What is brain plasticity
Brain plasticity also known as neuroplasticity or neural plasticity, is the ability of the brain to form and reorganize neurons and neural connections, especially in response to learning or experience or following injury 1. Brain plasticity (neuroplasticity) is defined as the ability of the nervous system to change its activity in response to intrinsic or extrinsic stimuli by reorganizing its structure, functions, or connections after injuries, such as a stroke or traumatic brain injury (TBI) 2. The brain is continuously plastic and without this ability, any brain, not just the human brain, would be unable to develop from infancy through to adulthood or recover from brain injury. Not so many years ago, mainstream neuroscience and neurological medicine contended that brain plasticity was limited to an early childhood epoch—a “critical” or “sensitive period.” Now scientists know that brain remodeling can be induced on a large scale at any age in life 3. What differs as a function of age is the way in which the brain regulates plasticity. In the very young brain, almost all inputs continuously engage competitive plasticity processes. In older brains, plasticity is regulated as a function of behavioral context and outcomes. As the brain grows, individual neurons mature, first by sending out multiple branches (axons, which transmit information from the neuron, and dendrites, which receive information) and then by increasing the number of synaptic contacts with specific connections. Whether the brain retains the ability to increase synaptogenesis is debatable, but it could explain why aggressive treatment after a stroke can appear to reverse the damage caused by the lack of blood supply to an area of the brain by reinforcing the function of undamaged connections.
The term “plastic” originates from the Latin word “plasticus,” which ultimately comes from the Greek term “plastikós” or “plastos,” originally meaning “molded, formed” 4. During the twentieth century, the question of how information is stored in the brain stimulated an enormous body of work that focused on the properties of synaptic transmission. The first mention of the term plasticity in regards to the nervous system was by William James in 1890 5. However, the term neural plasticity is credited to Jerzy Konorski in 1948 2 and was popularized by Donald Hebb in 1949 with the publication of “The Organization of Behavior” 6. In his book, Donald Hebb articulated a theory, the so-called “Hebb’s postulates,” regarding the possible neural mechanisms of learning and memory that have since had an enormous influence on studies into neurophysiology. Although Donald Hebb himself admitted that he did not propose anything new, the reality is that the terms “Hebbian postulates” and “Hebbian plasticity” are now widely used in the literature. A year before the publication of Hebb’s book, the Polish Neurophysiologist Konorski 7 postulated that morphological changes in neural connections could be the substrate of learning 4.
Clinically, brain plasticity is the process of brain changes after injury, such as a stroke or traumatic brain injury (TBI) 8. These changes can either be beneficial (restoration of function after injury), neutral (no change), or negative (can have pathological consequences).
Brain plasticity or neuroplasticity is a complicated process that is still being clarified; however, the concept can be applied in the setting of injury to the brain. Brain plasticity is traditionally thought of as occurring in 3 phases or epochs 9:
- First 48 hours: Depending on the mechanism of the injury (such as stroke or TBI), there is initial damage that cumulates as cell death with the loss of certain cortical pathways associated with the lost neurons. The brain attempts to use secondary neuronal networks to maintain function.
- The following weeks: Recruitment of support cells occurs in this period as the cortical pathways shift from inhibitory to excitatory. Synaptic plasticity and new connections are made during this period.
- Weeks to months afterward: The brain continues to remodel itself via axonal sprouting and further reorganization around the damage.
Brain plasticity or neuroplasticity can be broken down into two major mechanisms 8:
- Neuronal regeneration or collateral sprouting: This includes concepts such as synaptic plasticity and neurogenesis.
- Functional reorganization: This includes concepts such as equipotentiality, vicariation, and diaschisis
Synaptic plasticity, functional reorganization, and diaschisis demonstrate unique processes that the brain utilizes in response to damage and the restoration of function. As research continues exploring the functional connections in the brain and what influences those connections, scientists will be able to develop more targeted therapies to help the brain regain function more quickly and more completely.
Neuroplasticity studies
Fundamental studies of brain plasticity mechanisms have shown that the representations of inputs and actions are competitively sorted on the basis of the temporal distributions of inputs 10, that is every brief change cycle invokes a synapse-strengthening moment (e.g., strengthening all inputs whose coordinated actions moment by moment in time are correlated with a positive behavioral outcome), followed by a synapse-weakening moment (e.g., weakening all inputs occurring within a brief, following epoch of time) 11. Following these principles, it is just as easy to degrade the brain’s processing abilities as it is to strengthen or refine them. The synapse weakening can be viewed as an electrically homeostatic process that contributes to the ongoing weakening of behaviorally non-meaningful intrinsic activities or inputs—that is, to a normalization of internal or background external (environmental) noise.
Viewed from another perspective, brain plasticity processes can be viewed as continuously competitive. Through these two-way plasticity processes, neurons in coupled “mini-columns” are continuously competing with their neighbors for the domination on neurons on their mutual boundaries 3. By giving one coupled group the competitive advantage over their neighbors, it is easy to expand their team a 1000-fold or, if they are a competitive loser, to reduce its “membership” many times over. By giving any one source of input a competitive advantage or disadvantage, the territory it comes to dominate in the brain can be dramatically enlarged, or contracted; every moment of gain for the “winner” is a moment of loss for “losers.” By these two-way processes, one can easily both refine (for some inputs) and degrade (other inputs)—even the most-fundamental aspects of representation of visual or auditory or somatosensory inputs in the adult brain.
Nueroplasticity engages fundamentally reversible neurological change processes. Scientists have conducted a number of studies that have demonstrated that neuroplasticity follows Hebbian principles: “What fires together, wires together” 12. This coincident-input-dependent co-strengthening of synaptic connections occurring moment by moment in time in a learning context is achieved through both a multiplicity of physical changes in synapses that amplify connection strength, and by synaptogenesis. The magnitude of such changes under near-optimum learning contexts can be remarkable: a large proportion of synapses in any directly engaged cortical zone (commonly, many millions to billions of synapses) are altered in their connection strengths as you acquire any significant skill or ability 13. As you master any skill or ability through experience or progressive learning, these changes in brain circuitry result in the specialization of the brain as a master receiver and master controller of all of the inputs and actions supporting that mastery.
Through Hebbian network plasticity, the extensively cross-wired neurons in the cerebral cortex also strengthen their connections with their nearest neighbors. When the brain is engaged behaviorally, inputs that are activated nearly simultaneous in time strengthen together, increasing their cooperativity to generate more salient (i.e., more collectively powerful, more reliable) responses. That plasticity-driven growth in local “teamwork” is a critical aspect of the improvement in the processing of information supporting learning-based advances in behavior 3.
Learning-driven increases in neuronal response coordination are a primary determinant of the feed-forward power of any plastically strengthening cortical process. Cortical neurons at all “higher” system levels are integrators operating with short time constants. Their plasticity processes are also coincident-input dependent. The greater the coordination of neurons in the lower levels of the network that feeds them, the greater their selective powers and selectivity, and the greater the power of that input to drive plastic remodeling at higher system levels. Moreover, at the “top” of your great brain systems, coordination of activity is a primary determinant of the ability of cortical networks to sustain the reverberant activities that are selective for behavioral targets or goals (i.e., working memory) 14. The strengths of these key plasticity-gating processes “at the top” are crucially dependent upon the strengths, i.e., collective coordination, of the inputs that feed them.
Nonetheless, the same processes that confer growth in synaptic power for inputs that contribute to neurobehavioral advance are also driven backward, for other non-behaviorally-contributing synapses, in a synaptic weakening and synapse elimination direction. This “normalization” of collective synaptic input power has been extensively studied in other experimental models by depriving neurons of a major source of their inputs; in that event, synaptic strengths are rapidly adjusted to sustain neurons within a narrow electrical potential window that assures their ongoing functional viability 15.
Scientists have recently conducted a number of studies in animals that show that plasticity processes are very broadly reversible. For example, after documenting many aspects of the function, anatomy and chemistry in the brains of aged vs. young adult animals, it was shown that every measure differed markedly 16. In the aged rats’ auditory cortices, time and space constants were longer and greater; response selectivity was poorer; reliability of sound feature representation was poorer; response correlation was weaker; the neuron populations representing sensory inputs were less strongly coupled, operating with far weaker cooperativity; inhibitory processes controlling “top-down” modulation were weaker; local and long range connections were more poorly myelinated; level-to-level (system) coordination (in gamma and theta frequency ranges) was less sharply localized and more weakly persistent; representational topographies were degraded; trophic factors contributing to metabolic and physical maintenance and plasticity were only weakly expressed; the normally strong adaptation to repeated identical stimuli and responses to unexpected stimuli against a continuous or repeated background were sharply reduced; the strong suppression of non-attended distractors was reduced; receptor subunits for inhibitory and excitatory processes were altered in a degrading direction; and the modulatory control processes controlling plasticity were all more weakly operating in very old vs. prime-of-life animals. After recording these manifold, significant differences between aged and young rats’ brains, animals were intensively behaviorally trained in operant tasks to determine which of these operational characteristics of the brain could be “rejuvenated.” Somewhat to our surprise, with training limited in these aged rats to approximately 1 h/day for about 1 month, all of these degraded operational and physical-chemical characteristics of the aged brain could be substantially if not completely restored to a “youthful” state, in aged animals 16.
Given its reversible nature, plasticity processes can just as easily be engaged in a young prime-of-life animal in ways that drive their brains in an increasingly uncorrelated pattern activity (as seen in aged animals). That has also been achieved for the auditory brain by a simpler environmental exposure strategy. By housing young, vigorous adults in an environment of non-correlated noise (believed to increase the level of internal noise in the hearing brain) for a period of several weeks, all of the functional and physical characteristics of the machinery of the brain noted above altered as if the animal had advanced over those several weeks to an “old age” status 17.
Because these reversible change processes can drive neurological changes in either an advancing or degrading direction, driving the processing and physical characteristics of the brain rapidly “forward” to simulate aging is equivalent to driving the animal backward in age: The physical and functional properties of the brain near the end of life closely correspond to those same characteristics in the brain recorded near the beginning of life 18. That conclusion is supported by documenting the operational and physical characteristics of the machinery of the brain in very old and very young animals: they closely match one another. It is also manifested by the fact that key accelerated changes leading to “premature aging” achieved by noise exposure or by “negative” training, carried forward far enough, similarly result in the re-opening of the “critical period” 18.
Brain plasticity at the synaptic level
Brain plastic changes are age-, gender-, time-, experience-, and region-dependent 19. Among all regions, the cortex is the most widely reported as the most plastic region after injury. These plastic changes can be seen as reorganization in regions surrounding the damaged area and recruitment of new regions or use of alternative networks 20. In experimental animal models, extensive synaptic remodeling can be seen in the ipsilateral neocortex at the acute stage, as shown by a decrease in the density of pedunculated spines on apical dendrites 21. Depending on the injury type, animals with focal injury show reinnervation of damaged tissue by surrounding tissue, and animals with diffuse injury show regenerative responses in damaged tissue areas at the acute stage 22. At the chronic stage, there is a drag during neuroinflammatory microglial activation, which could be detrimental to brain recovery, along with continuous expression of plasticity-relevant proteins in promotion of cortical plasticity, including microtubule-associated protein-2 (MAP2) and synaptophysin (SYN) 23.
Another important region of brain plasticity investigation is the striatum, including the thalamus. The thalamus serves as a relay station of somatosensory and motor function, and the striatum is full of dopaminergic neurotransmitters and plays an important role in motor control, motivation, arousal, cognition, and reward. Using functional MRI (fMRI), manganese-enhanced MRI and histological validation, Yu et al. 24 demonstrated that thalamocortic inputs may represent a major site for adult plasticity, in contrast to the consensus that adult plasticity mainly occurs at cortico-cortical connections. Synaptic plasticity in the striatum is also linked to the activation of dopamine receptors with DARPP-32 as well as glutamatergic transmission 25, and tyrosine hydroxylase serves to catalyze the formation of dopamine precursors 26. van Bregt et al. 26 reported lasting alterations in dopamine metabolism in association with neuronal degeneration in the substantia nigra at the acute stage but no tyrosine hydroxylase change. At the chronic stage, 28 days after injury in rodent traumatic brain injury models, both van Bregt et al. 26 and Yan et al. 27 reported alternations of tyrosine hydroxylase in the striatum as a compensatory mechanism to counteract dopamine defficiency after traumatic brain injury.
Additionally, the hippocampus in particular has been an important region of interest for plasticity investigation in animal models, due to its theorized role in memory modulation, which are widely reported across different populations and can persist for long time. Early after traumatic brain injury, at 24 hours, a substantial increase in spine density on dendrites bilaterally in CA1 and CA3 and the dorsal dentate gyrus could be seen, indicative of an increase in excitatory synapses 21. Later, from the first week up to 2 months after injury, aberrant mossy fiber prouting in the dentate gyrus at the ipsilateral side of brain injury can be seen, which is associated with spontaneous convulsive seizures 28. Therefore, mossy fiber sprouting may play a role, along with neuron loss, neurogenesis, gliosis, and morphological changes that alter sensitivity to stimulation after traumatic brain injury in animal models 28.
Brain plasticity at microstructural level
It has been reported that brain injury in early life, at the perinatal stage, can trigger structural and functional reorganization of the brain, which can be seen in both structural and functional MRI. Among structural MRI, diffusion tensor imaging (DTI) has been reported being sensitive to white matter (white matter) injury at a microstructural level that is invisible on conventional MRI 29, 30.
Evidence of brain plasticity includes reorganizations of the motor system, somatosensory system, and language regions detected by structural T1, diffusion tensor imaging tractography and task oriented functional MRI (fMRI) 31. In the early childhood, a patient who was born prematurely with serious injury on the arcuate fasciculus, the critical pathway for language processing, could still have preserved language and reading functions 32. Instead, diffusion tensor imaging (DTI) tractography demonstrated intact ventral connections between the temporal and frontal lobes through the extreme capsule fiber system and uncinate fasciculus. This connection is likely to take over the language processing functionality as a compensational mechanism 32. In adult brain injury patients, Yogarajah et al. 33 conducted a diffusion tensor imaging study of 46 epilepsy patients with anterior temporal lobe resection. They reported increased fractional anisotropy (FA) in the ventro-medial language network, in suggestion of structural reorganization in response to the resection 33. In a case study of a patient suffering from tuberous meningitis at the age of 12 years, Jan et al. 34 reported a new motor pathway posterior to the lesion in the midbrain and upper pons, evidenced by diffusion tensor tractography, task oriented functional MRI (fMRI), and transcranial magnetic stimulation. Laitinen et al. 35 also reported increased fractional anisotropy in the hippocampal dentate gyrus in ananimal model induced with status epilepticus, and the fractional anisotropy (FA) changes are correlated with histologically verified axonal plasticity of myelinated and non-myelinated neuronal fibers. This evidence demonstrated that morphological structural is the foundation of brain plasticity. In case of injury at one location, other possible alternative routes must be intact to compensate the impaired function. Zhou et al. 36 demonstrated that rats with partial corpus callosotomy can have restored functional connectivity between hemispheres at 28 days after injury, but rats with complete corpus callosotomy cannot, likely due to the compensation that occurred through the remaining interhemispheric axonal pathways. Their data suggest that axonal connections are the indispensable foundation for resting state functional connectivity.
Figure 1. Adult neurogenesis brain regions
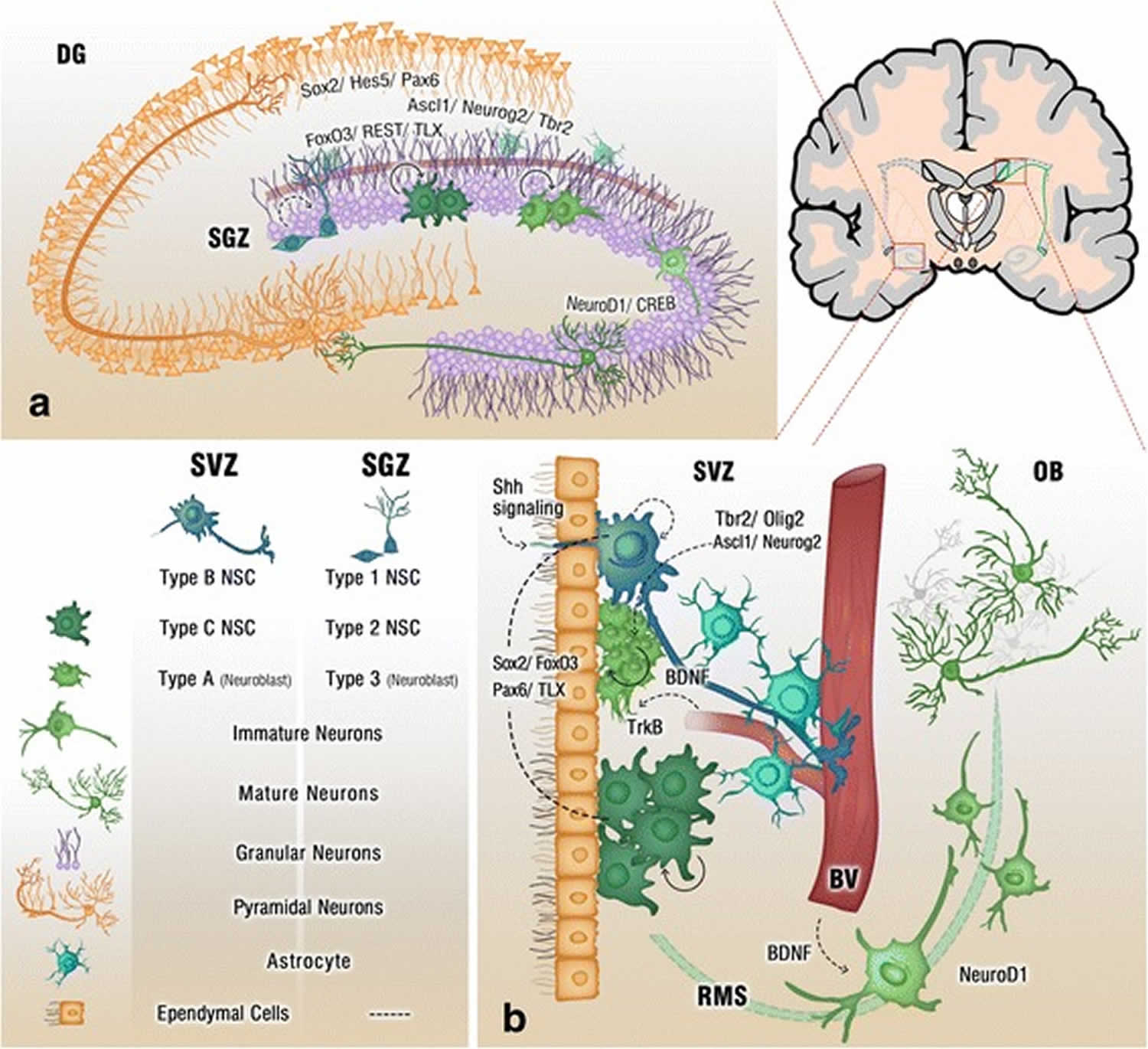
Footnote: Cross section of the adult brain showing regions of subgranular zone (SGZ) and subventricular zone (SVZ), where neurogenesis takes place. The schematic illustrates neurogenesis involving neural stem cells (NSCs) development into mature neurons and the neurogenic niches of Blood Vessels (BV), astrocytes and cilia, as well as transcription programs in the SGZ (a) and SVZ (b). Neuronal migration from the SVZ to the olfactory bulb (OB) via the rostral migratory stream (RMS) is also shown in (b)
[Source 37 ]Figure 2. Neurogenesis brain regions
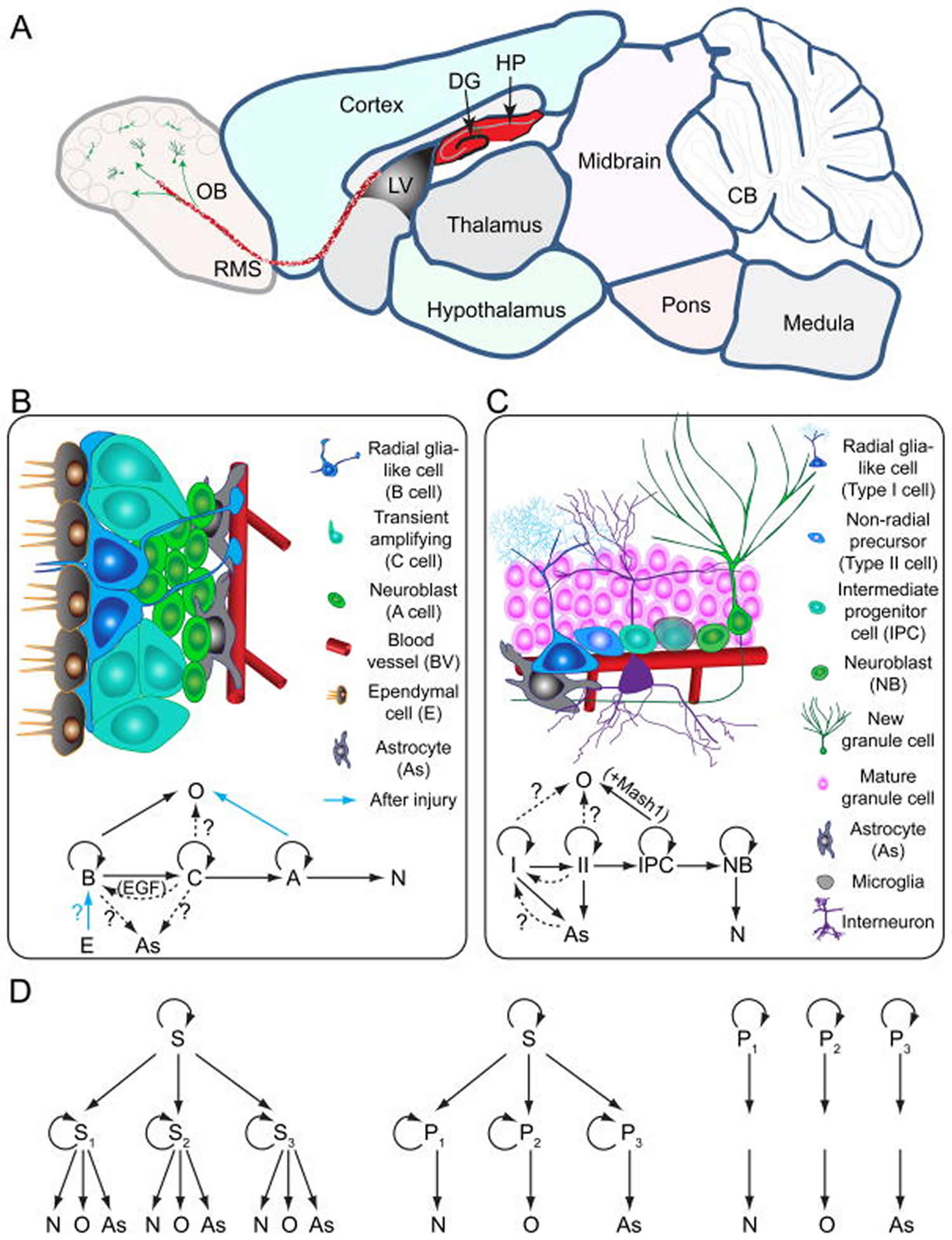
Footnotes: Models of neural stem cells and lineage relationship in the adult dentate gyrus and subventricular zone
(A) A sagittal section view of an adult rodent brain highlighting the two restricted regions that exhibit active adult neurogenesis: dentate gyrus (DG) in the hippocampal formation (HP); the lateral ventricle (LV) to the rostral migratory stream (RMS) to the olfactory bulb (OB).
(B) A schematic illustration of the neural stem cell niche in the subventricular zone (SVZ) and a model of potential lineage relationship under basal (solid arrows) and injury conditions (blue arrows). N: immature neurons
(C) A schematic illustration of the neural stem cell niche in the subgranular zone (SGZ) in the dentate gyrus and a model of potential lineage relationship.
(D) Three lineage models of neural precursors in the adult mammalian brain. In the first model (left), adult neural stem cells (S1,2,3…) generated from primitive neural stem cells (S) are intrinsically diverse, exhibiting vastly different developmental potential depending on their regions of distribution and developmental origins. In the second model (middle), adult neural stem cells (S) are relatively homogenous and give rise to a heterogeneous population of lineage-restricted progenitors (P1,2,3…). In the third model (right), only lineage-restricted neural progenitors (P1,2,3…) are present in the adult brain; self-renewal and multi-lineage differentiation represent a collective property of a mixture of different lineage-restricted neural progenitors.
Abbreviations: N = neurons; O = oligodendrocytes; As = astrocytes.
[Source 38 ]Not all neuroplasticity is good
While neuroplasticity can be beneficial (i.e., restoring function), not all brain plasticity is beneficial to the brain function 39. Maladaptive plasticity is when a connection that is made in the brain produces aberrant or negative symptoms. This can be seen in the examples of use-dependent dystonia (writer’s cramp) and phantom limb pain 40. Both of these examples have shown abnormal primary sensory cortex changes in association with painful symptoms.
Moreover, some brain plasticity can induce pathological conditions called pathological plasticity 41. Examples include pathological pain 42, pathological response to sickness 43, epilepsy 44, schizophrenia 45 and dementia 46. One well-known example of pathological brain plasticity is that some drug treatments can cause maladaptive brain plasticity for drug addiction 39. Another example is the mossy fiber sprouting and reorganization in hippocampus after traumatic brain injury that may lead to epilepsy due to pathological firing of neurons and reduced synaptic inhibition in the region 47. In addition, maladaptive changes during plasticity can have detrimental effects on the brain itself 48 and interfere with regaining of normal functions 49. In stroke, recruitment of the contralateral region is compensatory in the early stage but may become maladaptive in later stages 48. One challenge during pharmacological treatment or in the designing of rehabilitation programs for traumatic brain injury patients is to minimize the side effects of treatment so that it will not induce maladaptive plastic changes that could actually interfere with recovery. Particular caution has to be taken during drug treatment to avoid maladaptive behavior of drug addiction 39.
Neuroplasticity mechanisms
Synaptic plasticity is change that occurs at synapses, the junctions between neurons that allow them to communicate. Synaptic plasticity is the major component of neural plasticity 50. The existence of synaptic plasticity in response to the pattern of electrical activity has been postulated by Canadian psychologist Donald Hebb 51, who thought it might underlie associative learning. Donald Hebb introduced the concept that the pattern of electrical activity is important to induce plastic changes in synaptic efficacy, and in particular the notion that the presence of a correlated activity between pre- and postsynaptic neurons promotes the potentiation of synaptic transmission; as a corollary, decorrelated activity promotes depression of synaptic transmission. In Hebb’s statement, it is already present also the notion that synaptic plasticity involves both functional and structural changes. Homosynaptic long-term potentiation at excitatory synapses consists indeed of a prolonged increase in synaptic efficacy which involves both functional and structural modifications of synaptic contacts and is induced by repeated bouts of correlated pre- and postsynaptic activity. The temporal requirements for the pattern of pre- and postsynaptic activity has been now refined, since several studies have shown how synaptic efficacy can be increased (long-term potentiation) or decreased (long-term depression) according to the relative spike timing and firing rate of pre- and postsynaptic neurons (spike-timing-dependent plasticity) 52. Also, the level of previous activity in the circuit affects the probability that a given change of activity in the same circuit induces long-term potentiation rather than long-term depression 53.
Non-Hebbian forms of synaptic plasticity have also been observed: for instance, monocular neurons in the visual cortex responding exclusively to the deprived eye actually increase their responsiveness following monocular deprivation 54, 55 as if they were trying to preserve their original drive. Therefore, in addition to plastic changes driven by synapse-specific electrical activity which follow hebbian rules, there seem to be forms of global feedback plasticity aimed at preserving neuronal total excitatory drive.
Long-term potentiation and long-term depression have been observed in many brain areas, both cortical and subcortical 56. The sequence of events leading to induction, expression, and consolidation of long-term changes of the long-term potentiation and long-term depression type has been recently reviewed. The first step, the trigger for induction of long-term potentiation and long-term depression at glutamatergic synapses is, in most cases, dependent on the activation of glutamate N-methyl-d-aspartate receptors (NMDARs), which have classically been thought of as coincidence detectors of pre- and postsynaptic activity 57. Activity of inhibitory inputs is also important for synaptic plasticity induction, since it will both regulate the pattern of activity in the circuit and the activation of N-methyl-d-aspartate receptors (NMDAR) 58.
The opening of NMDARs during the induction of long-term potentiation leads to calcium entry that triggers a biochemical cascade involving intracellular kinases such as extracellular signal regulated kinase (ERK), cAMP-dependent protein kinase (PKA), calcium-calmodulin kinase type II (CaMKII), the end product of which are the first, rapid post- and presynaptic effects towards the expression of synaptic potentiation, which include potentiation of the 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA) receptor-mediated excitatory postsynaptic currents (EPSC) 59. The resulting Ca2+ transient determines the polarity of the induced plasticity, with low and prolonged Ca2+ transients inducing long-term depression and brief, steeper transients inducing long-term potentiation 60. A crucial step towards consolidation of long-term changes of synaptic efficacy is the synthesis of new proteins, either from existing mRNA 61 or following the activation of transcription factors such as the cAMP-responsive element-binding protein (CREB) by intracellular mechanisms mediated by kinase signaling 62, 63. This allows local morphological modifications in synaptic contacts, for instance, at the level of dendritic spines, and provides consolidation of changes in synaptic efficacy. Neurotrophic factors such as the brain-derived neurotrophic factor (BDNF) are also strongly involved in expression and consolidation of synaptic plasticity 64.
The presence of structural changes at synaptic sites has suggested the involvement of modifications of extracellular matrix (ECM) in synaptic plasticity. Indeed, ECM molecules regulate various aspects of synaptic plasticity 65 and proteases acting on ECM molecules are involved in hippocampal long-term potentiation 66 and in visual cortex plasticity 58.
It is important to underline that neuromodulators such as norepinephrine, acetylcholine, dopamine, or serotonin, which reflect the level of arousal, motivation, attention, affection, and emotion of a subject, are strongly involved in the induction and maintenance of synaptic plasticity 67. Striking examples from the visual system include the work by Bear and Singer 68 showing that ocular dominance plasticity in kittens is disrupted by pharmacological manipulations that simultaneously destruct cortical noradrenergic and cholinergic innervations, and by Dan studies 69 demonstrating that stimulation of the rat nucleus basalis decorrelates local cortical neuron responses via activation of muscarinic acetylcholine receptors, improving cortical representation of natural stimuli. An essential instructional role of the nucleus basalis has been also shown by Kilgard and Merzenich 70 for the reorganization of primary auditory cortex responses in the rat.
Finally, a crucial role in the consolidation of long-term neural plasticity is played by epigenetic mechanisms, including histone acetylation, which regulate gene expression 71. For instance, mice with deficient histone acetylation activity exhibit induction but not maintenance of hippocampal long-term potentiation 72. Epigenetic processes are now thought of as mechanisms through which environmental dynamic experiences are inscribed on a fixed genotype, producing a stable alteration of the phenotype; as an example, levels of maternal licking and grooming experienced by rat pups during the first days of life produce long-term effects on the feedback control of the hypothalamic-pituitary-adrenal (HPA) axis, an effect which involves the neuromodulator serotonin, thus programming anxiety-like behavior of pups once they become adult 73. In this case, the neural plastic event, i.e., memory of the experience, is due to epigenetic changes in the transcriptibility of the gene for the glucocorticoid receptors in hippocampal neurons, which modifies the response of the hypothalamic-pituitary-adrenal (HPA) axis to circulating glucocorticoids 74.
Neuronal regeneration and collateral sprouting
Synaptic plasticity
Synaptic plasticity is change that occurs at synapses, the junctions between neurons that allow them to communicate. Synaptic plasticity is the ability to make experience-dependent long-lasting changes in the strength of neuronal connections 2. This is best expressed with the concept of long-term potentiation. First discovered in 1973 by Bliss and Lomo 75 while studying the rabbit hippocampus, repetitive stimulation of presynaptic fibers resulted in high responses of granule cells of postsynaptic neurons 76. As the postsynaptic potential continued for a much longer time than expected, they termed this long-term potentiation. What is theorized to occur is that when the presynaptic neuron stimulates the postsynaptic neuron, the postsynaptic neuron responds by adding more neurotransmitter receptors, which lowers the threshold that is needed to be stimulated by the presynaptic neuron. This enhances the synapse over time in accordance with the idea by Konorski and Hebb. Synaptic plasticity can be positively influenced by several things, including, but not exclusively, exercise, the environment, repetition of tasks, motivation, neuromodulators (such as dopamine), and medications/drugs 77, 78. Aging and neurodegenerative diseases have been associated with a decrease of neuromodulators and may contribute to a reduction in the ability of synaptic plasticity 79. The theory of synaptic plasticity has also grown to include more of the evolving complexity of synaptic communication 2.
These include:
- Spike-timing-dependent plasticity (STDP): This incorporates the timing of action potentials generated by presynaptic and postsynaptic neurons to explain how some synapses are strengthened and others are weakened.
- Metaplasticity: This broadens the concept to include networks and involves the activity-dependent changes in synapses and how they respond.
- Homeostatic plasticity: Mechanisms that maintain homeostasis of the synaptic network over time.
As research continues to grow, these concepts will flesh out more of how synaptic plasticity can influence learning and aid in regaining function in the brain.
Adult neurogenesis
Adult neurogenesis is the concept that the brain continues to make new neurons. Studies by Ramon Cajal 80 had failed to find any evidence of new neuron development in adults, which led to his ‘harsh decree’ that there were no new neurons after the development of the brain stopped. This view continued until Josef Altman was able to find evidence of neurogenesis in adult rats Owji S, Shoja MM. The History of Discovery of Adult Neurogenesis. Clin Anat. 2020 Jan;33(1):41-55. doi: 10.1002/ca.23447. Since then, neurogenesis has been able to be discovered in birds and other small mammals. It has not been convincingly demonstrated in humans 81.
There have been two proposed sites of adult neurogenesis in humans, one in the olfactory bulb and the other in the hippocampus. Studies using specific biomarkers associated with developing neurons have been used to support the idea of adult neurogenesis in humans. These biomarkers are complicated as they have also been found in immature neurons, cells that can be found in the human brain that are not newborn nor migrating cells 81. Coupled with no evidence of a recognizable niche-like structure on histological examination (something seen in other species that exhibit adult neurogenesis), the evidence is inconclusive. More specific biomarkers will most likely need to be developed to identify newborn neurons from immature neurons to elucidate what role they may play in the plasticity of the brain 81.
Functional reorganization
Equipotentiality and vicariation
Equipotentiality is the concept that when one area of the brain is damaged, the opposing side of the brain would be able to sustain the lost function. This concept stretches back at least to Galen, which was a way to explain why the brain appeared ‘twinned.’ This ‘redundancy theory’ remained until famed researchers such as Pierre Paul Broca demonstrated that unilateral lesions to an area of the left side of the brain caused loss of speech, even though the opposing side was intact. Broca postulated that the relearning of certain functions, such as speech, were easier for a child than if an adult suffered loss. This concept morphed into equipotentiality, meaning that if the damage occurred very early, then the brain has the potential to be able to overtake lost functions 82.
This is slightly different from the thought of vicariation, which is that the brain can reorganize other portions of the brain to overtake functions that they were not intended to. Broca developed this theory after seeing that some of his patients had a sparing of function even though they had damage to the left hemisphere 82. In the strictest sense, vicariation is when a part of the brain overtakes a new and unrelated function. With the advent of advanced imaging techniques, it has been shown that neither theory is quite correct.
Graveline, Mikulis, Crawley, and Hwang 83 were able to show that after a hemispherectomy (where one-half of the cerebral cortex is removed, typically due to intractable seizures at a young age), the brain can reorganize the remaining half to restore lost function. Using functional magnetic resonance imaging (MRI), they were able to show that the remaining supplemental motor and sensory areas were able to be reorganized to take over the function of the affected side 83. Jaillard et al. 84 were able to demonstrate similar findings in 4 adult patients who had had an ischemic stroke of their right primary motor cortex. By performing serial functional MRI, they were able to show that the brain was able to show increased activity initially in the bilateral premotor cortex, which shifted over time to the right hemisphere supplemental motor cortex 84. These clinical examples highlight that the brain uses both equipotentiality and vicariation.
Diaschisis
Diaschisis is a concept that damage to one part of the brain could cause a loss of function in another area due to some connected pathway 85. Constantin von Monakow 86 proposed this concept in an attempt to explain why some people lost specific functions (such as speech) but did not have a lesion in the area of the brain thought to supply that function. An example of this is the hypoperfusion of the ipsilateral thalamus after an acute middle cerebral artery (MCA) stroke. The thalamus, which receives its blood supply from branches of the posterior cerebral artery and a branch of the posterior communicating artery, should be unaffected during an ipsilateral middle cerebral artery ischemic stroke. Surprisingly, in approximately 20% of acute middle cerebral artery strokes, there is noted hypoperfusion of the ipsilateral thalamus upon computed tomography (CT) perfusion imaging 87. Other studies have shown that the incidence increases in the subacute and chronic phases of stroke up to 86%, and while the cause is still unknown, a predominant theory is that there is disinhibition from the loss of gamma-aminobutyric acid (GABA-energic) neurons that leads to a combination of neurotoxicity and retrograde degeneration 87. While this phenomenon has been noted, it has not been shown to influence any major clinical outcomes at this time.
The concept of diaschisis has broadened over time and is used to explain several different concepts about the functional connections of the brain and what ensues when damage occurs. While these are discussed by Carrera and Tononi 85, a brief explanation is given:
- Diaschisis ‘at rest’: The classic von Monakow type such as ipsilateral thalamic hypoperfusion in middle cerebral artery stroke.
- Functional diaschisis: This is when an area of diaschisis is found when another part of the brain is activated. An example of this is when lesions affected the putamen, when given a functional task of their ipsilateral hand, causes hypoactivation of the ipsilateral cerebellums, which had no signs of hypoactivation at rest. Dynamic diaschisis can also be used and has been used when areas of the brain can be both hypoactive and hyperactive, depending on the task.
- Connectional diaschisis: This is when a loss of a part of the brain forces the rerouting of information. This has been seen in rat models where subcortical lesions can cause a decrease in interhemispheric connectivity of the motor strips 88.
- Connectome diaschisis: As advanced imaging has shown the vast complexity of connections between neurons, a map can be generated, called a connectome. This map shows clusters of high connected nodes, which are then linked by a limited number of nodes (hubs). If damage is done to a hub, this can cause much more severe damage than a non-hub node.
As scientists learn more about the functional connections of the brain, the concept and role of diaschisis will continue to evolve and change.
Brain plasticity in infants
At birth, each infant neuron in the cerebral cortex has about 2,500 synapses. By two or three-years-old, the number of synapses per neuron increases to about 15,000 as the infant explores its world and learns new skills – a process called synaptogenesis. But by adulthood the number of synapses halves, so-called synaptic pruning.
In the perinatal and early-childhood “critical period,” plasticity-enabling conditions are always “on” 89. In the older child and adult brain, changes in the control of the release of “neuro-modulatory neurotransmitters” and in the properties of the receptors in the brain that govern their actions—enable the older brain’s moment-by-moment control of change; it is permitted only when the specific contextual conditions that enable or trigger plasticity are met, with changes arising under those special contextual-enabling conditions “saved” (driving enduring changes in connection strengths) as a function of behavioral outcome 3.
For example, under conditions of focused attention, any stimulus excites acetylcholine (ACh) releasing neurons in the basal nucleus of Meynert 90. In the cortex, acetylcholine (ACh) inputs positively enable plasticity by (a) selectively amplifying only anticipated (“selectively attended”) and (b) selectively weakening non-anticipated inputs—including those at any given cortical location that may have most effectively excited neurons before learning-induced changes were initiated 91. By this action, brain circuits enable plasticity by advantaging input strengths for those specific activities that the brain can gain in ability by changing to, and disadvantage behaviorally non-contributing inputs that they shall change from.
As a second example, noradrenaline (NA) releasing neurons in the locus coeruleus (LC) (and in nucleus accumbens and amygdala) broadly amplify neuronal activity, increasing the general level of excitability (arousal, or baseline level of attention) in sub-cortical and cortical structures in any closely-attended context (for example, in stimulus- or goal-seeking or other “motivated” states) 92. Noradrenaline (NA) is also released to selectively amplify the activities evoked by unexpected (novel) input 93, conferring special powers for the representation of “surprising” inputs or activities for driving enduring representational change.
Dopamine (DA) releasing neurons in the ventral tegmental area and substantia nigra are highly specific plasticity enablers 94. They are excited when the brain receives or first predicts the occurrence of a reward input (hedonic), or when the brain achieves or first predicts behavioral success (for which it “rewards itself”) in a learning cycle 95. With their release, inputs that “predict” that reward (i.e., are highly correlated with its occurrence) are selectively strengthened; competitive inputs uncorrelated with reward prediction arriving in a short post-reward epoch are selectively weakened 96.
Scientists now have a first-level understanding of the “rules” that control the release and the actions of these (and other) neuromodulators in learning, and of the modulator-specific ways that they nuance brain changes in experience and learning.
It should be noted that this crucial neuromodulatory machinery, controlling learning and memory abilities throughout life, is also plastic 97. The strengths, selectivity, and reliability of its actions can be significantly improved via intensive training in most individuals with neurological or psychiatric impairment or disability.
In the older brain, a context- and outcomes-dependent release of neuromodulators from subcortical limbic system nuclei enable and trigger brain change.
Why is brain plasticity important?
What makes the brain special is that, unlike a computer, it processes sensory and motor signals in parallel. It has many neural pathways that can replicate another’s function so that small errors in development or temporary loss of function through damage can be easily corrected by rerouting signals along a different pathway.
The problem becomes severe when errors in development are large, such as the effects of the Zika virus on brain development in the womb, or as a result of damage from a blow to the head or following a stroke. Yet, even in these examples, given the right conditions the brain can overcome adversity so that some function is recovered.
The brain’s anatomy ensures that certain areas of the brain have certain functions. This is something that is predetermined by your genes. For example, there is an area of the brain that is devoted to movement of the right arm. Damage to this part of the brain will impair movement of the right arm. But since a different part of the brain processes sensation from the arm, you can feel the arm but can’t move it. This “modular” arrangement means that a region of the brain unrelated to sensation or motor function is not able to take on a new role. In other words, neuroplasticity is not synonymous with the brain being infinitely malleable.
Part of the body’s ability to recover following damage to the brain can be explained by the damaged area of the brain getting better, but most is the result of neuroplasticity – forming new neural connections. In a study of Caenorhabditis elegans, a type of nematode used as a model organism in research, it was found that losing the sense of touch enhanced the sense of smell. This suggests that losing one sense rewires others. It is well known that, in humans, losing one’s sight early in life can heighten other senses, especially hearing.
As in the developing infant, the key to developing new connections is environmental enrichment that relies on sensory (visual, auditory, tactile, smell) and motor stimuli. The more sensory and motor stimulation a person receives, the more likely they will be to recover from brain trauma. For example, some of the types of sensory stimulation used to treat stroke patients includes training in virtual environments, music therapy and mentally practising physical movements.
The basic structure of the brain is established before birth by your genes. But its continued development relies heavily on a process called developmental plasticity, where developmental processes change neurons and synaptic connections. In the immature brain this includes making or losing synapses, the migration of neurons through the developing brain or by the rerouting and sprouting of neurons.
There are very few places in the mature brain where new neurons are formed. The exceptions are the dentate gyrus of the hippocampus (an area involved in memory and emotions) and the sub-ventricular zone of the lateral ventricle, where new neurons are generated and then migrate through to the olfactory bulb (an area involved in processing the sense of smell). Although the formation of new neurons in this way is not considered to be an example of neuroplasticity it might contribute to the way the brain recovers from damage.
Neuroplasticity treatment
Clinically, several treatment options can be used to help guide neuroplasticity in restoring function and treating unwanted symptoms. An example is mirror therapy, a technique used in phantom limb pain. In a basic premise, the patent uses a mirror to cover their amputation and focuses on watching their intact limb perform activities while imaging that both limbs are performing the same activity. This has been shown to have increased activation and functional connectivity in the frontoparietal network 78.
One of the most studied rehabilitation techniques is constraint-induced movement therapy (CIMT). Used in patients with a stroke, the premise is that by constraining the functional limb, the affected limb is engaged in repetitive task practice and behavioral shaping. Using functional magnetic resonance imaging (fMRI) technology, patients who engage in this therapy have been shown to have increased activity in their contralateral premotor and secondary somatosensory cortex in association with improved function 98.
While therapies can be used to help guide neuroplasticity, multiple medications can also be used to influence brain healing. These include selective serotonin reuptake inhibitors (SSRIs) like fluoxetine, serotonin and noradrenergic reuptake inhibitors (SNRIs) like duloxetine, cholinergic agonists such as donepezil, glutaminergic partial antagonists like amantadine, and several others 99. Amantadine has been shown to improve recovery in patients in a minimally conscious or vegetative state after a severe traumatic brain injury 100. Amantadine has also been shown to have an increase in left prefrontal cortex activation in association with improved cognitive functioning in patients with chronic traumatic brain injury 101. As research continues, scientists will be able to utilize pharmacological treatments further to help guide the brain back to health.
Lastly, a great deal of research has been focused on influencing neuroplasticity through the modification of environmental factors. Music therapy has been shown to influence neuroplasticity positively. It has been shown to improve cognition and other executive functions 102. Exercise has been shown to improve episodic memory and processing speed in addition to decrease age-related atrophy of the hippocampus 103. A healthy diet has also been shown to be helpful for this. Different dietary supplements are being studied to see if they could help trigger neuroplasticity. Lastly, reducing stress and avoiding sleep deprivation have been shown to be helpful to improve memory, attention span, and other domains of cognition 104.
How to increase brain plasticity
Brain plasticity exercises
Following injury to the brain or spinal cord that induces motor deficits, many physical rehabilitation interventions have been reported to induce functional improvements. Constraint-induced movement therapy for the arm and hand 105, body weight-supported treadmill training 106, robotic devices 107, behavioral shaping, bilateral arm training 108 and task-oriented physical therapy 109 are all examples of interventions that have led to improved recovery following stroke. In some cases, functional neuroimaging studies have provided insights into mechanisms of treatment effects; for example, constraint-induced movement therapy of the upper extremity has been associated with an enlarged motor cortex map for the hand 110 as well as with bilateral increases in sensorimotor grey matter 111. However, the more complex training interventions, such as the use or robotic devices and treadmill locomotor training, generally have not improved outcomes more than conventional task related and strengthening therapies that also aim to optimize activity dependent plasticity. Likewise, among patients with incomplete spinal cord injury, training through robotic-assisted and body weight-supported techniques have improved walking only to a similar degree as standard over-ground training 112. Further research is needed to better understand how these therapies can be coordinated and optimized, especially across diverse patient groups with varied functional limitations. Practice strategies include increased repetition, sensory priming, visualization, modulation of attentional valence and reward, contextual interference, variable demand and intensity levels, blocked versus intermittent practice and various forms of feedback 113. Although these practice paradigms may enhance both skills and declarative learning in healthy subjects, their additive benefits for patients with impaired movement or cognition has been difficult to demonstrate. Note that physical rehabilitation training is not only a stand-alone therapy, but serves as an adjunct to other forms of therapy such as pharmacological and behavioral.
Aerobic exercise is a specific extension of activity-based therapies for promoting plasticity. Although the benefits of aerobic exercise in preventing or reversing cognitive and neural deterioration have yet to be fully investigated, substantial human and preclinical data support the utility of such exercise for promoting brain plasticity and improving CNS function in many conditions 114, including normal ageing and early dementia. Aerobic exercise is associated with increased neurogenesis and angiogenesis, as well as the production of neurotrophic molecules such as brain-derived neurotrophic factor and other growth factors involved in neuroprotection and the promotion of cell survival, neurite outgrowth and synaptic plasticity 115. In humans, neuroimaging studies have described a range of anatomic and functional correlates of such effects 116. Furthermore, these plasticity-promoting strategies are able to produce clinically significant changes. For example, aerobic exercise programmes lasting even just a few months significantly benefit cognitive functioning in both healthy ageing and early dementia, may be of benefit in schizophrenia 117, and have been shown to increase brain volume in a variety of regions 116 and to enhance brain network functioning 118.
Combining diet and exercise
Feeding and exercising are complementary aspects of regulating energy balance that have influenced the evolution of the modern brain over thousands of years 119. Given that the brain possess the largest demand on oxygen consumption, it is not surprising that energy metabolism has a profound influence on brain function 120. In particular, both food consumption and physical activity stimulate mitochondrial activity and thus energy provision to the brain which in turn modulates the signaling pathways linked to neuronal function and brain plasticity 120.
In addition to its role in enhancing brain plasticity, brain-derived neurotrophic factor (BDNF) has also been implicated in modulating brain energy metabolism, as evidenced by studies that demonstrate that perturbed BDNF signaling can manifest in metabolic disorders such as obesity 121. Together, these studies reveal that BDNF plays a key role in both brain energy metabolism and plasticity, demonstrating a strong and influential relationship between diet, exercise, and brain function 121.
Similar to diet, exercise entails a “broad spectrum of action” and also effectively promotes brain plasticity through the increases of neurogenesis, neurotrophins levels, and synaptic plasticity 122. It is possible that exercise potentiates the health-promoting effects of diet components and vice versa at the cellular and molecular levels. For example, it has recently been demonstrated that exercise works in tandem with a docosahexaenoic acid (an omega-3 fatty acid) enriched diet to enhance cognitive function 123. In particular, exercise appears to act on mechanisms that preserve docosahexaenoic acid (an omega-3 fatty acid) on the plasma membrane and in turn enhance neurotransmission 124. In addition, the concurrent effects of docosahexaenoic acid (an omega-3 fatty acid) diet and exercise engage BDNF-mediated synaptic plasticity 125.
Further to this, in a rodent model of traumatic brain injury (TBI), the combination of docosahexaenoic acid (an omega-3 fatty acid) supplementation and voluntary exercise restored membrane homeostasis to counteract the detrimental effects of traumatic brain injury on many parameters of synaptic plasticity and cognition 126. With regard to synaptic plasticity, exercise greatly enhanced the action of docosahexaenoic acid (an omega-3 fatty acid) supplementation on levels of BDNF and TrkB activation following traumatic brain injury 126. Together, these data reveal a strong and novel interaction between diet and exercise, whereby aspects of these lifestyles intersect at the molecular level under pathological conditions to promote brain plasticity 126.
Similarly, exercise and flavonoid-enriched diets together promote the elevation of genes that promote brain plasticity whilst decreasing expression of markers known to compromise this plasticity, including those related to inflammation and cell death 127. Moreover, exercise has also been shown to markedly reduce the effects of a diet rich in saturated fats through the counteracting of declines in BDNF-mediated synaptic plasticity in the hippocampus 128.
The combination of exercise and caloric restriction (a consistent reduction of total daily food intake) is particularly noteworthy. In this paradigm, typically, 12.5% of the energy restriction comes from adherence to a restricted diet and the other 12.5% comes from increased energy expenditure via exercise 129. The principal advantage of combining caloric restriction with exercise is that individuals may find it easier to comply with energy restriction if this split between caloric restriction and exercise-induced expenditure 130. Therefore caloric restriction plus exercise may well prove viable and effective means of promoting brain plasticity. Some studies, however, have reported that caloric restriction plus exercise does not elicit positive changes to health other than those elicited by caloric restriction. For example, animals maintained on an 80% caloric restriction plus exercise regimen demonstrated no significant changes in oxidative stress, in proinflammatory markers 131 or upon extension of life span 132.
Neuroplasticity training
Cognitive training can be thought of as a direct extension of physical therapy to the non-motor aspects of the human brain and so has been examined across a number of disease conditions. However, the complexities of the distributed neural systems that underlie human behavioural syndromes introduce unique challenges for the design of effective interventions. In depression and anxiety disorders, a long tradition of evidence-based cognitive-behaviour therapy is based on the principle of identifying and modifying top-down responses to maladaptive cognition, affect and behaviour 133. Evidence suggests that, as individuals learn to modify their cognitive representations and behavioural responses to distressing stimuli, widespread changes occur in frontal cognitive control systems and in limbic system activation 134.
New neuroscience-driven approaches to cognitive training are emerging and directly build on decades of animal research that have identified principles of harnessing plasticity mechanisms in the adult brain 135. For example, the cognitive deficits of schizophrenia—particularly in verbal learning and memory—are associated with illness severity and predict long-term functioning, but do not respond to currently available medications. Vinogradov et al. 136, guided by an understanding of the neurobiology of schizophrenia, performed a double-blind randomized controlled trial of intensive computerized cognitive training exercises that focus on the components of auditory and verbal processing that underlie verbal encoding. This intervention was associated with improved verbal memory in patients, as well as magnetoencephalographic evidence of increased amplitude of the M100 response to auditory stimuli, indicating plasticity in auditory cortex 137.
These findings require replication with a larger, preferably multi-site, study. Another cognitive-based, neuroscience-driven training approach aims to stimulate specific dysfunctional circuits, possibly in association with pharmacological intervention, in order to restore the integrity of frontostriatal circuitry in addiction 138.
Neuroimaging can contribute to cognitive training in a number of ways. Functional brain imaging can help to identify the neural correlates of various core mental processes such as interference control that can be targeted by cognitive training and that are relevant for a number of psychiatric disorders 139. Neuroimaging data can also be used as biomarkers, i.e. surrogate markers. A surrogate marker has been defined as ‘a laboratory measurement. . . used as a substitute for a clinically meaningful endpoint’ 140. For example, changes in functional MRI brain activation have been shown to correlate with functional gains in studies that employed cognitive training in the setting of schizophrenia 134, dyslexia 141 and depression 134, consistent with observations in studies that employed direct instructional approaches 142. Such imaging biomarkers might prove useful as predictors of clinical outcome, and a number of MRI and PET measures are under study in this regard 143. Neuroimaging can also provide molecular insights into treatment mechanism. For example, McNab et al. 144 found that cognitive training in healthy subjects was associated with changes in the density of cortical dopamine D1 receptors on PET scanning, a finding relevant to the treatment of children with attention deficit disorder 145. PET has also been used to describe changes in glucose metabolism associated with cognitive-behavioural or pharmacological treatment of depression 146. Though promising on a number of fronts, a number of issues remain to be addressed to maximize the impact of neuroimaging on cognitive training. Most imaging work has been performed on small samples, with differing approaches across labs, such as in relation to the underlying hypotheses of mechanisms of training-induced change, and further study is needed regarding the validity and reliability of neuroimaging data. Critically important is the question of whether changes in circuit strength demonstrated using neuroimaging are paralleled by meaningful behavioral changes.
The ultimate goal of cognitive training is to improve behavior by systematically harnessing neuroplasticity and driving adaptive changes in dysfunctional neural systems through carefully designed exercises. Note that cognitive training approaches have particularly broad potential, for example, as part of rehabilitation therapy of patients with focal brain injury such as stroke, where myriad cognitive syndromes exist with few treatment options, as well as in the treatment of numerous neuropsychiatric disorders such as depression and schizophrenia. Systems neuroscienceinformed cognitive training appears to be a promising treatment approach for a number of brain disorders. A key future direction for this field will be to maximize the extent to which cognitive training in one domain generalizes to others, and the extent to which such training has a meaningful impact on real-world functioning as well as the subjective experience of the individual 147.
Feedback using real-time functional magnetic resonance imaging
A central challenge to creating neuroplastic change is determining how to target plasticity to a particular brain system. Such targeting might be enabled by the ability to monitor changes in brain activation within localized brain regions in real time. Recent advances in neuroimaging and computing have enabled the development of such methods based on functional MRI-based measures of regional brain activation 148. These methods now offer the possibility of allowing individuals to view real-time measures of their own regional brain activation 149. Rapid feedback of regional activation level or of distributed patterns of brain activation might provide a novel means of instructing subjects on how to modulate their own brain function. Goals of such feedback include modulation of activity in specific brain regions in response to intrinsic or extrinsic cues, as well evaluation of the effects of various interventions.
Data suggest that subjects can indeed learn volitional control over a specific brain region. For example, healthy subjects can be taught to control brain activity within the anterior insula (Caria et al., 2007). In another study, both healthy subjects and patients with chronic pain were able to use real time functional MRI training to learn to control activation of brain regions involved in modulation of pain, which was associated with a concomitant decrease in pain perception 150. The long-term goal is to improve patient outcomes by modulating brain activity in those selected neural circuits that are most related to the target symptoms.
- Voss, P., Thomas, M. E., Cisneros-Franco, J. M., & de Villers-Sidani, É. (2017). Dynamic Brains and the Changing Rules of Neuroplasticity: Implications for Learning and Recovery. Frontiers in psychology, 8, 1657. https://doi.org/10.3389/fpsyg.2017.01657[↩]
- Mateos-Aparicio, P., & Rodríguez-Moreno, A. (2019). The Impact of Studying Brain Plasticity. Frontiers in cellular neuroscience, 13, 66. https://doi.org/10.3389/fncel.2019.00066[↩][↩][↩][↩]
- Merzenich M. M. (2013). Soft-Wired: How the New Science of Brain Plasticity Can Change Your Life. San Francisco: Parnassus Publishing[↩][↩][↩][↩]
- Markram, H., Gerstner, W., & Sjöström, P. J. (2011). A history of spike-timing-dependent plasticity. Frontiers in synaptic neuroscience, 3, 4. https://doi.org/10.3389/fnsyn.2011.00004[↩][↩]
- Berlucchi G, Buchtel HA. Neuronal plasticity: historical roots and evolution of meaning. Exp Brain Res. 2009 Jan;192(3):307-19. doi: 10.1007/s00221-008-1611-6[↩]
- Hebb D. (1949). The Organization of Behavior: A Neuropsychological Theory. New York, NY: Wiley.[↩]
- Konorski J. (1948). Conditioned Reflexes and Neuron Organization. Cambridge: Cambridge University Press.[↩]
- Puderbaugh M, Emmady PD. Neuroplasticity. [Updated 2021 Jul 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2022 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557811[↩][↩]
- Sophie Su YR, Veeravagu A, Grant G. Neuroplasticity after Traumatic Brain Injury. In: Laskowitz D, Grant G, editors. Translational Research in Traumatic Brain Injury. Boca Raton (FL): CRC Press/Taylor and Francis Group; 2016. Chapter 8. Available from: https://www.ncbi.nlm.nih.gov/books/NBK326735[↩]
- Merzenich M. M., de Charms C. (1996). Neural representations, experience and change, in The Mind-Brain Continuum, eds Llinas R., Churchland P. (Boston, MA: MIT Press; ), 61–81[↩]
- Cooper L. N., Bear M. F. (2012). The BCM theory of synapse modification at 30: interaction of theory with experiment. Nat. Rev. Neurosci. 13, 798–810 10.1038/nrn3353[↩]
- Hebb D. O. (1949). The Organization of Behavior. New York, NY: J Wiley[↩]
- Kleim J. A., Barbay S., Cooper N. R., Hogg T. M., Reidel C. N., Remple M. S., et al. (2002). Motor learning-dependent synaptogenesis is localized to functionally reorganized motor cortex. Neurobiol. Learn. Mem. 77, 63–77 10.1006/nlme.2000.4004[↩]
- Compte A. (2006). Computational and in vitro studies of persistent activity: edging towards cellular and synaptic mechanisms of working memory. Neuroscience 139, 135–151 10.1016/j.neuroscience.2005.06.011[↩]
- Feldman D. E. (2012). The spike-timing dependence of plasticity. Neuron 75, 55–71 10.1016/neuron.2012.08.001[↩]
- de Villers-Sidani E, Merzenich MM. Lifelong plasticity in the rat auditory cortex: basic mechanisms and role of sensory experience. Prog Brain Res. 2011;191:119-31. doi: 10.1016/B978-0-444-53752-2.00009-6[↩][↩]
- Kamal, B., Holman, C., & de Villers-Sidani, E. (2013). Shaping the aging brain: role of auditory input patterns in the emergence of auditory cortical impairments. Frontiers in systems neuroscience, 7, 52. https://doi.org/10.3389/fnsys.2013.00052[↩]
- Zhou, J., Gennatas, E. D., Kramer, J. H., Miller, B. L., & Seeley, W. W. (2012). Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron, 73(6), 1216–1227. https://doi.org/10.1016/j.neuron.2012.03.004[↩][↩]
- Kolb, B., & Gibb, R. (2011). Brain plasticity and behaviour in the developing brain. Journal of the Canadian Academy of Child and Adolescent Psychiatry = Journal de l’Academie canadienne de psychiatrie de l’enfant et de l’adolescent, 20(4), 265–276. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3222570/[↩]
- Chen H, Epstein J, Stern E. Neural plasticity after acquired brain injury: evidence from functional neuroimaging. PM R. 2010 Dec;2(12 Suppl 2):S306-12. doi: 10.1016/j.pmrj.2010.10.006[↩]
- Campbell JN, Register D, Churn SB. Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J Neurotrauma. 2012 Jan 20;29(2):201-17. doi: 10.1089/neu.2011.1761[↩][↩]
- Hall, K. D., & Lifshitz, J. (2010). Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain research, 1323, 161–173. https://doi.org/10.1016/j.brainres.2010.01.067[↩]
- Morris GP, Clark IA, Zinn R, Vissel B. Microglia: a new frontier for synaptic plasticity, learning and memory, and neurodegenerative disease research. Neurobiol Learn Mem. 2013 Oct;105:40-53. doi: 10.1016/j.nlm.2013.07.002[↩]
- Yu, X., Chung, S., Chen, D. Y., Wang, S., Dodd, S. J., Walters, J. R., Isaac, J. T., & Koretsky, A. P. (2012). Thalamocortical inputs show post-critical-period plasticity. Neuron, 74(4), 731–742. https://doi.org/10.1016/j.neuron.2012.04.024[↩]
- Calabresi, P., Gubellini, P., Centonze, D., Picconi, B., Bernardi, G., Chergui, K., Svenningsson, P., Fienberg, A. A., & Greengard, P. (2000). Dopamine and cAMP-regulated phosphoprotein 32 kDa controls both striatal long-term depression and long-term potentiation, opposing forms of synaptic plasticity. The Journal of neuroscience : the official journal of the Society for Neuroscience, 20(22), 8443–8451. https://doi.org/10.1523/JNEUROSCI.20-22-08443.2000[↩]
- van Bregt, D. R., Thomas, T. C., Hinzman, J. M., Cao, T., Liu, M., Bing, G., Gerhardt, G. A., Pauly, J. R., & Lifshitz, J. (2012). Substantia nigra vulnerability after a single moderate diffuse brain injury in the rat. Experimental neurology, 234(1), 8–19. https://doi.org/10.1016/j.expneurol.2011.12.003[↩][↩][↩]
- Yan, H. Q., Ma, X., Chen, X., Li, Y., Shao, L., & Dixon, C. E. (2007). Delayed increase of tyrosine hydroxylase expression in rat nigrostriatal system after traumatic brain injury. Brain research, 1134(1), 171–179. https://doi.org/10.1016/j.brainres.2006.11.087[↩]
- Hunt, R. F., Scheff, S. W., & Smith, B. N. (2009). Posttraumatic epilepsy after controlled cortical impact injury in mice. Experimental neurology, 215(2), 243–252. https://doi.org/10.1016/j.expneurol.2008.10.005[↩][↩]
- Niogi SN, Mukherjee P. Diffusion tensor imaging of mild traumatic brain injury. J Head Trauma Rehabil. 2010 Jul-Aug;25(4):241-55. doi: 10.1097/HTR.0b013e3181e52c2a[↩]
- Niogi, S. N., Mukherjee, P., Ghajar, J., Johnson, C., Kolster, R. A., Sarkar, R., Lee, H., Meeker, M., Zimmerman, R. D., Manley, G. T., & McCandliss, B. D. (2008). Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. AJNR. American journal of neuroradiology, 29(5), 967–973. https://doi.org/10.3174/ajnr.A0970[↩]
- Staudt M. Brain plasticity following early life brain injury: insights from neuroimaging. Semin Perinatol. 2010 Feb;34(1):87-92. doi: 10.1053/j.semperi.2009.10.009[↩]
- Yeatman, J. D., & Feldman, H. M. (2013). Neural plasticity after pre-linguistic injury to the arcuate and superior longitudinal fasciculi. Cortex; a journal devoted to the study of the nervous system and behavior, 49(1), 301–311. https://doi.org/10.1016/j.cortex.2011.08.006[↩][↩]
- Yogarajah, M., Focke, N. K., Bonelli, S. B., Thompson, P., Vollmar, C., McEvoy, A. W., Alexander, D. C., Symms, M. R., Koepp, M. J., & Duncan, J. S. (2010). The structural plasticity of white matter networks following anterior temporal lobe resection. Brain : a journal of neurology, 133(Pt 8), 2348–2364. https://doi.org/10.1093/brain/awq175[↩][↩]
- Ndode-Ekane XE, Hayward N, Gröhn O, Pitkänen A. Vascular changes in epilepsy: functional consequences and association with network plasticity in pilocarpine-induced experimental epilepsy. Neuroscience. 2010 Mar 10;166(1):312-32. doi: 10.1016/j.neuroscience.2009.12.002. Epub 2009 Dec 23. Erratum in: Neuroscience. 2010 Sep 1;169(3):1486.[↩]
- Laitinen T, Sierra A, Pitkänen A, Gröhn O. Diffusion tensor MRI of axonal plasticity in the rat hippocampus. Neuroimage. 2010 Jun;51(2):521-30. doi: 10.1016/j.neuroimage.2010.02.077[↩]
- Zhou IY, Liang YX, Chan RW, Gao PP, Cheng JS, Hu Y, So KF, Wu EX. Brain resting-state functional MRI connectivity: morphological foundation and plasticity. Neuroimage. 2014 Jan 1;84:1-10. doi: 10.1016/j.neuroimage.2013.08.037[↩]
- Shohayeb B, Diab M, Ahmed M, Ng DCH. Factors that influence adult neurogenesis as potential therapy. Transl Neurodegener. 2018;7:4. Published 2018 Feb 21. doi:10.1186/s40035-018-0109-9 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5822640/[↩]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70(4):687-702. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3106107/[↩]
- Kolb B, Teskey GC, Gibb R. Factors influencing cerebral plasticity in the normal and injured brain. Front Hum Neurosci. 2010 doi:10.3389/fnhum.2010.00204[↩][↩][↩]
- Johnston MV. Plasticity in the developing brain: implications for rehabilitation. Dev Disabil Res Rev. 2009;15(2):94-101. doi: 10.1002/ddrr.64[↩]
- Kou Z, Iraji A. Imaging brain plasticity after trauma. Neural Regen Res. 2014;9(7):693-700. doi:10.4103/1673-5374.131568 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4146265[↩]
- Baranauskas G. Pain-induced plasticity in the spinal cord. In: Shaw CA, McEachern J, editors. Toward a Theory of Neuroplasticity. Philadelphia: PA: Psychology Press; 2001.[↩]
- Raison C, Capuron L, Miller AH. Cytokines sing the blues: inflammation and thepathogenesis of depression. Trends Immunol. 2010;27:24–31.[↩]
- Teskey GC. Using kindling to model the neuroplastic changes associated with learning and memory, neuropsychiatric disorders, and epilepsy. In: Shaw CA, McEachern J, editors. Toward a Theory of Neuroplasticity. Philadelphia, PA: Psychology Press; 2001.[↩]
- Black JE, Kodish IM, Grossman AW, Klintsova AY, Orlovskaya D, Vostrikov V, Uranova N, Greenough WT. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744.[↩]
- Mattson MP, Duan W, Chan SL, Guo Z. Modification of brain aging and neurodegenerative disorders by genes, diet, and behavior. In: Shaw CA, McEachern J, editors. Toward a Theory of Neuroplasticity. Philadelphia, PA: Psychology Press; 2001.[↩]
- Hunt RF, Scheff SW, Smith BN. Post traumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol. 2009;215:243–252.[↩]
- Chen H, Epstein J, Stern E. Neural plasticity after acquired brain injury: evidence from functional neuroimaging. PM R. 2010;2:S306–312.[↩][↩]
- Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861–872.[↩]
- Environment and Brain Plasticity: Towards an Endogenous Pharmacotherapy. Alessandro Sale, Nicoletta Berardi, and Lamberto Maffei. Physiological Reviews 2014 94:1, 189-234. https://doi.org/10.1152/physrev.00036.2012[↩]
- Stent G. S. (1973). A physiological mechanism for Hebb’s postulate of learning. Proceedings of the National Academy of Sciences of the United States of America, 70(4), 997–1001. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC433410/pdf/pnas00067-0035.pdf[↩]
- Graupner, M., & Brunel, N. (2012). Calcium-based plasticity model explains sensitivity of synaptic changes to spike pattern, rate, and dendritic location. Proceedings of the National Academy of Sciences of the United States of America, 109(10), 3991–3996. https://doi.org/10.1073/pnas.1109359109[↩]
- Deisseroth K, Bito H, Schulman H, Tsien RW. Synaptic plasticity: A molecular mechanism for metaplasticity. Curr Biol. 1995 Dec 1;5(12):1334-8. doi: 10.1016/s0960-9822(95)00262-4[↩]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002 Aug;5(8):783-9. doi: 10.1038/nn878[↩]
- Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hübener M. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 2007 Jun 21;54(6):961-72. doi: 10.1016/j.neuron.2007.05.028[↩]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004 Sep 30;44(1):5-21. doi: 10.1016/j.neuron.2004.09.012[↩]
- Hunt, D. L., & Castillo, P. E. (2012). Synaptic plasticity of NMDA receptors: mechanisms and functional implications. Current opinion in neurobiology, 22(3), 496–508. https://doi.org/10.1016/j.conb.2012.01.007[↩]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005 Nov;6(11):877-88. doi: 10.1038/nrn1787[↩][↩]
- Lisman, J., Yasuda, R., & Raghavachari, S. (2012). Mechanisms of CaMKII action in long-term potentiation. Nature reviews. Neuroscience, 13(3), 169–182. https://doi.org/10.1038/nrn3192[↩]
- Bliss, T. V., & Cooke, S. F. (2011). Long-term potentiation and long-term depression: a clinical perspective. Clinics (Sao Paulo, Brazil), 66 Suppl 1(Suppl 1), 3–17. https://doi.org/10.1590/s1807-59322011001300002[↩]
- Liu-Yesucevitz, L., Bassell, G. J., Gitler, A. D., Hart, A. C., Klann, E., Richter, J. D., Warren, S. T., & Wolozin, B. (2011). Local RNA translation at the synapse and in disease. The Journal of neuroscience : the official journal of the Society for Neuroscience, 31(45), 16086–16093. https://doi.org/10.1523/JNEUROSCI.4105-11.2011[↩]
- Lyons, M. R., & West, A. E. (2011). Mechanisms of specificity in neuronal activity-regulated gene transcription. Progress in neurobiology, 94(3), 259–295. https://doi.org/10.1016/j.pneurobio.2011.05.003[↩]
- Leslie, J. H., & Nedivi, E. (2011). Activity-regulated genes as mediators of neural circuit plasticity. Progress in neurobiology, 94(3), 223–237. https://doi.org/10.1016/j.pneurobio.2011.05.002[↩]
- Yoshii, A., & Constantine-Paton, M. (2010). Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Developmental neurobiology, 70(5), 304–322. https://doi.org/10.1002/dneu.20765[↩]
- Pizzorusso, T., Medini, P., Landi, S., Baldini, S., Berardi, N., & Maffei, L. (2006). Structural and functional recovery from early monocular deprivation in adult rats. Proceedings of the National Academy of Sciences of the United States of America, 103(22), 8517–8522. https://doi.org/10.1073/pnas.0602657103[↩]
- Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998 Oct;21(4):813-25. doi: 10.1016/s0896-6273(00)80597-8[↩]
- Gu Q. Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience. 2002;111(4):815-35. doi: 10.1016/s0306-4522(02)00026-x[↩]
- Bear MF, Singer W. Modulation of visual cortical plasticity by acetylcholine and noradrenaline. Nature. 1986 Mar 13-19;320(6058):172-6. doi: 10.1038/320172a0[↩]
- Goard, M., & Dan, Y. (2009). Basal forebrain activation enhances cortical coding of natural scenes. Nature neuroscience, 12(11), 1444–1449. https://doi.org/10.1038/nn.2402[↩]
- Kilgard MP, Merzenich MM. Cortical map reorganization enabled by nucleus basalis activity. Science. 1998 Mar 13;279(5357):1714-8. doi: 10.1126/science.279.5357.1714[↩]
- Sultan, F. A., & Day, J. J. (2011). Epigenetic mechanisms in memory and synaptic function. Epigenomics, 3(2), 157–181. https://doi.org/10.2217/epi.11.6[↩]
- Alarcón JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/- mice: a model for the cognitive deficit in Rubinstein-Taybi syndrome and its amelioration. Neuron. 2004 Jun 24;42(6):947-59. doi: 10.1016/j.neuron.2004.05.021[↩]
- Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005 Sep;28(9):456-63. doi: 10.1016/j.tins.2005.07.006[↩]
- Weaver IC, Cervoni N, Champagne FA, D’Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci. 2004 Aug;7(8):847-54. doi: 10.1038/nn1276[↩]
- Bliss, T. V., & Lomo, T. (1973). Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. The Journal of physiology, 232(2), 331–356. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1350458/pdf/jphysiol00958-0128.pdf[↩]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100(5):433-42. doi: 10.1254/jphs.cpj06007x[↩]
- Brzosko Z, Mierau SB, Paulsen O. Neuromodulation of Spike-Timing-Dependent Plasticity: Past, Present, and Future. Neuron. 2019 Aug 21;103(4):563-581. doi: 10.1016/j.neuron.2019.05.041[↩]
- Maier, M., Ballester, B. R., & Verschure, P. (2019). Principles of Neurorehabilitation After Stroke Based on Motor Learning and Brain Plasticity Mechanisms. Frontiers in systems neuroscience, 13, 74. https://doi.org/10.3389/fnsys.2019.00074[↩][↩]
- Mora F. (2013). Successful brain aging: plasticity, environmental enrichment, and lifestyle. Dialogues in clinical neuroscience, 15(1), 45–52. https://doi.org/10.31887/DCNS.2013.15.1/fmora[↩]
- Owji S, Shoja MM. The History of Discovery of Adult Neurogenesis. Clin Anat. 2020 Jan;33(1):41-55. doi: 10.1002/ca.23447[↩]
- La Rosa, C., Parolisi, R., & Bonfanti, L. (2020). Brain Structural Plasticity: From Adult Neurogenesis to Immature Neurons. Frontiers in neuroscience, 14, 75. https://doi.org/10.3389/fnins.2020.00075[↩][↩][↩]
- Finger S. Chapter 51: recovery of function: redundancy and vicariation theories. Handb Clin Neurol. 2010;95:833-41. doi: 10.1016/S0072-9752(08)02151-9[↩][↩]
- Graveline CJ, Mikulis DJ, Crawley AP, Hwang PA. Regionalized sensorimotor plasticity after hemispherectomy fMRI evaluation. Pediatr Neurol. 1998 Nov;19(5):337-42. doi: 10.1016/s0887-8994(98)00082-4[↩][↩]
- Jaillard A, Martin CD, Garambois K, Lebas JF, Hommel M. Vicarious function within the human primary motor cortex? A longitudinal fMRI stroke study. Brain. 2005 May;128(Pt 5):1122-38. doi: 10.1093/brain/awh456[↩][↩]
- Carrera E, Tononi G. Diaschisis: past, present, future. Brain. 2014 Sep;137(Pt 9):2408-22. doi: 10.1093/brain/awu101[↩][↩]
- Wiesendanger M. Constantin von Monakow (1853-1930): a pioneer in interdisciplinary brain research and a humanist. C R Biol. 2006 May-Jun;329(5-6):406-18. doi: 10.1016/j.crvi.2006.03.011[↩]
- Reidler P, Thierfelder KM, Fabritius MP, Sommer WH, Meinel FG, Dorn F, Wollenweber FA, Duering M, Kunz WG. Thalamic Diaschisis in Acute Ischemic Stroke: Occurrence, Perfusion Characteristics, and Impact on Outcome. Stroke. 2018 Apr;49(4):931-937. doi: 10.1161/STROKEAHA.118.020698[↩][↩]
- van Meer, M. P., van der Marel, K., van der Sprenkel, J. W., & Dijkhuizen, R. M. (2011). MRI of bilateral sensorimotor network activation in response to direct intracortical stimulation in rats after unilateral stroke. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 31(7), 1583–1587. https://doi.org/10.1038/jcbfm.2011.61[↩]
- Merzenich MM, Van Vleet TM, Nahum M. Brain plasticity-based therapeutics. Front Hum Neurosci. 2014;8:385. Published 2014 Jun 27. doi:10.3389/fnhum.2014.00385 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4072971[↩]
- Sarter M., Gehring W. J., Kozak R. (2006). More attention must be paid: the neurobiology of attentional effort. Brain Res. Rev. 51, 145–160 10.1016/j.brainresrev.2005.11.002[↩]
- Froemke R. C., Merzenich M. M., Schreiner C. E. (2007). A synaptic memory trace for cortical receptive field plasticity. Nature 450, 425–429 10.1038/nature06289[↩]
- Sara S. J., Bouret S. (2012). Orienting and reorienting: the locus coeruleus mediates cognition through arousal. Neuron 76, 130–141 10.1016/j.neuron.2012.09.011[↩]
- Aston-Jones J., Rajkowski J., Cohen J. (1999). Role of locus coeruleus in attention and behavioral flexibility. Biol. Psychiatry 46, 1309–1320 10.1016/S0006-3223(99)00140-7[↩]
- Lisman J., Grace A. A., Duzel E. (2011). A neoHebbian framework for episodic memory; role of dopamine-dependent late LTP. Trends Neursci. 34, 536–547 10.1016/j.tins.2011.07.006[↩]
- Schultz W. (2007). Multiple dopamine functions at different time courses. Annu. Rev. Neurosci. 30, 259–288 10.1146/annurev.neuro.28.061604.135722[↩]
- Bao S., Chan V. T., Zhang L. I., Merzenich M. M. (2003). Suppression of cortical representation through backward conditioning. Proc. Natl. Acad. Sci. U.S.A. 100, 1405–1408 10.1073/pnas.0337527100[↩]
- Smith B. A., Goldberg N. R., Meshul C. K. (2011). Effects of treatmill exercise on behavioral recovery and changes in the substantia nigra and striatum of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse. Brain Res. 1386, 60–80 10.1016/j.brainres.2011.02.003[↩]
- Johansen-Berg H, Dawes H, Guy C, Smith SM, Wade DT, Matthews PM. Correlation between motor improvements and altered fMRI activity after rehabilitative therapy. Brain. 2002 Dec;125(Pt 12):2731-42. doi: 10.1093/brain/awf282. Erratum in: Brain. 2003 Nov;126(Pt 11):2569.[↩]
- Carrillo-Mora P, Alcantar-Shramm JM, Almaguer-Benavides KM, Macías-Gallardo JJ, Fuentes-Bello A, Rodríguez-Barragán MA. Pharmacological Stimulation of Neuronal Plasticity in Acquired Brain Injury. Clin Neuropharmacol. 2017 May/Jun;40(3):131-139. doi: 10.1097/WNF.0000000000000217[↩]
- Giacino JT, Whyte J, Bagiella E, Kalmar K, Childs N, Khademi A, Eifert B, Long D, Katz DI, Cho S, Yablon SA, Luther M, Hammond FM, Nordenbo A, Novak P, Mercer W, Maurer-Karattup P, Sherer M. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med. 2012 Mar 1;366(9):819-26. doi: 10.1056/NEJMoa1102609[↩]
- Kraus MF, Smith GS, Butters M, Donnell AJ, Dixon E, Yilong C, Marion D. Effects of the dopaminergic agent and NMDA receptor antagonist amantadine on cognitive function, cerebral glucose metabolism and D2 receptor availability in chronic traumatic brain injury: a study using positron emission tomography (PET). Brain Inj. 2005 Jul;19(7):471-9. doi: 10.1080/02699050400025059[↩]
- Benz, S., Sellaro, R., Hommel, B., & Colzato, L. S. (2016). Music Makes the World Go Round: The Impact of Musical Training on Non-musical Cognitive Functions-A Review. Frontiers in psychology, 6, 2023. https://doi.org/10.3389/fpsyg.2015.02023[↩]
- Niemann, C., Godde, B., & Voelcker-Rehage, C. (2014). Not only cardiovascular, but also coordinative exercise increases hippocampal volume in older adults. Frontiers in aging neuroscience, 6, 170. https://doi.org/10.3389/fnagi.2014.00170[↩]
- Mueller AD, Meerlo P, McGinty D, Mistlberger RE. Sleep and adult neurogenesis: implications for cognition and mood. Curr Top Behav Neurosci. 2015;25:151-81. doi: 10.1007/7854_2013_251[↩]
- Wolf SL, Newton H, Maddy D, Blanton S, Zhang Q, Winstein CJ, et al.The Excite Trial: relationship of intensity of constraint induced move-ment therapy to improvement in the wolf motor function test. RestorNeurol Neurosci 2007; 25: 549–62[↩]
- Duncan PW, Sullivan KJ, Behrman AL, Azen SP, Wu SS, Nadeau SE,et al. Protocol for the Locomotor Experience Applied Post-stroke (LEAPS) trial: a randomized controlled trial. BMC Neurol2007; 7: 39[↩]
- Lo AC, Guarino PD, Richards LG, Haselkorn JK, Wittenberg GF,Federman DG, et al. Robot-assisted therapy for long-term upper-limbimpairment after stroke. N Engl J Med 2010; 362: 1772–83[↩]
- Lin KC, Chen YA, Chen CL, Wu CY, Chang YF. The effects of bilateralarm training on motor control and functional performance in chronicstroke: a randomized controlled study. Neurorehabil Neural Repair2010; 24: 42–51[↩]
- Jonsdottir J, Cattaneo D, Recalcati M, Regola A, Rabuffetti M,Ferrarin M, et al. Task-oriented biofeedback to improve gait in indi-viduals with chronic stroke: motor learning approach. NeurorehabilNeural Repair 2010; 24: 478–85[↩]
- Sawaki L, Butler AJ, Xiaoyan L, Wassenaar PA, Mohammad YM,Blanton S, et al. Constraint-induced movement therapy results inincreased motor map area in subjects 3 to 9 months after stroke.Neurorehabil Neural Repair 2008; 22: 505–13[↩]
- Gauthier LV, Taub E, Perkins C, Ortmann M, Mark VW, Uswatte G.Remodeling the brain: plastic structural brain changes produced bydifferent motor therapies after stroke. Stroke 2008; 39: 1520–5[↩]
- Dobkin BH. Confounders in rehabilitation trials of task-oriented training:lessons from the designs of the EXCITE and SCILT multicenter trials.Neurorehabil Neural Repair 2007; 21: 3–13[↩]
- Subramanian SK, Massie CL, Malcolm MP, Levin MF. Does provision ofextrinsic feedback result in improved motor learning in the upper limbpoststroke? A systematic review of the evidence. Neurorehabilitationand Neural Repair 2010; 24: 113–24[↩]
- Hillman CH, Erickson KI, Kramer AF. Be smart, exercise your heart: ex-ercise effects on brain and cognition. Nature Reviews Neuroscience2008; 9: 58–65.[↩]
- Rhyu IJ, Bytheway JA, Kohler SJ, Lange H, Lee KJ, Boklewski J, et al.Effects of aerobic exercise training on cognitive function and corticalvascularity in monkeys. Neuroscience 2010; 167: 1239–48[↩]
- Pajonk FG, Wobrock T, Gruber O, Scherk H, Berner D, Kaizl I, et al.Hippocampal plasticity in response to exercise in schizophrenia. ArchGen Psychiatry 2010; 67: 133–43[↩][↩]
- Kramer AF, Erickson KI. Capitalizing on cortical plasticity: influence ofphysical activity on cognition and brain function. Trends Cogn Sci2007; 11: 342–8.[↩]
- Colcombe SJ, Kramer AF, Erickson KI, Scalf P, McAuley E, Cohen NJ,et al. Cardiovascular fitness, cortical plasticity, and aging. Proc NatlAcad Sci USA 2004; 101: 3316–21[↩]
- Gomez-Pinilla F, Tyagi E. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Current Opinion in Clinical Nutrition and Metabolic Care. 2013;16(6):726–733.[↩]
- Gomez-Pinilla F, Nguyen TT. Natural mood foods: the actions of polyphenols against psychiatric and cognitive disorders. Nutritional Neuroscience. 2012;15(3):127–133.[↩][↩]
- Rothman SM, Griffioen KJ, Wan R, Mattson MP. Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Annals of the New York Academy of Sciences. 2012;1264:49–63.[↩][↩]
- Voss MW, Vivar C, Kramer AF, van Praag H. Bridging animal and human models of exercise-induced brain plasticity. Trends in Cognitive Sciences. 2013;17(10):525–544.[↩]
- Chytrova G, Ying Z, Gomez-Pinilla F. Exercise contributes to the effects of DHA dietary supplementation by acting on membrane-related synaptic systems. Brain Research. 2010;1341:32–40.[↩]
- Gomez-Pinilla F. The combined effects of exercise and foods in preventing neurological and cognitive disorders. Preventive Medicine. 2011;52(supplement 1):S75–S80.[↩]
- Wu A, Ying Z, Gomez-Pinilla F. Docosahexaenoic acid dietary supplementation enhances the effects of exercise on synaptic plasticity and cognition. Neuroscience. 2008;155(3):751–759.[↩]
- Wu A, Ying Z, Gomez-Pinilla F. Exercise facilitates the action of dietary DHA on functional recovery after brain trauma. Neuroscience. 2013;248:655–663.[↩][↩][↩]
- van Praag H, Lucero MJ, Yeo GW, et al. Plant-derived flavanol (-)epicatechin enhances angiogenesis and retention of spatial memory in mice. Journal of Neuroscience. 2007;27(22):5869–5878.[↩]
- Molteni R, Wu A, Vaynman S, Ying Z, Barnard RJ, Gómez-Pinilla F. Exercise reverses the harmful effects of consumption of a high-fat diet on synaptic and behavioral plasticity associated to the action of brain-derived neurotrophic factor. Neuroscience. 2004;123(2):429–440.[↩]
- Ribaric S. Diet and aging. Oxidative Medicine and Cellular Longevity. 2012;2012:20 pages.741468[↩]
- Trepanowski JF, Canale RE, Marshall KE, Kabir MM, Bloomer RJ. Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: a summary of available findings. Nutrition Journal. 2011;10(1, article 107).[↩]
- Deruisseau KC, Kavazis AN, Judge S, et al. Moderate caloric restriction increases diaphragmatic antioxidant enzyme mRNA, but not when combined with lifelong exercise. Antioxidants and Redox Signaling. 2006;8(3-4):539–547.[↩]
- Holloszy JO. Mortality rate and longevity of food-restricted exercising male rats: a reevaluation. Journal of Applied Physiology. 1997;82(2):399–403.[↩]
- Beck AT. The current state of cognitive therapy: a 40-year retrospective.Arch Gen Psychiatry 2005; 62: 953–9[↩]
- Farb NA, Anderson AK, Mayberg H, Bean J, McKeon D, Segal ZV.Minding one’s emotions: mindfulness training alters the neural expres-sion of sadness. Emotion 2010; 10: 25–33[↩][↩][↩]
- Buonomano DV, Merzenich MM. Cortical plasticity: from synapses tomaps. Annu Rev Neurosci 1998; 21: 149–86[↩]
- Vinogradov S, Luks TL, Schulman BJ, Simpson GV. Deficit in a neuralcorrelate of reality monitoring in schizophrenia patients. Cereb Cortex2008; 18: 2532–9[↩]
- Dale CL, Findlay AM, Adcock RA, Vertinski M, Fisher M, Genevsky A,et al. Timing is everything: neural response dynamics during syllableprocessing and its relation to higher-order cognition in schizophreniaand healthy comparison subjects. Int J Psychophysiol 2010; 75:183–93[↩]
- Packard MG. Anxiety, cognition, and habit: a multiple memory systemsperspective. Brain Res 2009; 1293: 121–8.[↩]
- Persson J, Reuter-Lorenz PA. Gaining control: training executive functionand far transfer of the ability to resolve interference. Psychol Sci 2008;19: 881–8[↩]
- Temple R. A regulatory authority’s opinion about surrogate endpoints.In: Nimmo W, Tucker G, editors. Clinical Measurement in DrugEvaluation. New York: J. Wiley; 1995.[↩]
- Temple E, Deutsch GK, Poldrack RA, Miller SL, Tallal P, Merzenich MM,et al. Neural deficits in children with dyslexia ameliorated by behavioralremediation: evidence from functional MRI. Proc Natl Acad Sci USA2003; 100: 2860–5.[↩]
- Keller TA, Just MA. Altering cortical connectivity: remediation-inducedchanges in the white matter of poor readers. Neuron 2009; 64:624–31[↩]
- Rosenberg PB, Hillis AE. Biomarkers for Alzheimer’s disease: ready for thenext step. Brain 2009; 132 (Pt 8): 2002–4[↩]
- McNab F, Varrone A, Farde L, Jucaite A, Bystritsky P, Forssberg H, et al.Changes in cortical dopamine D1 receptor binding associated withcognitive training. Science 2009; 323: 800–2[↩]
- Klingberg T, Fernell E, Olesen PJ, Johnson M, Gustafsson P, Dahlstrom K,et al. Computerized training of working memory in children withADHD–a randomized, controlled trial. J Am Acad Child AdolescPsychiatry 2005; 44: 177–86[↩]
- Kennedy SH, Konarski JZ, Segal ZV, Lau MA, Bieling PJ, McIntyre RS,et al. Differences in brain glucose metabolism between responders toCBT and venlafaxine in a 16-week randomized controlled trial. Am JPsychiatry 2007; 164: 778–88.[↩]
- Green CS, Bavelier D. Exercising your brain: a review of human brainplasticity and training-induced learning. Psychol Aging 2008; 23:692–701[↩]
- Cox RW, Jesmanowicz A, Hyde JS. Real-time functional magnetic reson-ance imaging. Magn Reson Med 1995; 33: 230–6.[↩]
- deCharms RC. Applications of real-time fMRI. Nat Rev Neurosci 2008; 9:720–9.[↩]
- deCharms RC, Maeda F, Glover GH, Ludlow D, Pauly JM, Soneji D,et al. Control over brain activation and pain learned by usingreal-time functional MRI. Proc Natl Acad Sci USA 2005; 102:18626–31[↩]


{kind=link}