Foreign body aspiration
Foreign body aspiration is when an object is inhaled and becomes lodged in a child’s airway or lungs. Foreign body aspiration remains a significant cause of death in children for anatomic as well as developmental reasons 1. Foreign body aspiration is the number one cause of accidental infantile deaths 2 and the fourth leading cause of death in preschool and younger age children less than five years of age 3. Foreign body aspiration accounts for a significant number of emergency department visits in the United States. The position of a child’s larynx or voice box, also makes small children susceptible to aspiration of foreign bodies into the airway. It’s natural for children to explore their environment by seeing, touching and tasting objects around them. Unfortunately, their tendency to put non-food objects in their mouth can be dangerous or even life threatening.
Children can also choke on foods given to them too early in their development, before they have the molars and coordinated chewing motions to safely break those foods down. Because infants and young children aged 6 months to 3 years lack molar teeth, have uncoordinated swallowing mechanisms, and, most importantly, engage with their surroundings by placing objects in their mouths, they are prone to foreign body aspiration 4.
Choking is typically defined as an aerodigestive foreign body causing varying amounts of obstruction to the airway. The obstruction can lead to difficulties with ventilation and oxygenation thus resulting in significant morbidity or mortality. The most commonly aspirated foreign bodies in children include vegetable matter, nuts and round foods such as hot dogs and grapes, coins, toys, and balloons. Less common, but more difficult to manage foreign bodies include beads, pins and small plastic toys, among an infinite number of other small objects. Aspirate objects, most commonly peanuts, lodge with the highest frequency within the right main stem bronchus due to its larger size and more vertical alignment as compared with the left. The main cause of death has been attributed to hypoxic-ischemic brain injury and less commonly, pulmonary hemorrhage 5.
According to the National Safety Council, in 2016 the rate of fatal choking in American children <5 years of age in the general population was 0.43 per 100,000. However, a previous study analyzing non-fatal choking data of children under the age of 14 has revealed a comparatively higher rate of 20.4 per 100,000 population. 55.2% of these non-fatal choking cases in children <4 years of age involved candy, with hot dogs and nuts being more likely to require hospitalization. Males accounted for 55.4% of cases but there was no statistically significant difference between the sexes. Analysis of the national trend of inorganic foreign body aspiration has revealed that aspirated coins have decreased in relative frequency with aspirated jewelry conversely becoming more common in the United States. Overall the rate of aspiration from foreign bodies resulting in emergency department visits has remained stable between the years of 2001 and 2014.
A recent meta-analysis of the worldwide literature on foreign body aspiration revealed a sex discrepancy with 60% of patients being males. Nuts were seen to be causative in 40% of cases in high-income and low-middle income nations. Among inorganic foreign bodies, a pooled proportion among the literature stemming from high-income countries revealed that magnets were causative in 34% of the cases. Diagnosis was delayed by more than 24 hours in 60% of those particular cases 6.
Often, diagnosing and treating foreign body aspiration requires special expertise and equipment.
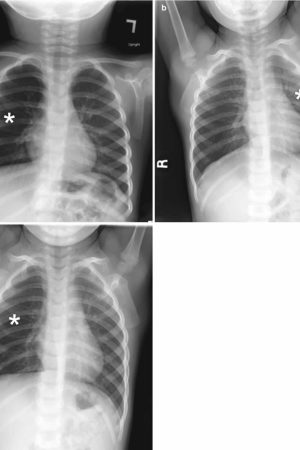
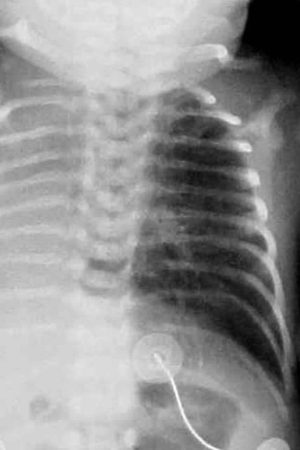
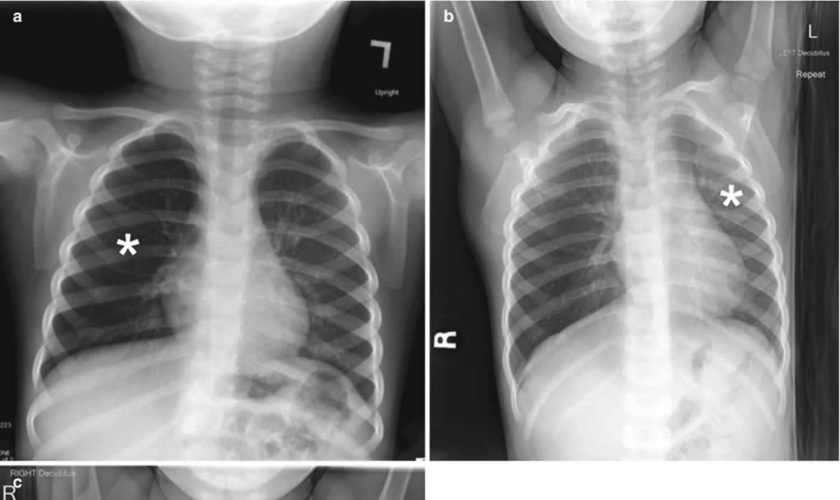
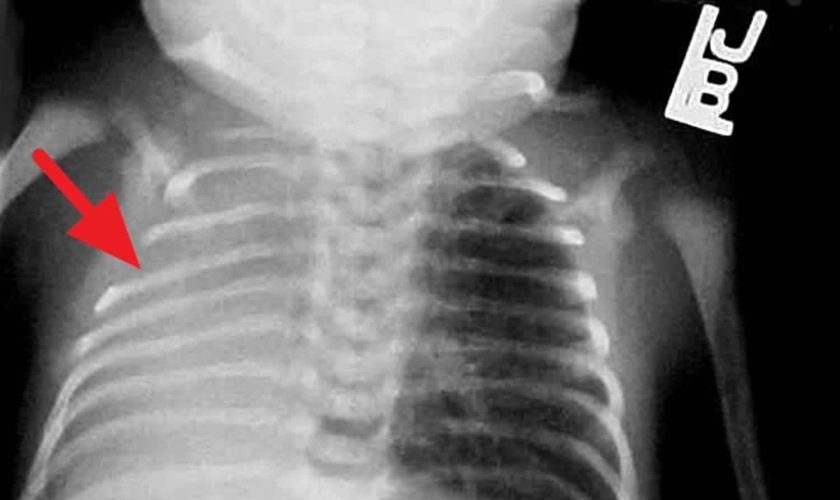
Chest radiography is the first-line imaging study in cases of suspected foreign body aspiration because it is readily available, low in cost, and associated with minimal radiation exposure 7. As only 10% of aspirated foreign bodies are radiopaque 8, however, indirect signs of aspiration including air trapping, focal airspace disease, pleural effusion, mediastinal shift, pneumothorax, or subcutaneous emphysema are important imaging surrogates. Unilateral hyperinflation 9 is the most commonly identified indirect sign of aspiration (Figure 3a) 10. This finding may be further evaluated with bilateral decubitus radiographs to assess for air trapping with the side of foreign body aspiration failing to deflate in the dependent position (Figure 3b and c). Lateral decubitus radiography, however, have only 68–74% sensitivity and 45–67% specificity 11. Furthermore, it has been shown to increase false-positive rates without increasing the rate of true-positive identification. As such, a negative chest radiography in the setting of high clinical suspicion should prompt further evaluation with CT.
CT scan has an accuracy close to 100% for the detection of aspirated foreign body 11 able to identify both an endoluminal mass as well as secondary signs of aspiration. Because of the increased radiation exposure of CT as compared with radiography, study benefits must be weighed. 3D virtual bronchoscopy reconstructions can be utilized to assess foreign body aspiration and may be used for preprocedural planning. Additionally, following bronchoscopic removal, CT can be used to assess for residual aspirated foreign body 12.
The definitive diagnosis and management of a foreign body typically involves bronchoscopy to remove the offending object. However, no universal standard of care has been thoroughly defined. Rigid bronchoscopy has been described to have several key benefits compared to flexible bronchoscopy for definitive management. Some cited reasons include: 1) the ability to ventilate via the rigid bronchoscope, 2) improved visualization with a rigid telescope, and 3) greater versatility to accommodate various sizes of suctioning and optical forceps. Additionally, the rigid scope offers a wider space to manipulate the offending object and to facilitate removal while avoiding obstruction at the level of the glottis. In cases of diagnostic uncertainty or complexity such as in recurrent pneumonia with no clear history of an aspiration, a flexible bronchoscopy may be the preferred initial test.
According to one study, when a combination of three or more highly suggestive clinical and radiological diagnostic data were noted, the risk of identifying a foreign body in the airway was 91%. Diagnostic clinical criteria significantly correlated with the presence of a foreign body included: a history of sudden choking, cyanosis, apnea, and decreased breath sounds. Diagnostic radiological criteria included: atelectasis, mediastinal shift, or signs of air trapping. The absence of the aforementioned criteria was very predictive of a negative flexible bronchoscopy. Thus, it was deemed safe to refer these patients for outpatient follow-up as opposed to initial bronchoscopy 13.
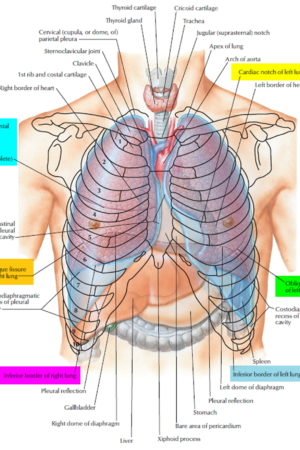
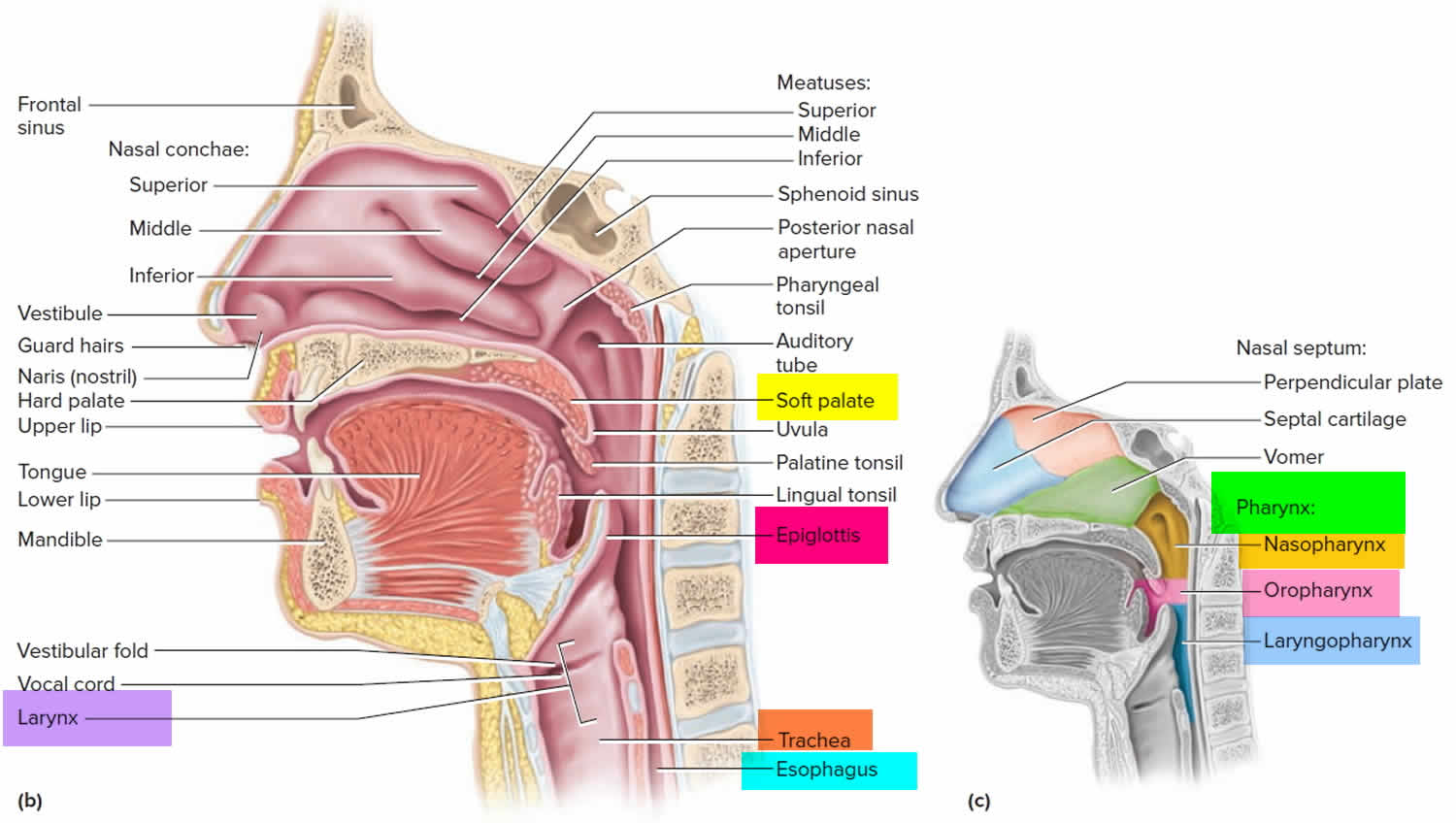
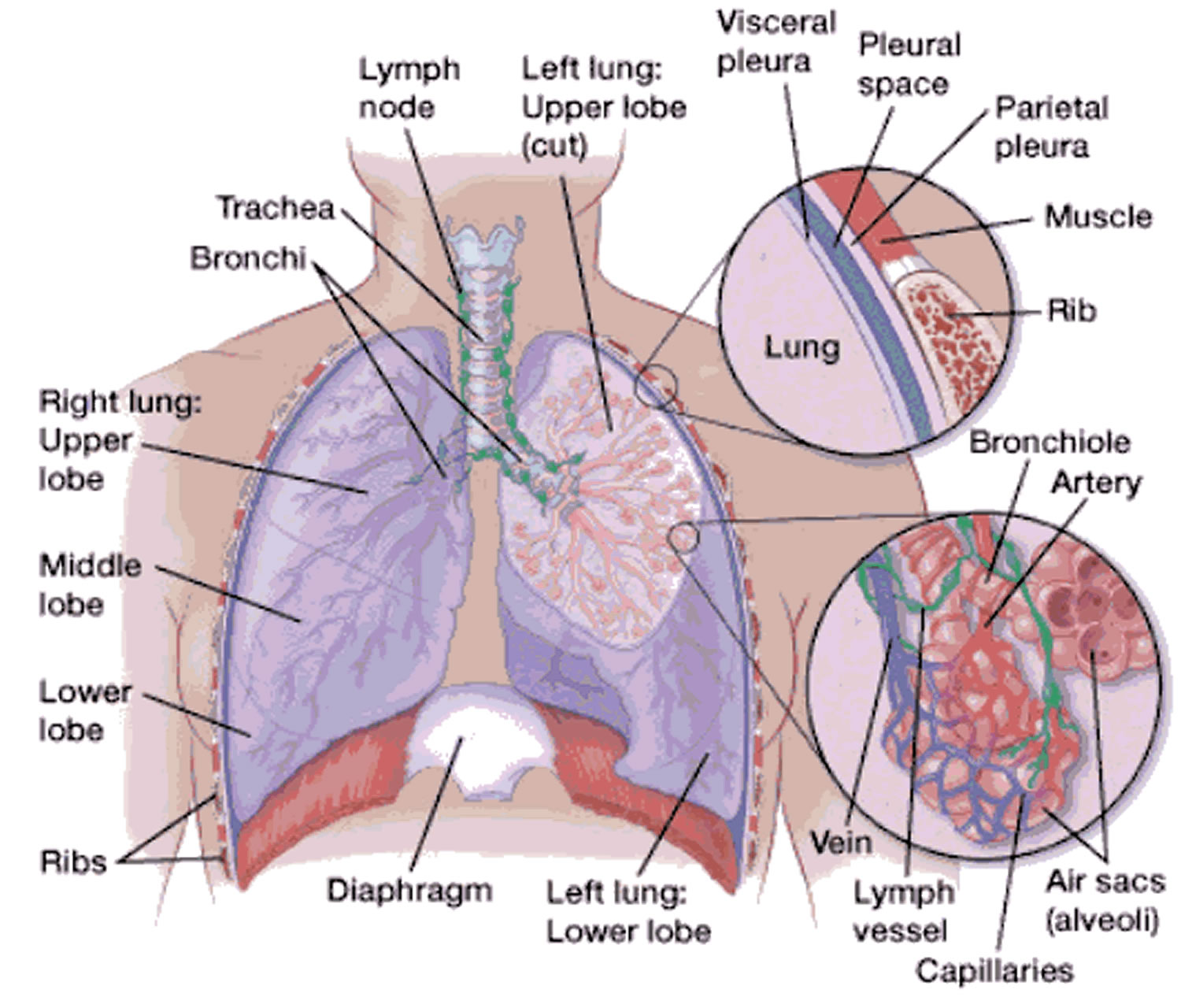
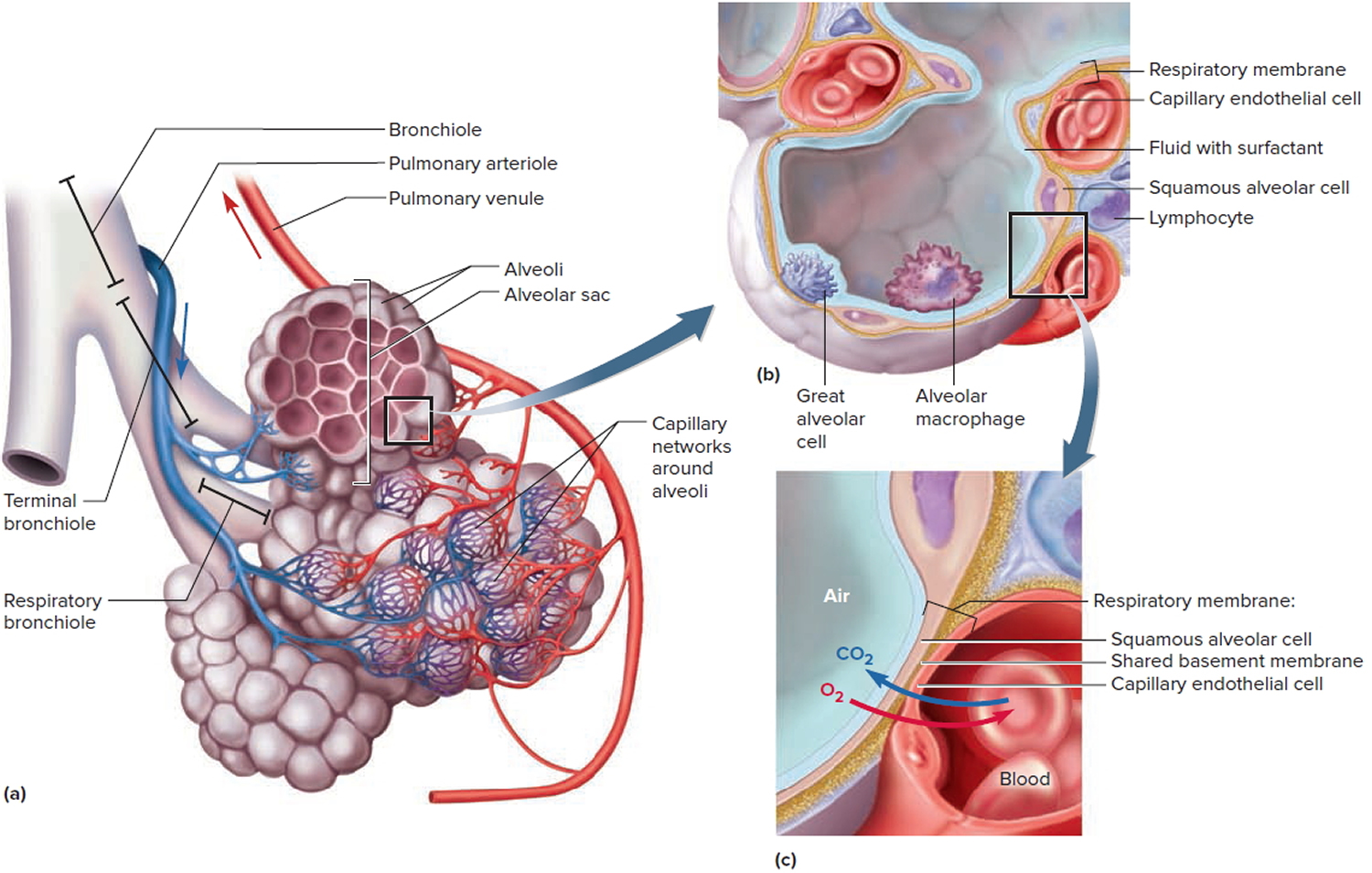
Lungs anatomy and function of the airway
An infant is developmentally able to suck and swallow and is equipped with involuntary reflexes (gag, cough, and glottic closure) that help to protect against aspiration during swallowing. Dentition initially develops at approximately 6 months with eruption of the incisors. Molars are required for chewing and grinding food and do not erupt until approximately 1.5 years of age. However, mature mastication abilities take longer to develop and remain relatively incomplete throughout early childhood 14. Young children and children with developmental and neurologic impairment also do not have the overall cognitive skills, behavioral control, or experience to chew well and eat slowly.
Despite a strong gag reflex, a young child’s airway is more vulnerable to obstruction than that of an adult in several ways. The smaller diameter is more likely to experience significant blockage by small foreign bodies. Resistance to air flow is inversely related to the radius of the airway to the fourth power, so even small changes in the cross-section of the airway of a young child can lead to dramatic changes in airway resistance and air flow. Mucus and secretions around a foreign body in the airway will reduce the radius of the airway even further and may also form a seal around the foreign body, making it more difficult to dislodge by forced air, such as with a cough or Heimlich maneuver. The force of air generated by a cough in an infant or young child is less than that in an adult; therefore, a cough may be less effective in dislodging a complete or partial airway obstruction during early childhood 15.
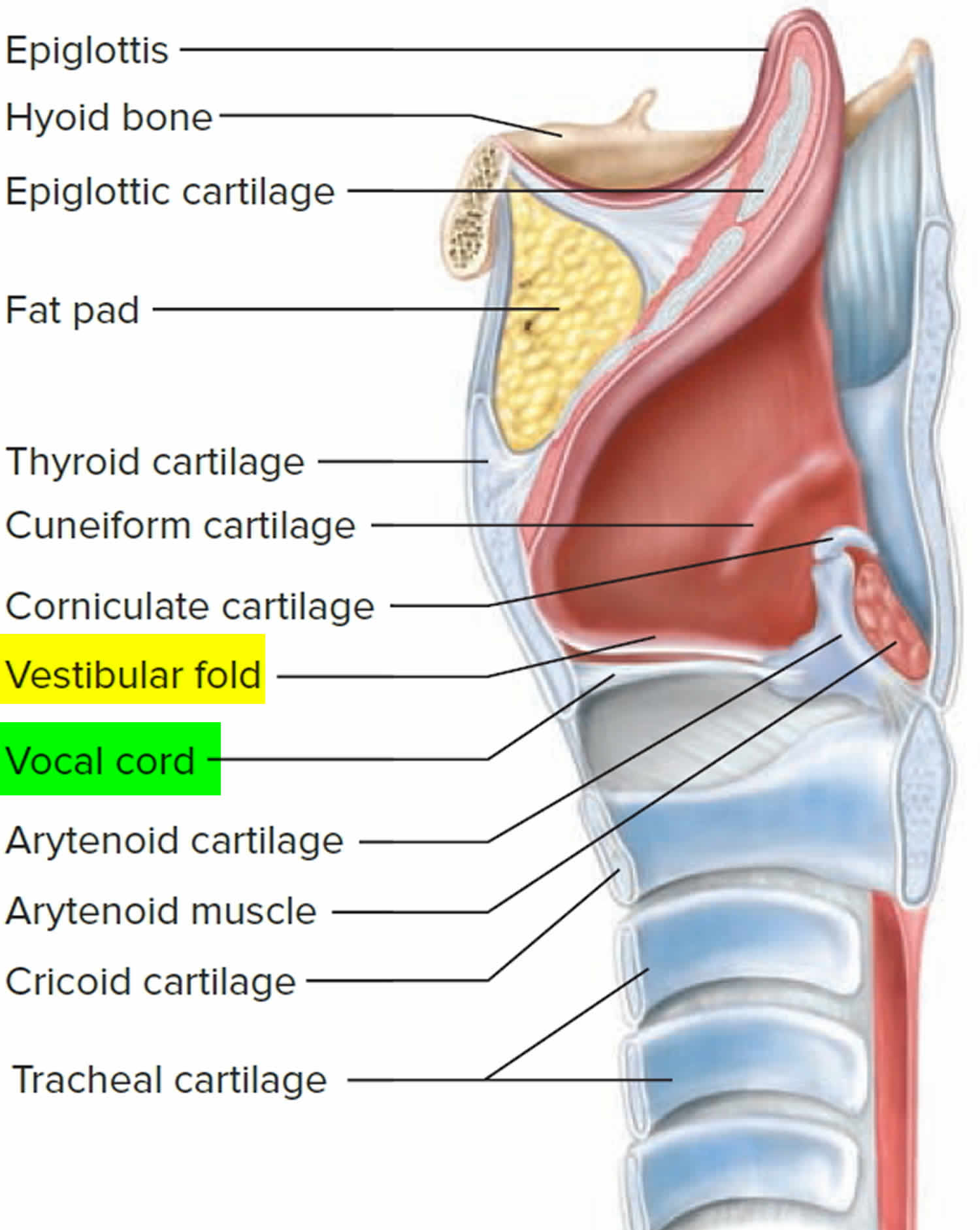
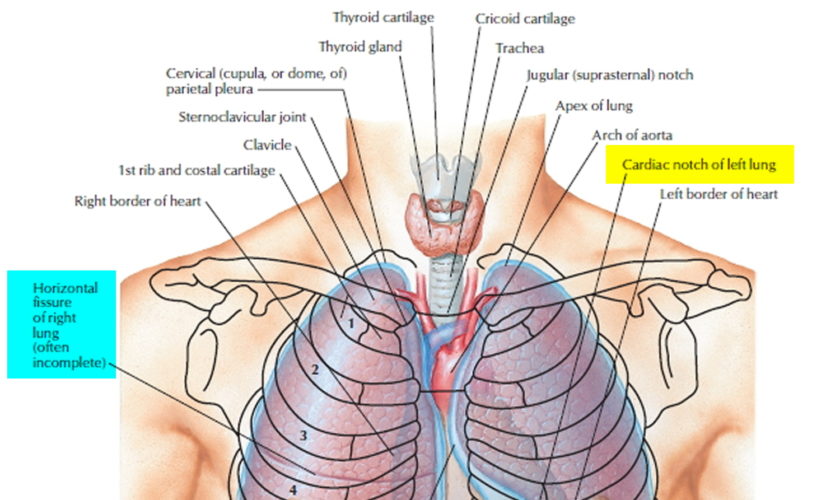
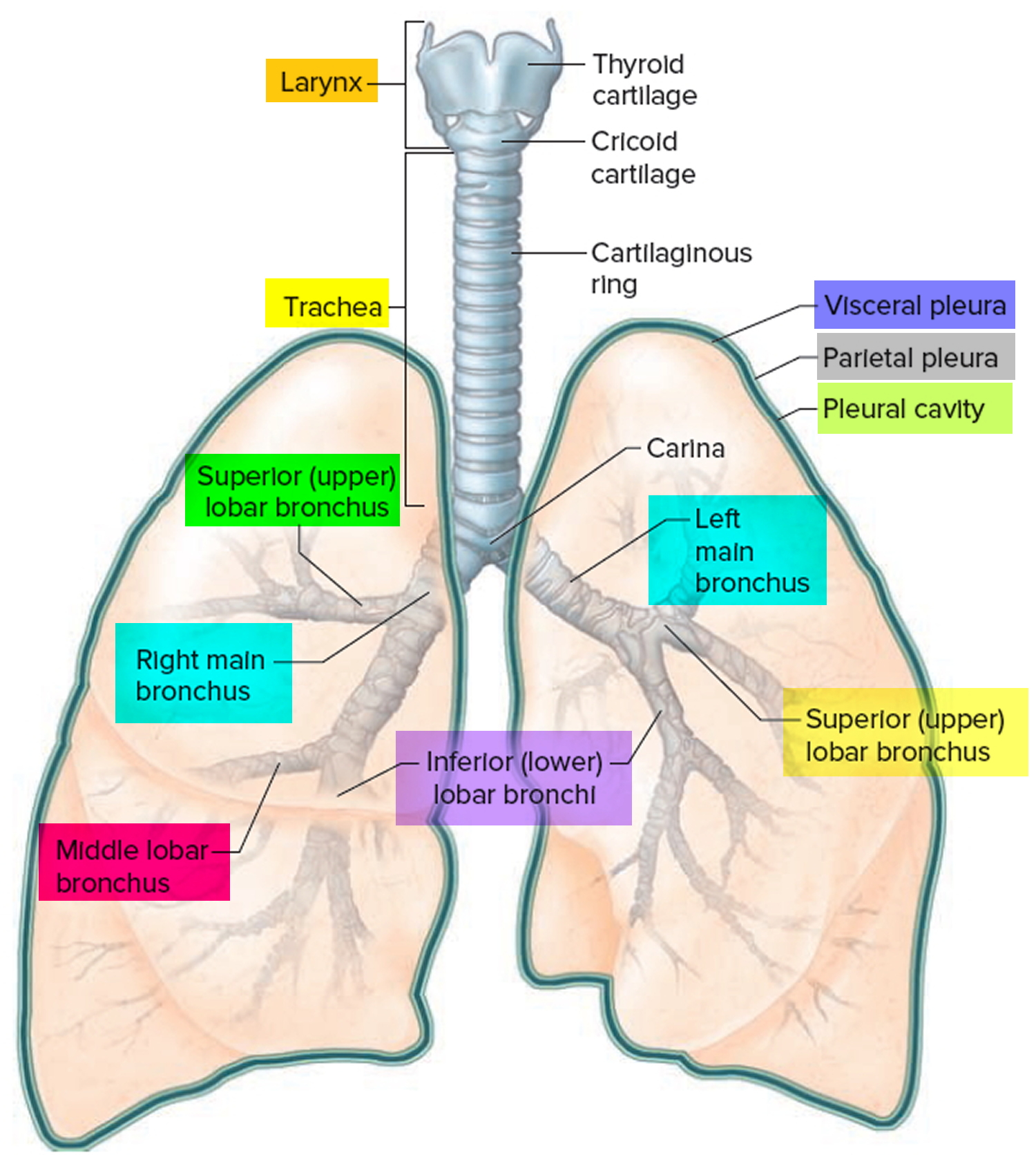
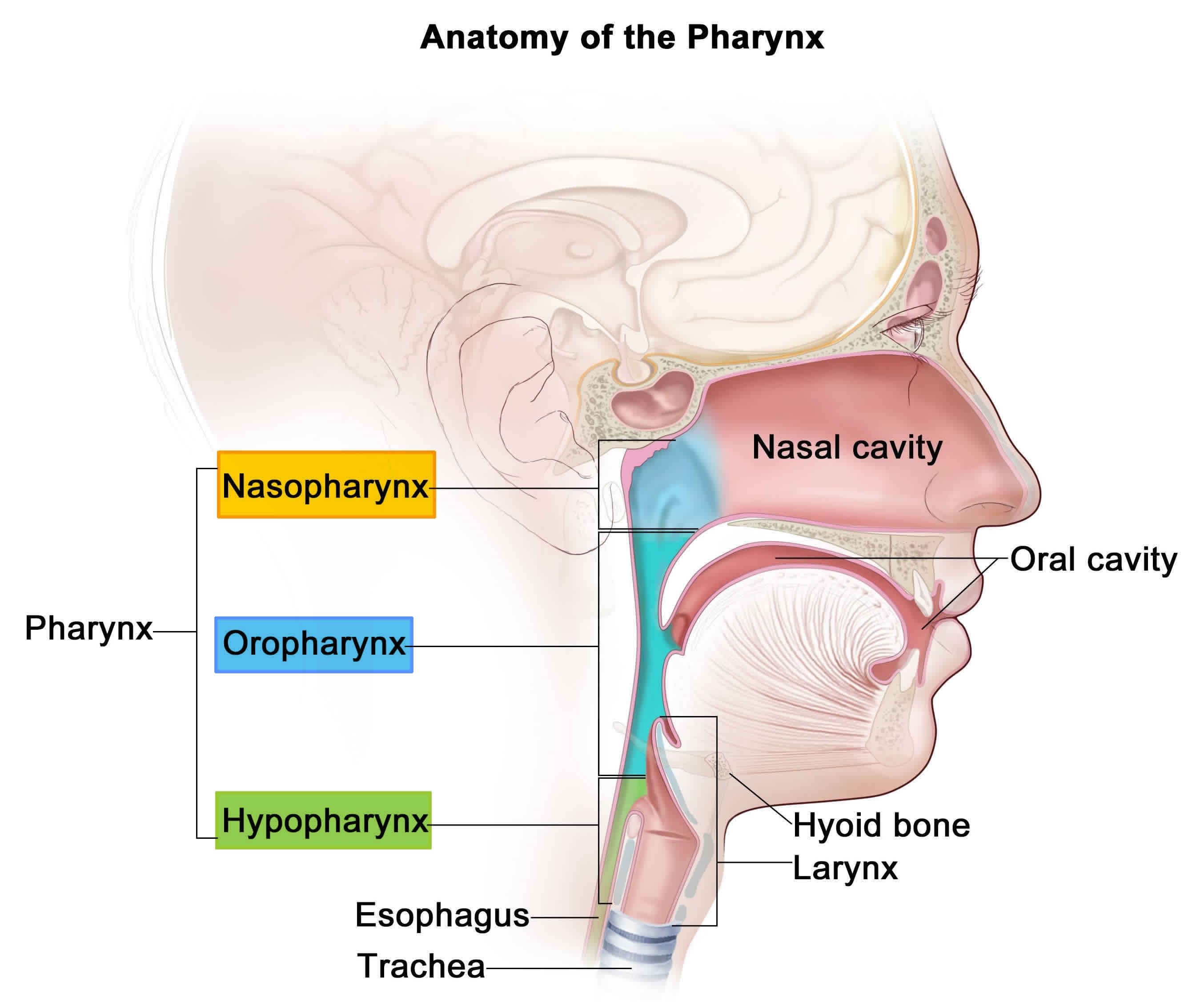
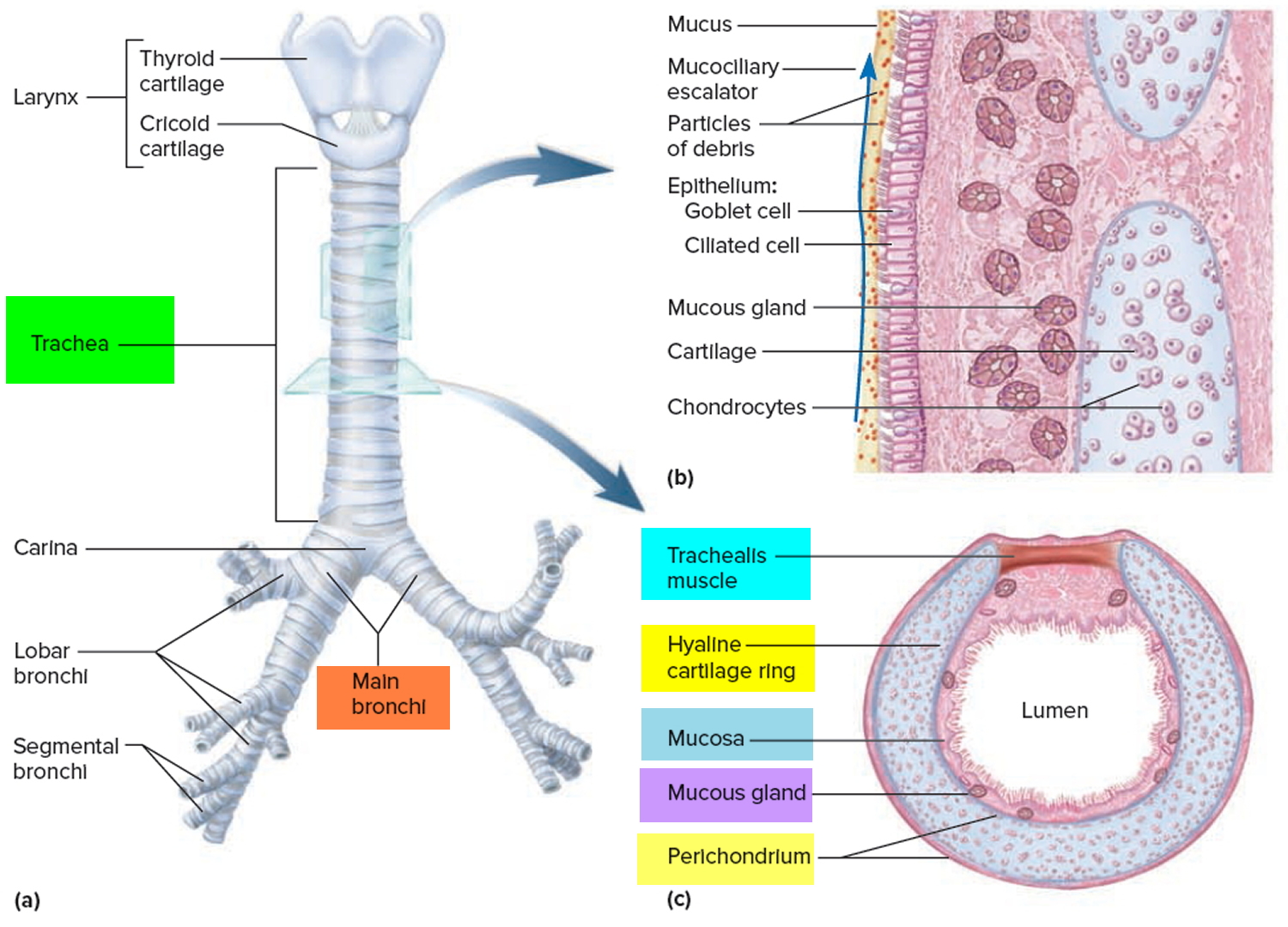
Airway anatomy in children differs from anatomy in adults. The narrowest portion of the pediatric airway is the cricoid, while in adults the narrowest portion is the glottis. Thus particles may be large enough to be aspirated past the vocal cords (glottis) in children only to become lodged in the subglottis at the area of the cricoid, to potentially devastating effect. When considering aspiration of foreign objects, children have a slight predominance for aspiration into the right mainstem bronchus, but this proclivity increases with age owing to a more vertical orientation of the right mainstem in adults that parallels the orientation of the trachea – it becomes the most dependent and direct portion of the adult airway.
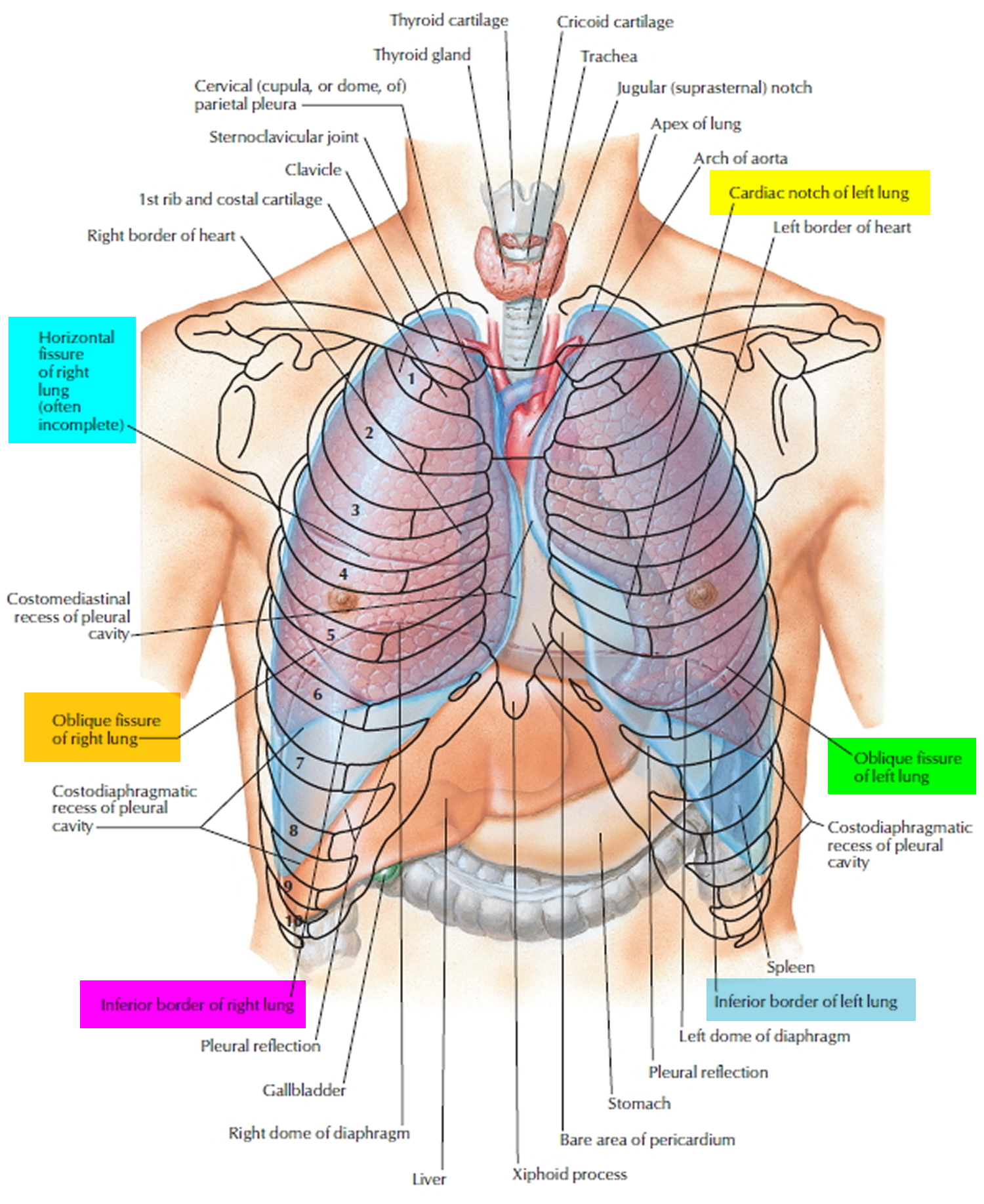
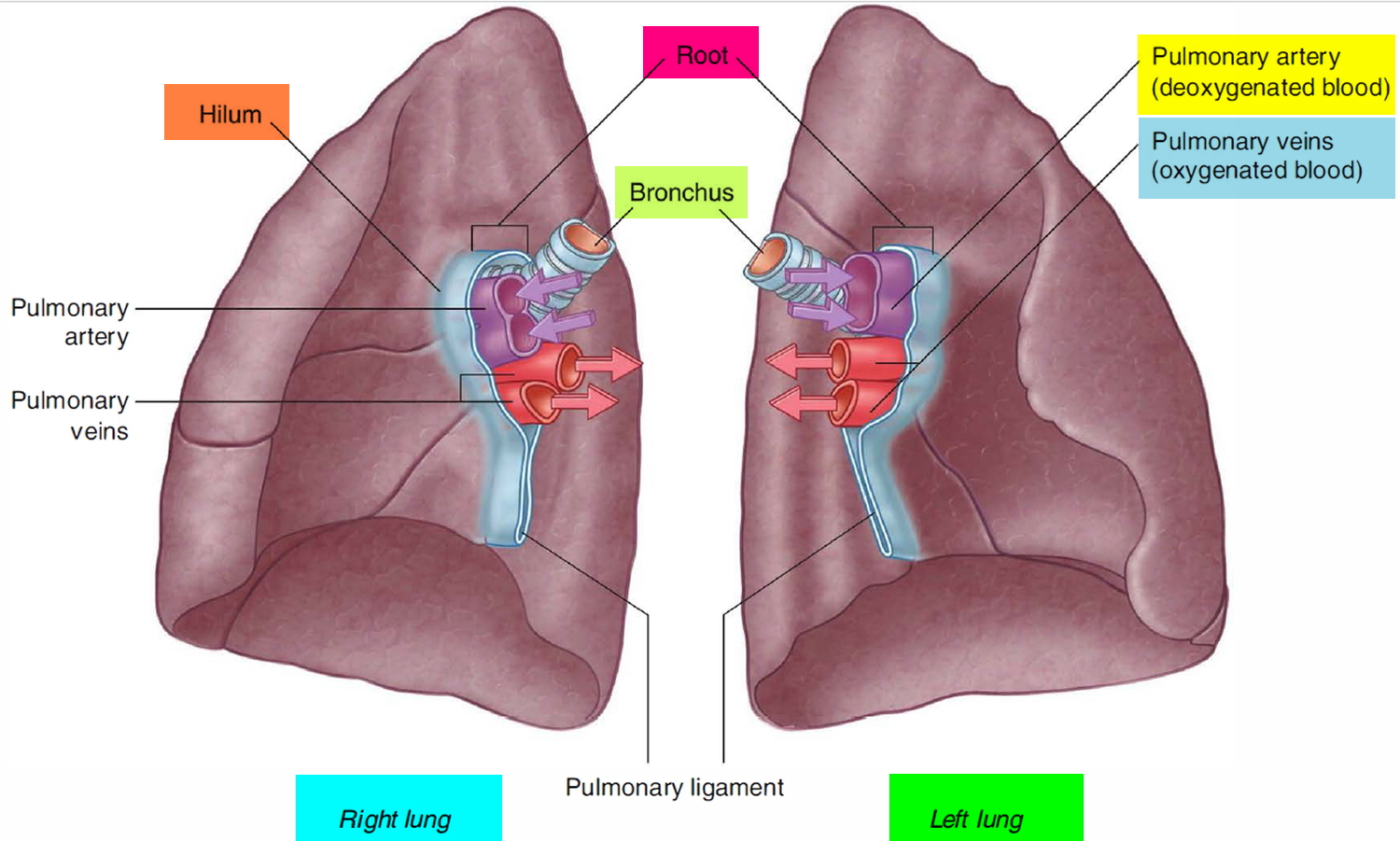
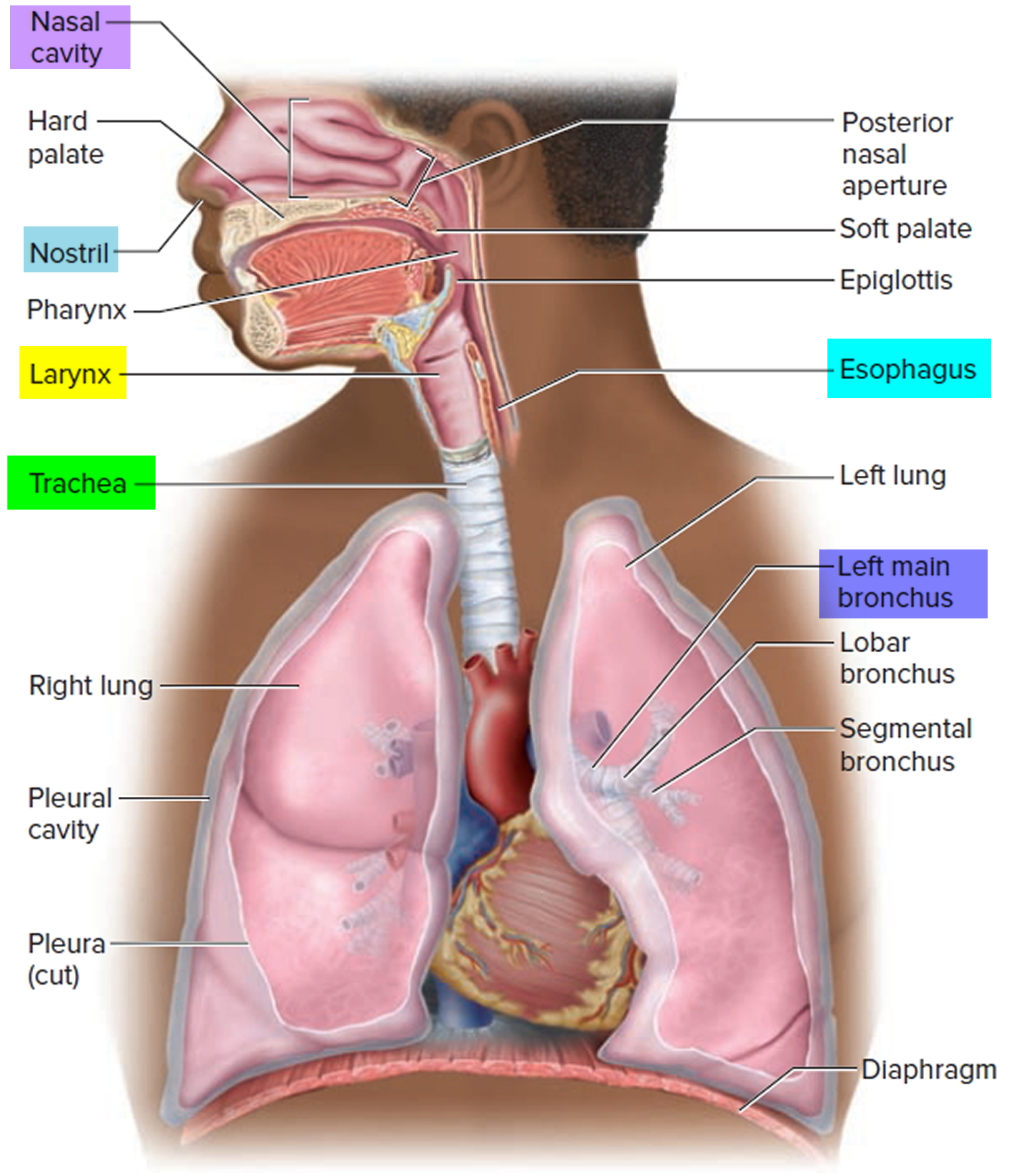
Figure 1. Lung anatomy
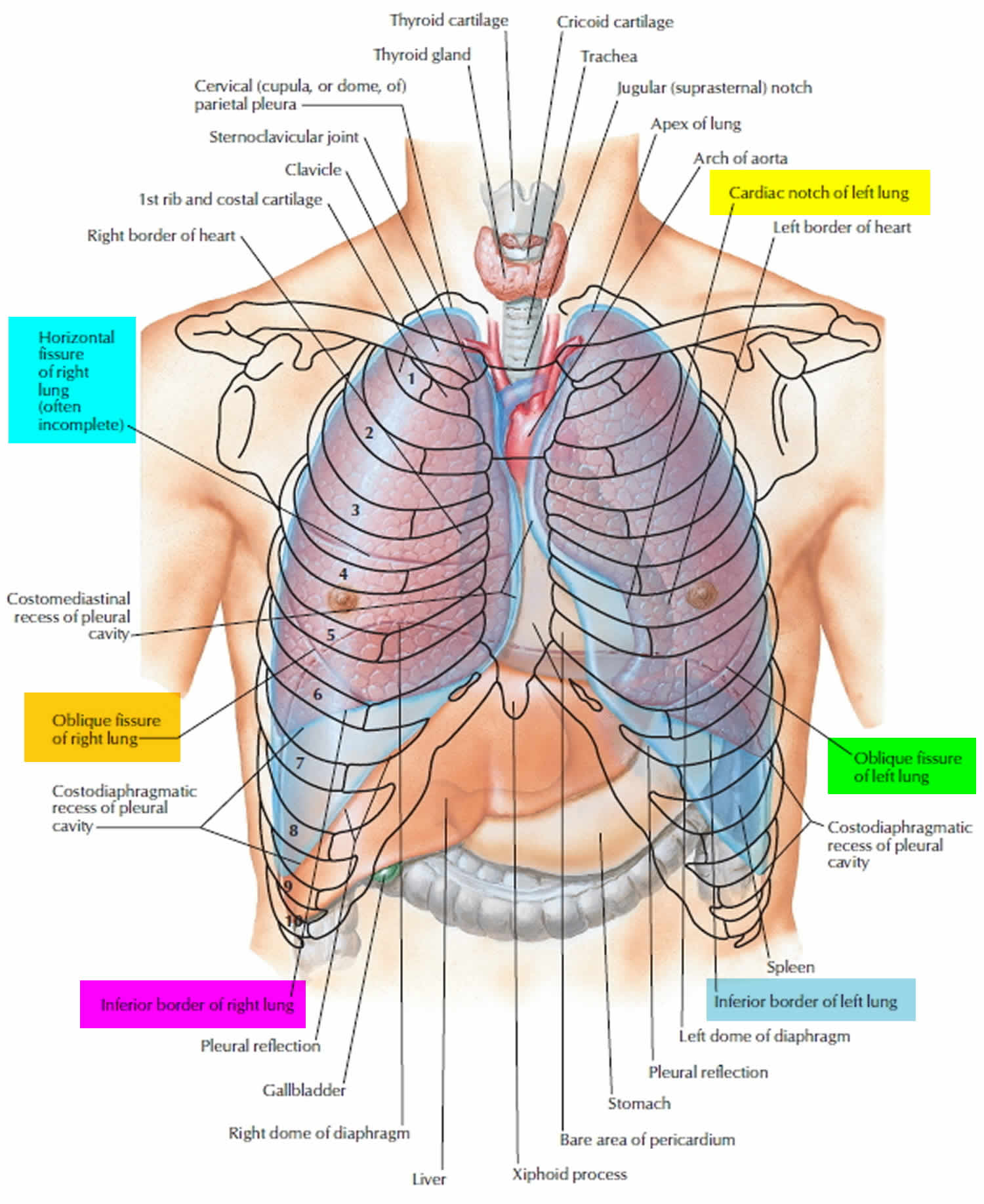
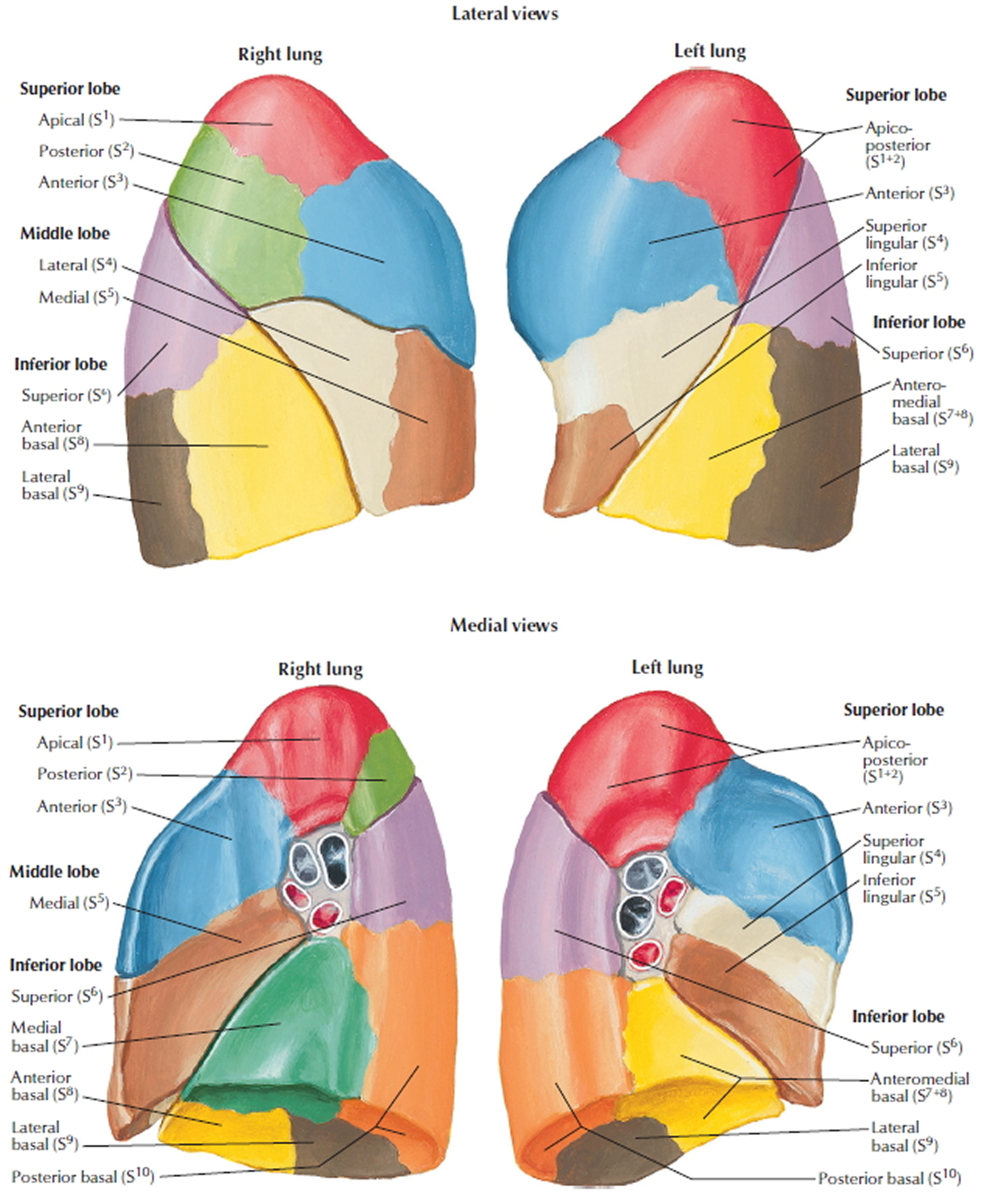
Figure 2. Bronchial tree of the lungs
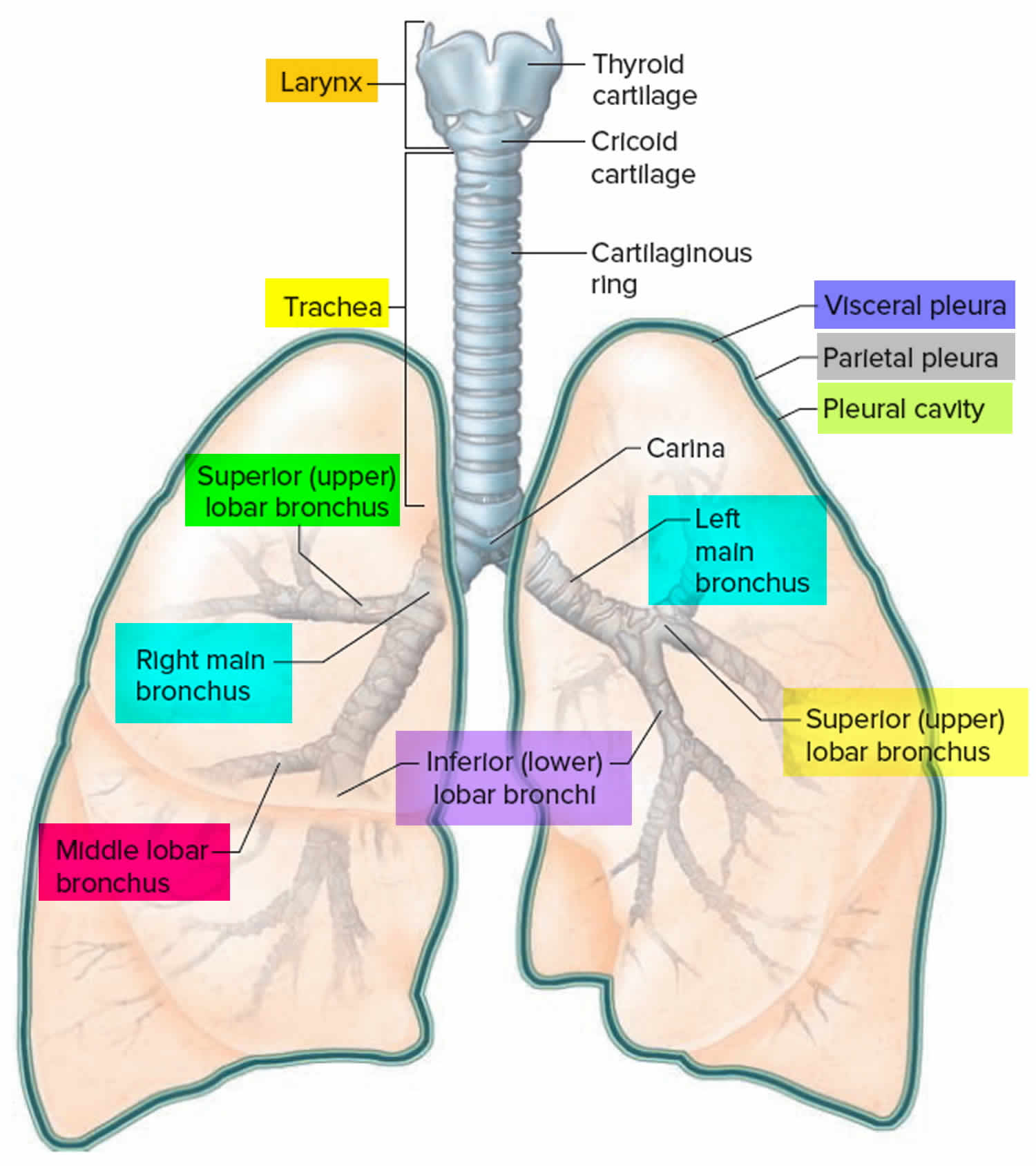
Figure 3. Foreign body aspiration
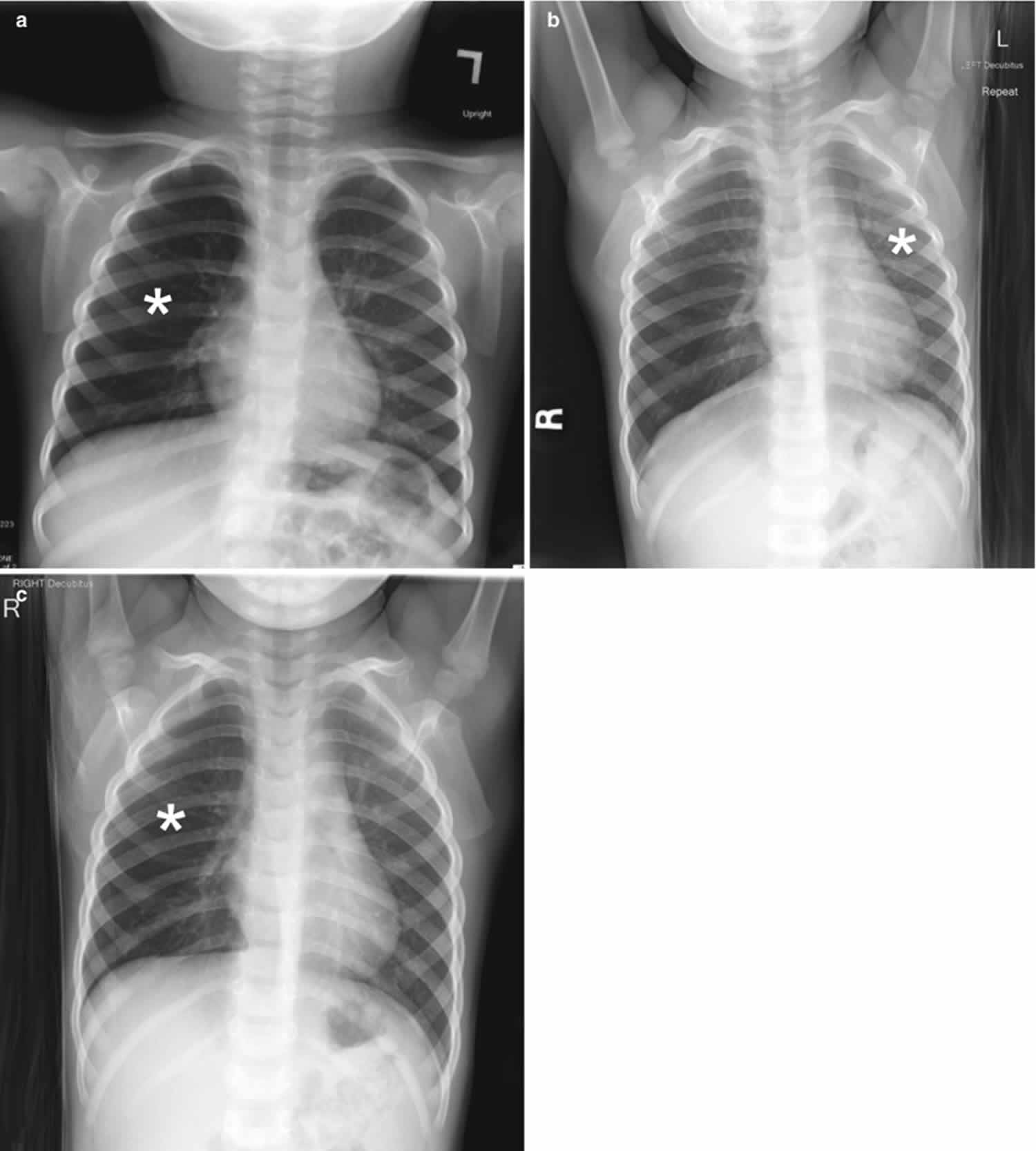
Footnote: A 30-month-old boy with an endobronchial non-radiopaque foreign body aspiration who presented with acute onset of wheezing and respiratory distress. (a) Frontal chest radiograph shows asymmetric hyperinflation of the right lung (asterisk) as compared with the normally aerated left lung. (b) Left lateral decubitus radiograph demonstrates expected deflation of the left lung (asterisk) in the dependent portion. (c) Right lateral decubitus radiograph shows persistent relative hyperinflation representing air trapping of the right lung (asterisk) despite dependent positioning. Radiographic findings suspicious for aspirated non-radiopaque foreign body in the right main stem bronchus. Subsequently obtained bronchoscopy demonstrated a nearly obstructive foreign body (likely almond) located within the right main stem bronchus
[Source 16 ]Foreign body aspiration causes
Any object that can be placed into the mouth can potentially be aspirated. This is of particular concern in infants and young children who explore and interact with their environment by placing objects into their mouth; parental vigilance regarding which objects are available to an unsupervised child is paramount. Similarly, infants’ and young children’s swallowing coordination has not fully developed, and there is a proclivity to inhale or aspirate foods when eating. The lack of molars to chew food is also a contributing factor in children 17. Peanuts being the most commonly aspirated food, followed by hotdogs and hard candy, with hotdogs causing the most mortality. Male children are more likely to aspirate than female children 17. Food or other objects with a smooth, round shape are the highest risk for aspiration (nuts, beans, grapes, hotdogs/sausages, etc), and a primary prevention strategy is to prepare such foods in a way so as to change this shape to something more angular and easier to chew and swallow (by quartering grapes, for example).
Foreign body aspiration pathophysiology
Young children are particularly at risk for foreign body aspiration. One study has shown the mean age to be 24 months with 98% of cases involving children < 5 of age. As airway resistance is inversely related to the cross sectional radius by a power of four, the relatively smaller diameter of pediatric airways means that they are more prone to significant airflow obstruction with even small foreign bodies. Dental development also contributes to the risk of foreign body aspiration, as molars typically are not present before the age of 2; thus children in this age group are able to bite pieces of food with their incisors but not effectively able to grind food into smaller pieces. Additionally, young children tend to explore the world with their mouths while playing and exhibit high levels of activity and distractibility while eating, further putting them at risk. Due to the relative anatomical narrowing of the tracheobronchial tree in children, the proximal airway is typically the site of obstruction. In fact, in one retrospective review, 96% of foreign bodies aspirated were found in this location. In children <15 years of age, foreign bodies lodge within the left lung almost as often as in the right lung. This is due to the symmetric tracheal take-off angle found between the two bronchi in many children prior to the development of a prominent aortic indent affecting the trachea and left main bronchus. Regardless of age, if there is noticeable aortic indentation on the trachea when examining radiographs, then the right bronchial angle will be less distinct compared to the left side and aspiration will be more common in the right lung 18.
Foreign body aspiration symptoms
Patients may be completely asymptomatic and the only evidence of an aspiration event may be found during history taking. Upon aspiration of a foreign body into the larynx or proximal trachea, there is always the potential for respiratory compromise or for further inhalation into the distal airways causing subacute symptoms including shortness of breath, wheezing, or coughing. However, sudden onset of cough, choking, and/or shortness of breath (dyspnea) have been found to be the most common symptoms. One prospective study has cited a sensitivity of 91.1% and specificity of 45.2% for choking and acute cough. Wheeze on auscultation has been found to be a major physical finding and in one study was documented in 60% of cases. In the same study, 32% of patients had asymmetric breath sounds. An abnormal physical exam has been seen to have a sensitivity of 80.4% and specificity of 59.5% for foreign body aspiration 19.
Chronic shortness of breath can be related to aspiration of a foreign body, especially in children and developmentally delayed individuals who are unable to articulate the event reliably. This is more likely to be in the smaller, more distal airways and symptoms can relate either to complete occlusion of a terminal bronchus and development of pneumonia or to partial obstruction leading to wheezing, coughing, stridor, and progressive symptoms as the surrounding respiratory epithelium becomes more reactive and edematous. The patient may have several weeks of coughing, shortness of breath, or even complain of chest discomfort. Such patients may present weeks to months later, and the initial aspiration event may have been unknown or forgotten by patients and families.
Foreign body aspiration complications
A myriad of complications including recurrent pneumonia, bronchiectasis, lung abscess, and atelectasis can occur from a missed foreign body aspiration. Bronchial stenosis is also a well-known complication of chronic foreign bodies in the airway. However, nearly universal, tracheal lacerations are the most commonly reported complication among affluent countries. Pneumonia is the most common complication among countries with a poorer socioeconomic status. In a retrospective study evaluating risk factors associated with a missed diagnosis of foreign body aspiration, the incidence of a major complication was often seen to be increased the longer a foreign body was present in the airways. Obstructive emphysema was found to be the most common complication for foreign bodies discovered >3 days after the initial event. Importantly, it should be noted that a normal radiograph with absent physical findings does not exclude the possibility of an aspirated foreign body. Furthermore, patients on bronchodilators and steroid may suppress reactive respiratory symptoms 19.
Foreign body aspiration diagnosis
The diagnosis of an aspirated foreign body is based on a combination of the history of the child’s illness, the child’s presenting symptoms, and chest X-rays. If a foreign body in the airway is strongly suspected, a child needs to go to the operating room and have an airway examination performed under anesthesia. This examination is called a microlaryngoscopy and bronchoscopy (a look at the voice box and windpipe, or airway).
A child may be diagnosed with foreign body in the airway when a family member has seen the child swallow food or a small object then noticed signs of airway distress, like coughing or difficulty breathing. Children with a persistent segmental pneumonia, especially right lower lobe, should also be considered for foreign body aspiration.
There are three primary ways to see if a child has inhaled something into the airway or lungs:
- Chest X-ray. Some non-food items can be seen in the airway or lungs using a traditional X-ray. However, most food, vegetable matter and plastic toys won’t appear on chest X-ray films.
- Inspiratory and expiratory phase X-ray. These are X-rays taken when the child has inhaled and then exhaled the air out of their lungs. If a foreign body cannot be seen with a traditional X-ray, then inspiratory and expiratory phase films may show hyperinflation or air-trapping which suggests an aspirated foreign body.
- Bronchoscopy. When suspicion of aspiration is high enough but the physical exam and X-rays are not definitive for a diagnosis, an instrument called a bronchoscope is inserted through the mouth and used to look at the inside of the airways under anesthesia. Bronchoscopy can be used both to locate the foreign body and to remove it.
Chest radiographs are often utilized as initial tool of investigation for foreign body aspiration. Radiological findings consistent with a foreign body aspiration include atelectasis, pneumothorax, and air trapping. However, chest radiographs have been seen in multiple studies to be frequently normal, with one study citing a normal chest radiograph in 35% of cases of foreign body aspiration. This same study indicated that the most common abnormality found on radiograph was air trapping, present in 53% of cases. The sensitivity of chest radiograph has been documented as 67.9% and the specificity has been seen to be 71.4%. In one study, children with a concerning history of sudden cough, abnormal pulmonary exam, and abnormal chest x-ray have been shown to have a risk of 88.6% for foreign body aspiration. It is important to note that rarely a single finding or historical piece of information can definitively diagnose a foreign body aspiration. Rather, direct identification via bronchoscope is often needed.
In a review of common imaging modalities for diagnosis of foreign body aspiration, a simple algorithm based on feasibility and utility was generated at one institution. Recommendations included initial diagnostic work-up with frontal and lateral chest films with additional neck films if warranted on exam. Abnormalities at this stage would be sufficient to necessitate bronchoscopy. However, if the radiographs are unrevealing in the symptomatic patient with a suggestive history, a CT of the chest should subsequently be performed. If CT is unavailable and the child is > 5 years of age, then inspiratory-expiratory films can be obtained. If the child is younger than 5 years old, bilateral decubitus films are preferred. Fluoroscopy and MRI can be used as adjunctive measures. It should be noted that this was an algorithm formulated at one particular institution and is not universal practice.
To mitigate the potential need for CT scan, one study has evaluated a quantitative method to increase the sensitivity of radiographs to detect foreign bodies, in particular aspirated objects which are radiolucent. In a case-control study, radiodensity ratios were compared between the affected lung and the contralateral lung in patients with definitively diagnosed foreign body aspiration and healthy controls. The radiodensity ratio was seen to be significantly higher in in patients with aspiration as compared to controls. Radiodensity was calculated for each lung by measuring total Hounsfield units and then the ratio developed involved comparison between the affected lung and the normal lung. The authors of this study suggest a radiodensity ratio cutoff of 1.10 is sufficiently positive to warrant further investigation with bronchoscopy, whereas a ratio below this value would warrant further work-up with a CT scan. As there is a relative paucity evaluating this method in the literature, further studies are needed for validation 20.
Foreign body aspiration treatment
Bronchoscopy is the standard method for removal of an aspirated foreign body. An anesthesiologist puts the child into a deep sleep and then topical lidocaine spray is used to further anesthetize the child’s larynx. An instrument called a laryngoscope is inserted into the airway to view the larynx; then a rigid, ventilating bronchoscope is passed beyond it, into the airway, and used to examine the trachea and right and left bronchi to locate the foreign body.
When the object is found, specially designed forceps are inserted into the airway through the bronchoscope to retrieve the object. This procedure requires training, delicacy and skill.
There are three kinds of forceps that may be used to remove airway foreign bodies:
- Optical forceps with an attached telescope to view the retrieval of the object
- Non-optical forceps used to remove beads, nails, screws, tacks and other objects that are in a distant tiny space
- Biopsy forceps used to remove new or granulating tissue or tissue masses, which can form as the body attempts to enclose a foreign body that has been present for an extended period
Very rarely, the doctor may make a tracheotomy incision (an incision that opens the child’s airway) to extract a foreign body that is difficult to remove due to its size or shape.
The child will typically stay in the hospital overnight after the procedure for observation. There may be airway swelling, increased secretions, infection or difficulty breathing after the foreign body is removed. Occasionally, if a foreign body has been left in a child for a long period of time, the child may require additional bronchoscopies to make sure that all of the foreign body has been removed and that there is no residual scarring or granulation tissue. Sometimes the child requires antibiotics, steroids or inhaled bronchodilators for a brief period of time after a foreign body is removed from the airway.
Foreign body aspiration prognosis
A large retrospective review identified the mortality rate among pediatric patients with foreign body aspiration to be 2.5%. Age was seen to be correlated with the anatomic location of the foreign body with increasing age positively correlated to increasing distal anatomic location of the aspiration. Unsurprisingly, neurologic disability was the most common condition among patients with aspiration. This particular co-morbidity was associated with increased odds of death and mechanical ventilation. A lodged foreign body lower in the respiratory tract carried a comparatively higher odds of mortality compared to those proximally positioned. It is believed by the authors that this is secondary to significant mucous plugging along with later dislodgement resulting in contralateral obstruction and hypoxia. Despite this, foreign bodies present in the trachea had the highest odds of requiring mechanical ventilation 21.
- Cramer N, Jabbour N, Tavarez MM, et al. Foreign Body Aspiration. [Updated 2020 Apr 27]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531480[↩]
- Cheng J, Liu B, Farjat AE, Routh J. National estimations of airway foreign bodies in children in the United States, 2000 to 2009. Clin Otolaryngol. 2019 May;44(3):235-239.[↩]
- Tsang JE, Sun J, Ooi GC, Tsang KW. Endobronchial Foreign Body Presenting as Exacerbation of Asthma. Case Rep Emerg Med. 2017;2017:6863083[↩]
- Green SS. Ingested and aspirated foreign bodies. Pediatr Rev. 2015;36(10):430–6.[↩]
- Wu X, Wu L, Chen Z, Zhou Y. Fatal choking in infants and children treated in a pediatric intensive care unit: A 7- year experience. Int. J. Pediatr. Otorhinolaryngol. 2018 Jul;110:67-69.[↩]
- Hanba C, Cox S, Bobian M, Svider PF, Gonik NJ, Shkoukani MA, Sheyn A. Consumer product ingestion and aspiration in children: A 15-year review. Laryngoscope. 2017 May;127(5):1202-1207.[↩]
- Laya BF, Restrepo R, Lee EY. Practical imaging evaluation of foreign bodies in children: an update. Radiol Clin N Am. 2017;55(4):845–67.[↩]
- Brian PS, Lim R, Avery LL. Review of ingested and aspirated foreign bodies in children and their clinical significance for radiologists. Radiographics. 2015;35(5):1528–38.[↩]
- Wasilewska E, Lee EY, Esenberg RL. Unilateral hyperlucent lung in children. AJR Am J Roentgenol. 2012;198(5):W400–14.[↩]
- Hegde SV, Hui PK, Lee EY. Tracheobronchial foreign bodies in children: imaging assessment. Semin Ultrasound CT MR. 2015;36(1):8–20.[↩]
- Lee EY, Restrepo R, Dillman JR, Ridge CA, Boiselle PM. Imaging evaluation of pediatric trachea and bronchi: systematic review and updates. Semin Roentgenol. 2012;47(2):182–96.[↩][↩]
- Shin SM, Kim WS, Cheon JE, Jung AY, Youn BJ, Kim IO, Yeon KM. CT in children with suspected residual foreign body in airway after bronchoscopy. AJR Am J Roentgenol. 2009;192(6):1744–51.[↩]
- Haller L, Barazzone-Argiroffo C, Vidal I, Corbelli R, Anooshiravani-Dumont M, Mornand A. Safely Decreasing Rigid Bronchoscopies for Foreign-Body Aspiration in Children: An Algorithm for the Emergency Department. Eur J Pediatr Surg. 2018 Jun;28(3):273-278.[↩]
- Carruth BR, Skinner JD. Feeding behaviors and other motor development in healthy children (2–24 months). J Am Coll Nutr. 2002;21(2):88–96[↩]
- Foltin GL, Tunik M, Cooper A, et al., eds. Teaching Resources for Instructors in Prehospital Pediatrics (TRIPP): Respiratory Emergencies. Vol 2.0. New York, NY: Center for Pediatric Emergency Medicine; 1988[↩]
- Hart A, Lee EY. Pediatric Chest Disorders: Practical Imaging Approach to Diagnosis. 2019 Feb 20. In: Hodler J, Kubik-Huch RA, von Schulthess GK, editors. Diseases of the Chest, Breast, Heart and Vessels 2019-2022: Diagnostic and Interventional Imaging [Internet]. Cham (CH): Springer; 2019. Chapter 10. Available from: https://www.ncbi.nlm.nih.gov/books/NBK553873[↩]
- Salih AM, Alfaki M, Alam-Elhuda DM. Airway foreign bodies: A critical review for a common pediatric emergency. World J Emerg Med. 2016;7(1):5-12.[↩][↩]
- Chapin MM, Rochette LM, Annest JL, Haileyesus T, Conner KA, Smith GA. Nonfatal choking on food among children 14 years or younger in the United States, 2001-2009. Pediatrics. 2013 Aug;132(2):275-81.[↩]
- Foltran F, Ballali S, Rodriguez H, Sebastian van As AB, Passali D, Gulati A, Gregori D. Inhaled foreign bodies in children: a global perspective on their epidemiological, clinical, and preventive aspects. Pediatr. Pulmonol. 2013 Apr;48(4):344-51.[↩][↩]
- Caliskan E, Aliyev S, Habibi HA, Bayramoglu Z, Yilmaz R, Adaletli I. Utility of lung radiodensity ratios in diagnosis of radiolucent foreign body aspiration in children: a practical approach. Clin Imaging. 2019 Mar – Apr;54:178-182.[↩]
- Johnson K, Linnaus M, Notrica D. Airway foreign bodies in pediatric patients: anatomic location of foreign body affects complications and outcomes. Pediatr. Surg. Int. 2017 Jan;33(1):59-64.[↩]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}