patients with ASXL1 Mutations and Bohring-Opitz Syndrome, Emphasizing the Need for Wilms Tumor Surveillance. American journal of medical genetics. Part A. 167. 10.1002/ajmg.a.37131[↩]
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, deVries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R,Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M,Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA,Brunner HG, de Vries BBBA. 2011.De novononsense mutations inASXL1cause Bohring–Opitz syndrome. Nat Genet 43:729–731.[↩]
- Brunner HG, Van Tintelen JP, De Boer RJ. 2000. Bohring syndrome. Am JMed Genet 92:366–368.[↩]
- Hastings R, Cobben JM, Gillessen-Kaesbach G, Goodship J, Hove H,Kjaergaard S, Kemp H, Kingston H, Lunt P, Mansour S, McGowan R, Metcalfe K, Murdoch-Davis C, Ray M, Rio M, Smithson S, Tolmie J,Turnpenny P, van Bon B, Wieczorek D, Newbury-Ecob R. 2011. Bohr-ing–Opitz (Oberklaid–Danks) syndrome: Clinical study, review of the literature, and discussion of possible pathogenesis. Eur J Hum Genet19:513–519.[↩]
- Hoischen A, van Bon BW, Rodriguez-Santiago B, Gilissen C, Vissers LE, deVries P, Janssen I, van Lier B, Hastings R, Smithson SF, Newbury-Ecob R,Kjaergaard S, Goodship J, McGowan R, Bartholdi D, Rauch A, Peippo M,Cobben JM, Wieczorek D, Gillessen-Kaesbach G, Veltman JA,Brunner HG, de Vries BBBA. 2011.De novo nonsense mutations in ASXL1 cause Bohring–Opitz syndrome. Nat Genet 43:729–731.[↩]
- Wang J, Li Z, He Y, Pan F, Chen S, Rhodes S, Nguyen L, Yuan J, Jiang L,Yang X, Weeks O, Liu Z, Zhou J, Ni H, Cai CL, Xu M, Yang FC. 2014. Loss of Asxl1 leads to myelodysplastic syndrome-like disease in mice. Blood 123:541–553.[↩][↩]
- Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S,Patel JP, Chung YR, Koche R, Perna F, Zhao X, Taylor JE, Park CY,Carroll M, Melnick A, Nimer SD, Jaffe JD, Aifantis I, Bernstein BE,Levine RL. 2012. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 22:180–193.[↩]





{kind=link}