Cerebrotendinous xanthomatosis
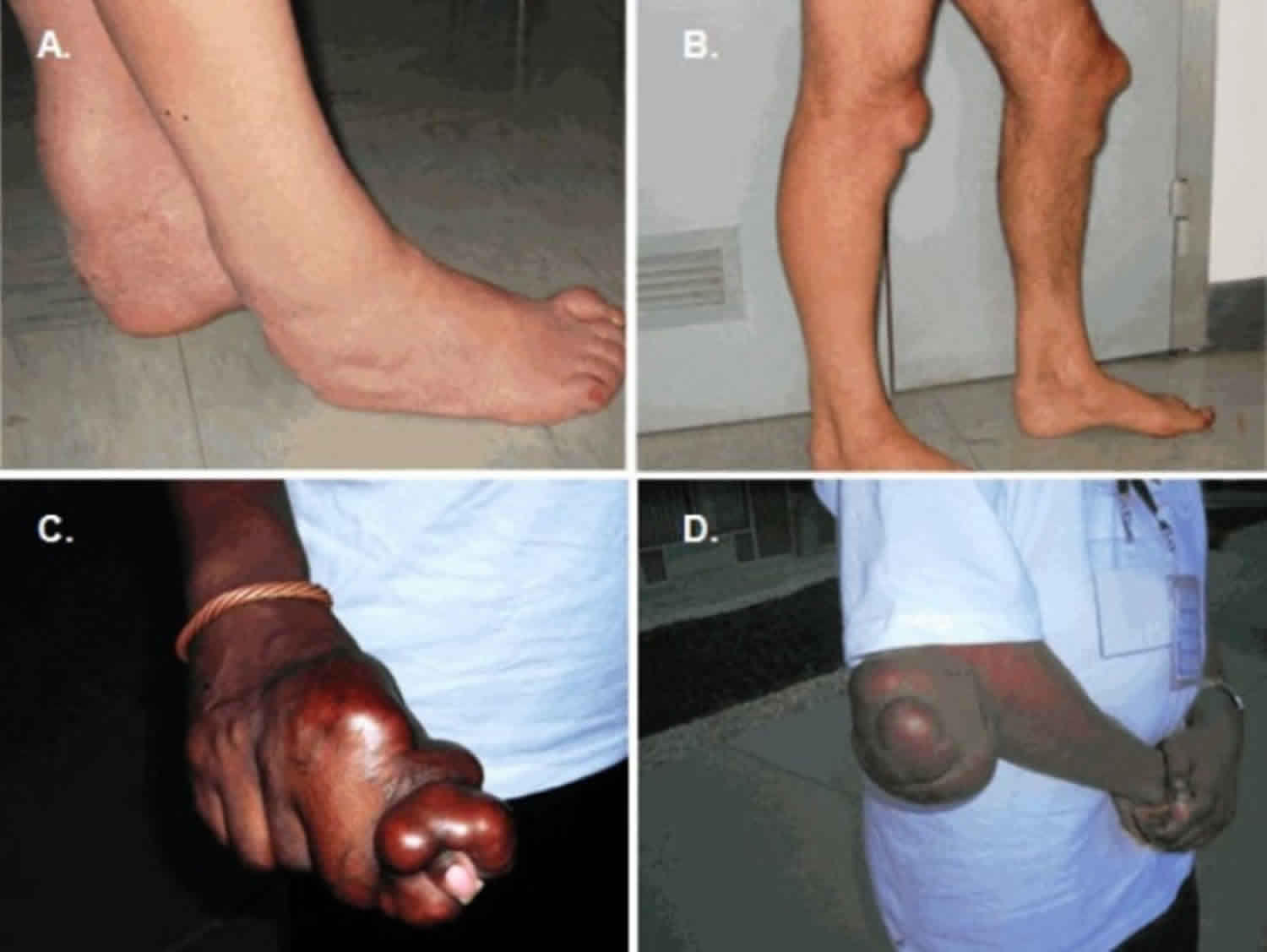
Cerebrotendinous xanthomatosis is a rare genetic metabolic disorder of cholesterol and bile acid metabolism that is characterized by abnormal storage of fats (lipids) in many areas of the body. People with cerebrotendinous xanthomatosis cannot break down certain lipids effectively, specifically different forms of cholesterol, so these fats accumulate in the body in the form of fatty yellow nodules called xanthomas. These xanthomas are most commonly found in the brain and in connective tissue called tendons that attach muscle to bone, which is reflected in the condition name (cerebro- meaning brain and -tendinous referring to tendons).
People with cerebrotendinous xanthomatosis often develop neurological problems in early adulthood that are thought to be caused by an abnormal accumulation of fats and an increasing number of xanthomas in the brain. These neurological problems include recurrent seizures (epilepsy), movement disorders, impaired speech (dysarthria), loss of sensation in the arms and legs (peripheral neuropathy), decline in intellectual function (dementia), hallucinations, and depression. Xanthomas can accumulate in the fatty substance that insulates and protects nerves (myelin), causing the destruction of myelin and disrupting nerve signaling in the brain. Degeneration (atrophy) of brain tissue caused by excess lipid deposits also contributes to the neurological problems.
Xanthomas in the tendons begin to form in early adulthood. The most common areas for xanthomas to develop are tendons in the hands, elbows, knees, neck, and in the Achilles tendon, which connects the heel of the foot to the calf muscles in the leg. Tendon xanthomas may cause discomfort and interfere with tendon flexibility. While many affected people develop tendon xanthomas, these nodules may not be easily visible underneath the skin.
Other features of cerebrotendinous xanthomatosis include clouding of the lenses of the eyes (cataracts) and chronic diarrhea in childhood; a reduced ability to produce and release a digestive fluid called bile (cholestasis), which can lead to a yellowing of the skin or whites of the eyes (jaundice); and progressively brittle bones that are prone to fracture (osteoporosis). People with cerebrotendinous xanthomatosis are also at an increased risk of developing cardiovascular disease or respiratory failure because of lipid accumulation in the heart or lungs, respectively. If untreated, the signs and symptoms related to cerebrotendinous xanthomatosis worsen over time; however, this condition varies greatly among those who are affected.
The incidence of cerebrotendinous xanthomatosis is estimated to be 1 per million individuals worldwide. Cerebrotendinous xanthomatosis is more common in the Moroccan Jewish population with an incidence of 1 in 108 individuals 1.
Cerebrotendinous xanthomatosis is caused by mutations in the CYP27A1 gene 2. And cerebrotendinous xanthomatosis is inherited in an autosomal recessive pattern 1.
Cerebrotendinous xanthomatosis may be treated with chenodeoxycholic acid (CDCA), which has been shown to normalize levels of cholestonal and improve neurologic symptoms. Inhibitors of HMG-CoA reductase may be used alone or in combination with CDCA. They are also effective in decreasing cholestanol concentration and improving clinical symptoms, however these treatments can lead to muscle damage. Coenzyme Q10 may improve muscle weakness, and cataract surgery may also be required 2.
Cerebrotendinous xanthomatosis causes
Mutations in the CYP27A1 gene located on the long arm (q) of chromosome 2 (2q35) cause cerebrotendinous xanthomatosis. The CYP27A1 gene provides instructions for producing an enzyme called sterol 27-hydroxylase. This enzyme works in the pathway that breaks down cholesterol to form acids used in the digestion of fats (bile acids), specifically a bile acid called chenodeoxycholic acid.
Mutations in the CYP27A1 gene lead to the production of a nonfunction or abnormal sterol 27-hydroxylase that cannot help form chenodeoxycholic acid. As a result, other molecules are formed by an alternative pathway. A molecule called cholestanol, which is similar to cholesterol, is produced as well as substances called bile alcohols. Cholestanol and bile alcohols are increased in the blood, while blood cholesterol levels are typically normal. In various tissues in the body, including the brain and heart, cholesterol and cholestanol levels are increased. These lipids make up much of the fats found in xanthomas. The accumulation of cholesterol and cholestanol throughout the body’s tissues causes the signs and symptoms of cerebrotendinous xanthomatosis.
Cerebrotendinous xanthomatosis inheritance pattern
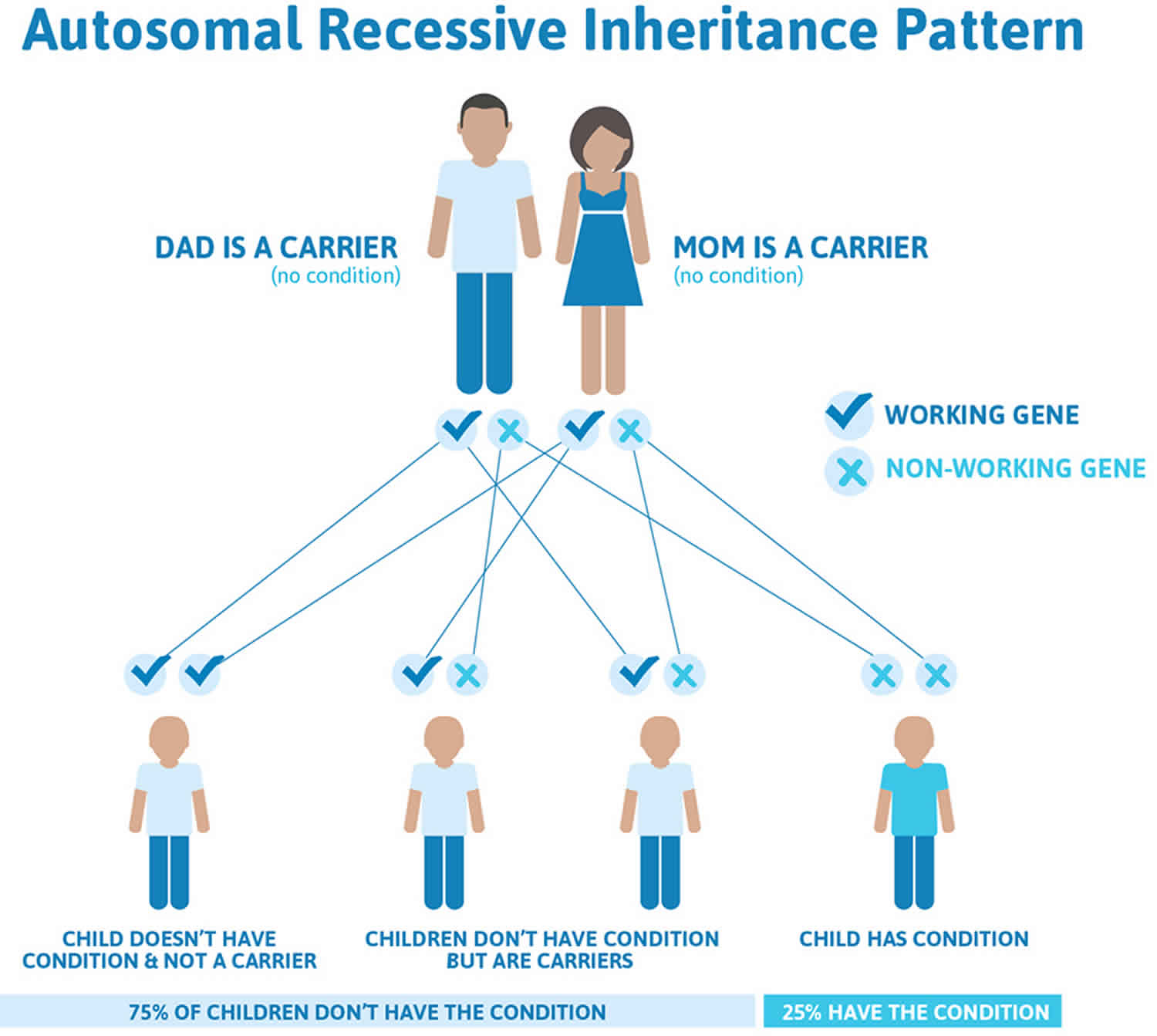
Cerebrotendinous xanthomatosis is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Cerebrotendinous xanthomatosis autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cerebrotendinous xanthomatosis symptoms
The presentation of cerebrotendinous xanthomatosis is highly variable and is associated with a wide range of potential abnormalities. The symptoms seen in cerebrotendinous xanthomatosis are listed below:
- Chronic diarrhea (infancy)
- Cataracts (early childhood)
- Mental impairment (infancy or at puberty)
- Xanthomas (adolescents to early adulthood)
- Dementia with slow deterioration in intellectual abilities (early adulthood)
- Spasticity (early adulthood)
- Cerebellar signs such as intention tremor, difficulty with fast hand movements, nystagmus, truncal ataxia, and rhomberg’s sign) (early adulthood)
- Behavioral changes (early adulthood)
- Hallucinations (early adulthood)
- Agitation (early adulthood)
- Aggression (early adulthood)
- Depression (early adulthood)
- Suicide attempt (early adulthood)
Other symptoms may include dystonia, atypical parkinsonism, seizures, and peripheral neuropathy.
Originally, cerebrotendinous xanthomatosis was believed only to be a neurological disorder of abnormal fat (lipid) storage not associated with liver disease. It is now known that cerebrotendinous xanthomatosis can occasionally present in childhood with cholestatic liver disease that can be severe or can be mild and resolve on its own in individuals who may later develop other complications of the disorder such as neurological disease. Cholestatic liver disease refers to the interruption or suppression of the flow of bile from the liver (cholestasis). Features of cholestasis include yellowing of the skin, mucous membranes and whites of the eyes (jaundice), failure to thrive, and growth deficiency. Enlargement of the liver (hepatomegaly) and/or spleen (splenomegaly) may also occur.
Generally, systemic symptoms develop earlier than neurologic symptoms. The first symptom may be chronic diarrhea in infancy. Diarrhea is often resistant to treatment (intractable). Infantile spasms have also been reported as a possible symptom. Cataracts in the first decades of life are the initial symptom in about 75% of those affected. Tendinous xanthomas (fatty tumors) appear in the second or third decade and can be located on the Achilles tendon, extensor tendons of the elbows and hands, and the knees.
Most affected individuals experience a decline in mental function beginning at puberty, but some show impairment beginning in childhood. Cognitive impairment can be mild to severe but becomes progressively worse without treatment. The mean age of diagnosis has been reported as between 35-37 years old at which time neurological involvement has often become significant and characteristic of cerebrotendinous xanthomatosis. Seizures and epilepsy have also been reported. Psychiatric abnormalities including behavioral changes, hallucinations, agitation, aggression, depression, and suicidal tendencies can also occur, although specific expression varies greatly. Increased muscle tone and stiffness (spasticity) can occur.
In some cases, additional neurologic findings may occur including impaired coordination of voluntary movements due to underdevelopment (hypoplasia) of the brain’s cerebellum (cerebellar ataxia); symptoms that resemble Parkinson disease (atypical parkinsonism); and dystonia, which is a general term for a large group of movement disorders that vary in their symptoms, causes, progression, and treatments. Dystonia is generally characterized by involuntary muscle contractions that force the body into abnormal, sometimes painful, movements and positions (postures). As the disorder progresses affected individuals can become incapacitated with motor dysfunction, and affected individuals may die prematurely due to advancing neurological deterioration.
Cardiovascular disease has been reported in individuals with cerebrotendinous xanthomatosis, although the exact prevalence of this finding is unknown. Hardening of the arteries (atherosclerosis) and coronary heart disease may occur. Additional symptoms that have been reported include underactivity of the thyroid (hypothyroidism) and skeletal abnormalities such as porous, brittle bones (osteoporosis) and a higher incidence of bone fractures.
Cerebrotendinous xanthomatosis diagnosis
Cerebrotendinous xanthomatosis is diagnosed based on a thorough clinical evaluation, a detailed patient and family history, identification of characteristic findings, and a variety of specialized tests including genetic testing and biochemical tests on blood and urine.
Molecular genetic testing can confirm a diagnosis of cerebrotendinous xanthomatosis by detecting mutations in the CYP27A1 gene known to cause the disorder. This type of testing can confirm the presence of mutations that have already been described in the literature to cause cerebrotendinous xanthomatosis. Sometimes novel (unknown) mutations are uncovered by genetic testing and in these cases biochemical testing will confirm the biochemical defect is present.
Certain specialized laboratories can conduct analysis to detect biochemical features that are indicative of cerebrotendinous xanthomatosis. Due to the nature of the biochemical defect, the cholestanol concentration in blood (or plasma derived from blood) is high, while the plasma cholesterol concentration is normal to low. In addition, the concentration of plasma bile acid precursors is high, and the concentration of bile alcohols in bile, urine and plasma is increased. Bile alcohols are formed in an abnormal pathway that generates some cholic acid in cerebrotendinous xanthomatosis. Due to the nature of the biochemical defect in cerebrotendinous xanthomatosis there is little or no formation of chenodeoxycholic acid.
Biochemical testing to measure plasma cholestanol is usually done through a procedure known as gas chromatography-mass spectrometry (GC-MS). Complex sample preparation and a lengthy analysis time make GC-MS testing a specialized and time-consuming technique.
Cerebrotendinous xanthomatosis is a candidate disorder to screen for in newborns. A faster testing technique than GC-MS is required to screen newborn dried bloodspots for cerebrotendinous xanthomatosis, such as liquid chromatography-tandem mass spectrometry (LC-MS/MS). Researchers have developed an LC-MS/MS test for cerebrotendinous xanthomatosis with potential to screen newborn dried bloodspots for the disorder. The LC-MS/MS test measures blood 7alpha, 12Alpha-dihydroxy-4-cholesten-3-one, a bile acid precursor that accumulates in cerebrotendinous xanthomatosis.
LC-MS/MS measurement of bile acid precursors may also be useful to discriminate between negative and cerebrotendinous xanthomatosis positive plasma samples, especially diagnostically challenging cerebrotendinous xanthomatosis positive samples with relatively low cholestanol concentrations. The LC-MS/MS test, with simple sample preparation and a rapid analysis time, can be performed by most laboratories, which can potentially provide wide availability of testing for cerebrotendinous xanthomatosis.
In some cases, specialized imaging techniques may include computerized tomography (CT) scanning of the head and magnetic resonance imaging (MRI) of the brain may assist in assessing disease progression in individuals suspected of cerebrotendinous xanthomatosis. During CT scanning, a computer and x-rays are used to create a film showing cross-sectional images of certain tissue structures. An MRI uses a magnetic field and radio waves to produce cross-sectional images of particular organs and bodily tissues. These tests may show cerebellar lesions and white matter damage in individuals with cerebrotendinous xanthomatosis.
Cerebrotendinous xanthomatosis treatment
Because oral bile acid replacement therapy can halt disease progression or prevent symptoms from occurring in asymptomatic individuals, early diagnosis of cerebrotendinous xanthomatosis is extremely important to prevent disease complications. Researchers have recently shown that cerebrotendinous xanthomatosis patients who started treatment after the age of 25 years had worse outcome and were significantly more limited in ambulation and more cognitively impaired than those that started treatment before the age of 25 years.
Successful long-term treatment of a number of affected individuals identified as children has been reported in the literature.
Treatment with chenodeoxycholic acid normalizes the production of cholestanol. The efficacy of treatment with chenodeoxycholic acid can be monitored with GC-MS testing to confirm a decrease in blood cholestanol. Treatment can prevent symptoms in asymptomatic individuals and stop the progression of disease symptoms in affected individuals. After significant disease progression, treatment does not readily reverse neurological deficits that have already occurred.
It may be effective to give a drug that inhibits HMG-CoA reductase, (an enzyme that plays a role in the creation of cholesterol in the liver) in conjunction with chenodeoxycholic acid. There are concerns that treatment with HMG-CoA reductase inhibitors (better known as statins) could boost the activity of receptors for low-density lipoprotein (LDL) cholesterol, thereby increasing cholesterol uptake and potentially worsening cerebrotendinous xanthomatosis. HMG-CoA reductase inhibitors can also cause muscle damage.
In 2009, the U.S. Food and Drug Administration (FDA) re-approved an artificially made (synthetic) form of chenodeoxycholic acid known as Chenodal® as a treatment for gallstones. However, this drug is also used as a first-line therapy to treat individuals with cerebrotendinous xanthomatosis. Chenodal received an orphan drug designation from the FDA for the treatment of cerebrotendinous xanthomatosis in the U.S. Chenodal is manufactured by Retrophin.
Cholic acid, another bile acid, has also been used to treat young children with cerebrotendinous xanthomatosis. However, cholic acid is generally not as effective as chenodeoxycholic acid, but does lack the potential toxic effects on the liver (hepatotoxicity) sometimes associated with chenodeoxycholic acid.
Genetic counseling will be of benefit for affected families and individuals. Additional treatment is symptomatic and supportive. For example, cataract surgery may be necessary before 50 years of age.
Cerebrotendinous xanthomatosis prognosis
If cerebrotendinous xanthomatosis is untreated, life expectancy is into the fifth and sixth decades; however, confirmed deaths have been reported as early as age 4 months. This is a progressive and terminal disease if left untreated. Treated patients may have a normal lifespan.
- Cerebrotendinous xanthomatosis. https://ghr.nlm.nih.gov/condition/cerebrotendinous-xanthomatosis[↩][↩]
- Federico A, Dotti MT, Gallus GN. Cerebrotendinous Xanthomatosis. 2003 Jul 16 [Updated 2016 Apr 14]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1409[↩][↩]




{kind=link}