Cooley’s anemia
Cooley’s anemia also called beta-thalassemia major, is the most severe form of beta-thalassemia in which the complete lack of beta protein in the hemoglobin causes a life-threatening anemia that requires regular blood transfusions and extensive ongoing medical care. The signs and symptoms of Cooley’s anemia (thalassemia major) appear within the first 2 years of life. Children develop life-threatening anemia. They do not gain weight and grow at the expected rate (failure to thrive) and may develop yellowing of the skin and whites of the eyes (jaundice). Affected individuals may have an enlarged liver and spleen (hepatosplenomegaly), and heart, and their bones may be misshapen. Some adolescents with Cooley’s anemia (thalassemia major) experience delayed puberty. In some cases, anemia is worsened if there is a nutritional deficiency such as with iron, folic acid or vitamin B12 1. Many people with thalassemia major have such severe symptoms that they need frequent blood transfusions to replenish their red blood cell supply. Over time, an influx of iron-containing hemoglobin from chronic blood transfusions can lead to a buildup of iron in the body (iron-overload), resulting in liver, heart, and hormone problems, which must be treated with iron chelation therapy to prevent early death from organ failure.
The incidence of children born with Cooley’s anemia (beta-thalassemia major) is about 68,000 worldwide 2. The prevalence rate is 1.5% (80-90 million carriers) across the globe. The exact prevalence rate in the United States is not known, however, it is speculated that the number is increasing dramatically due to immigration 2. In 1960, the prevalence rate was 5.6%, and it reached 13% in 2010, which was highest in California due to 26% of immigrant residents.
If Cooley’s anemia (thalassemia major) is not treated, it causes failure to thrive and a shortened life expectancy 3. Treatment involves regular blood transfusions and chelation therapy, aimed at reducing transfusion iron overload, allows for normal growth and development and may improve the overall prognosis. Treatment allows for normal growth and development. Bone marrow transplantation or cord blood transplantation may eliminate the need for regular treatment.
What is hemoglobin
Hemoglobin (Hb or Hgb) is the iron-containing protein found in all red blood cells (RBCs) that gives red blood cells their characteristic red color and it carries oxygen (O2) throughout the body. Hemoglobin enables red blood cells to bind to oxygen in the lungs and carry oxygen to tissues and organs throughout your body. Hemoglobin also helps transport a small portion of carbon dioxide (CO2), a product of cell metabolism, from tissues and organs to the lungs, where it is exhaled 4. Under normal circumstances, adult blood contains 4 types of hemoglobin: oxygenated hemoglobin or oxyhemoglobin, hemoglobin that contains no oxygen (reduced hemoglobin) or deoxyhemoglobin, hemoglobin transporting carbon dioxide or carboxyhemoglobin, and oxidized hemoglobin or methemoglobin. The last 2 are usually found in small amounts in blood.
Hemoglobin is made up of heme, which is the iron-containing portion, and globin chains, which are proteins. The globin protein consists of chains of amino acids, the “building blocks” of proteins. One portion of hemoglobin called heme is the molecule with iron at the center (see Figure 1). Another portion is made of up four protein chains called globins. Depending on their structure, the globin chains are designated as alpha (α), beta (ß), delta (δ), and gamma (γ). Each of the four globin chains holds a heme group containing one iron atom. .
Not all hemoglobin is the same. Different types of hemoglobin are classified according to the type of globin chains they contain. The type of globin chains present is important in hemoglobin’s ability to transport oxygen.
Normal hemoglobin types include:
- Hemoglobin A (Hb A) – this is the predominant type of hemoglobin (Hb) in adults, makes up about 95%-98% of hemoglobin found in adults; Hb A contains two alpha (α) protein chains and two beta (ß) protein chains.
- Hemoglobin A2 (Hb A2) – makes up about 2-3.5% of hemoglobin (Hb) found in adults; it has two alpha (α) and two delta (δ) protein chains.
- Hemoglobin F (Hb F, fetal hemoglobin) – makes up to 1%-2% of hemoglobin (Hb) found in adults; it has two alpha (α) and two gamma (γ) protein chains. Hemoglobin F (Hb F, fetal hemoglobin) is the primary hemoglobin produced by a developing baby (fetus) during pregnancy. Its production usually falls to a low level within a year after after birth and reaches adult level within 1-2 years.
For hemoglobin (Hb), there are four genes in your DNA that code for the alpha (α) globin chains and two genes (each) for the beta (β), delta (δ), and gamma (γ) globin chains. Globin polypeptides are synthesized from separate α-like and β-like globin gene clusters located on human chromosomes 16 and 11, respectively. Since everyone inherits a set of chromosomes from each parent, each person inherits two alpha globulin genes and one beta globulin gene from each parent. A person may inherit mutations in either the alpha or beta globin genes.
The hemoglobin test measures the amount of hemoglobin in a person’s sample of blood. Serum free hemoglobin is a blood test that measures the level of free hemoglobin in the liquid part of the blood (the serum). Free hemoglobin is the hemoglobin outside of the red blood cells. Most of the hemoglobin is found inside the red blood cells, not in the serum. A hemoglobin level can be performed alone or with a hematocrit, a test that measures the proportion of blood that is made up of red blood cells, to quickly evaluate an individual’s red blood cells. Red blood cells, which make up about 40% (ranging 37-49%) of the blood’s volume, are produced in the bone marrow and are released into the bloodstream when they are, or nearly are, mature. The typical lifespan of an red blood cell is 120 days, and the bone marrow must continually produce new red blood cells to replace those that age and degrade or are lost through bleeding.
Several diseases and conditions can affect red blood cells and consequently the level of hemoglobin in the blood. In general, the hemoglobin level and hematocrit rise when the number of red blood cells increases. The hemoglobin level and hematocrit fall to less than normal when there is a drop in production of red blood cells by the bone marrow, an increase in the destruction of red blood cells, or if blood is lost due to bleeding. A drop in the red blood cell count, hemoglobin and hematocrit can result in anemia, a condition in which tissues and organs in the body do not get enough oxygen, causing fatigue and weakness. If too many red blood cells are produced, polycythemia results and the blood can become thickened, causing sluggish blood flow and related problems.
Since a hemoglobin level is often performed as part of a complete blood count (CBC), results from other components are taken into consideration. A rise or drop in the hemoglobin level must be interpreted in conjunction with other parameters, such as red blood cell count, hematocrit, reticulocyte count, and/or red blood cell indices. Age, sex, and race are other factors to be considered.
Figure 1. Red blood cell hemoglobin
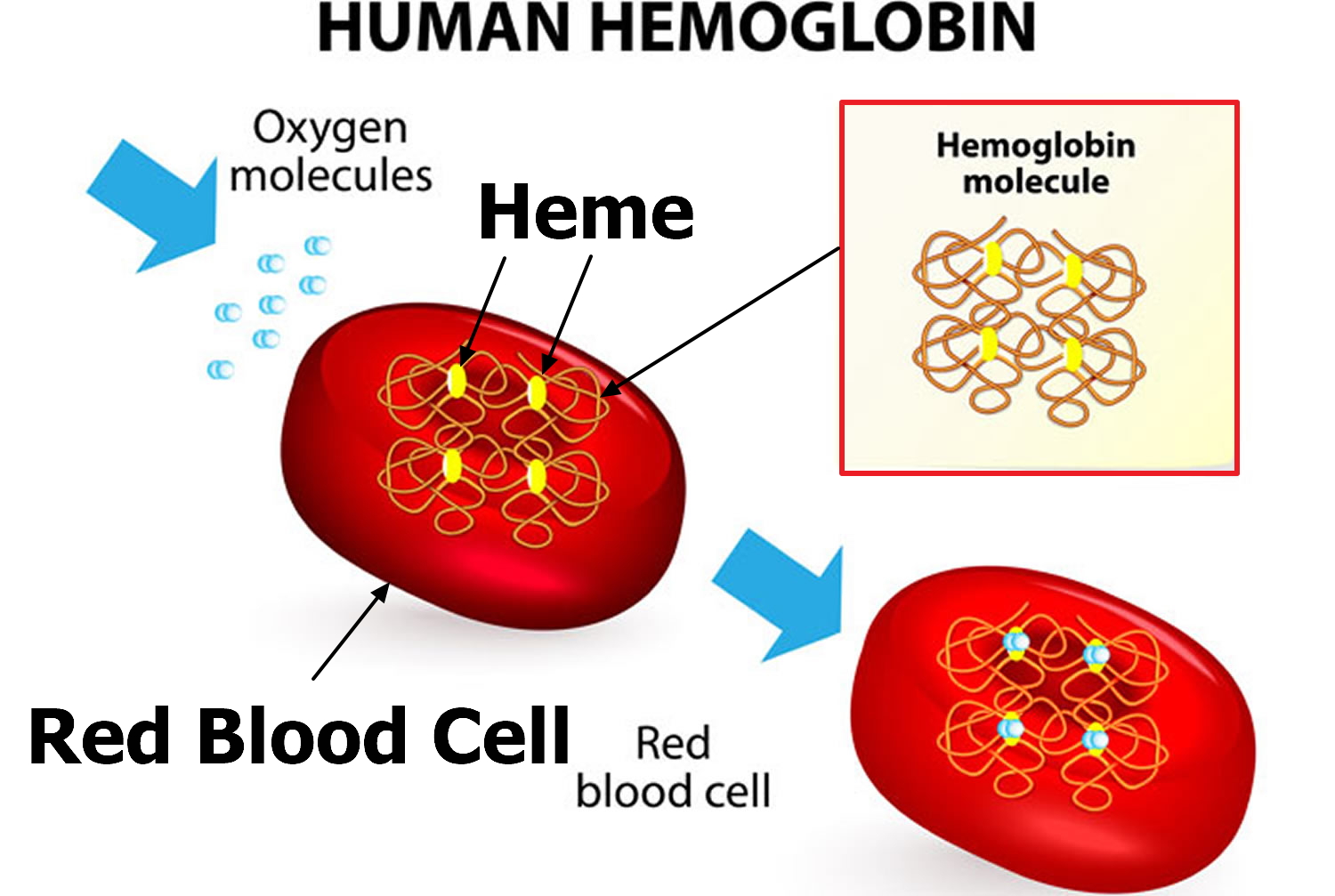
Figure 2. Structure of hemoglobin
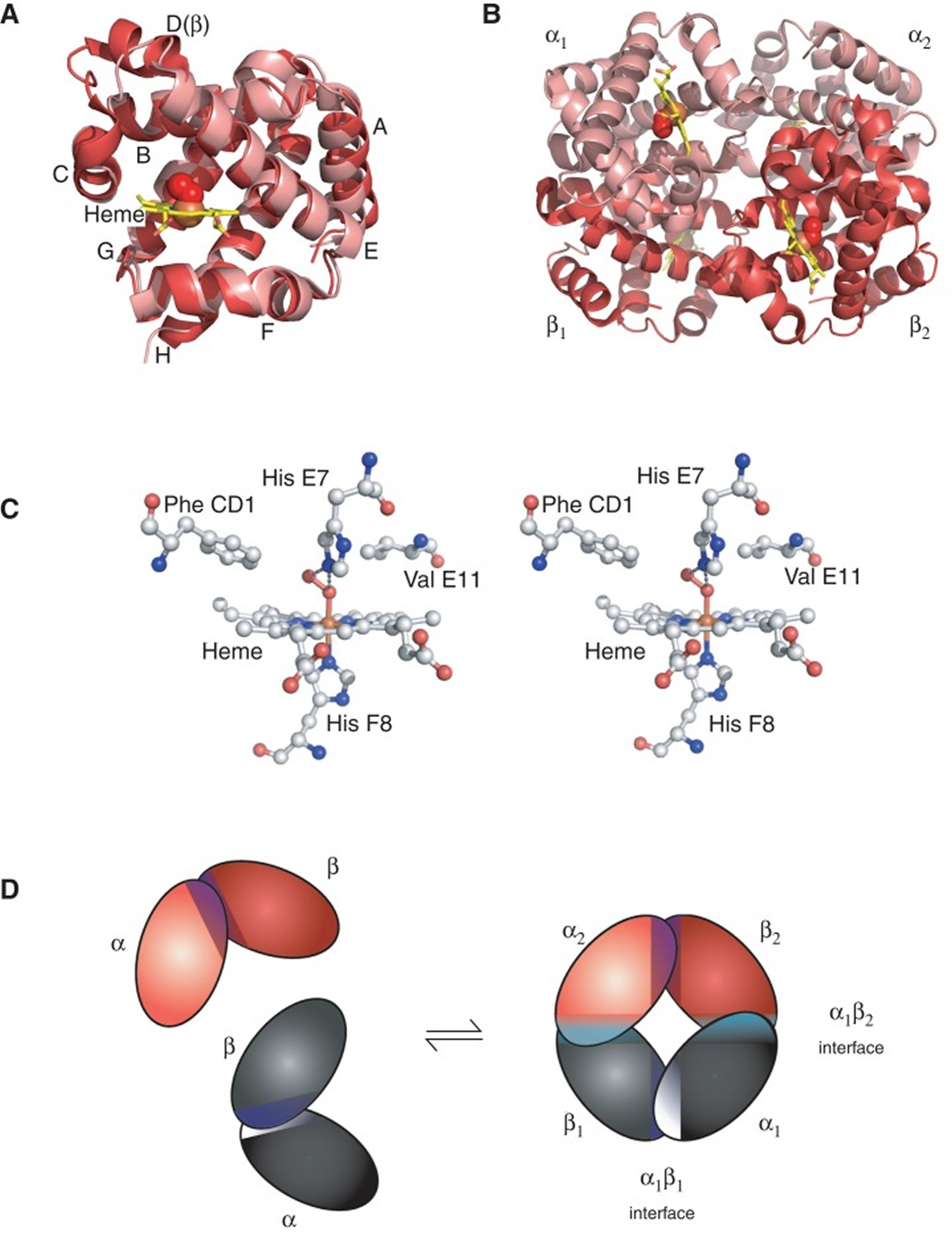
Footnotes: The structure of hemoglobin (Hb). (A) The α (pink) and β (red) hemoglobin (Hb) subunits have conserved α-helical folds. Helices are labeled A–H from the amino terminus. The α subunit lacks helix D. (B) The high O2 affinity R state quaternary structure of hemoglobin (Hb) with O2 (red spheres) bound at all four heme sites (protoporphyrin-IX as yellow sticks, with central iron atom as orange sphere). (C) Stereo (wall-eye) diagram of the heme pocket of β showing the proximal (F8) and distal (E7) histidines and selected residues in the distal heme pocket that influence ligand binding and autoxidation. (D) hemoglobin (Hb) tetramer is assembled from two identical αβ dimers (shown in red and gray for clarity). In the tetramer, each subunit makes contact with the unlike chain through a high affinity dimerization α1β1 interface and a lower affinity α1β2 dimer–tetramer interface (cyan).
Cooley’s anemia causes
Beta-thalassemia is caused by mutations in the hemoglobin subunit beta (HBB) gene and is typically inherited in an autosomal recessive manner. The HBB gene provides instructions for making a protein called beta-globin. Beta-globin is a component (subunit) of hemoglobin. Hemoglobin consists of four protein subunits, typically two subunits of beta-globin and two subunits of another protein called alpha-globin. This means that people with Cooley’s anemia (thalassemia major) or thalassemia intermedia (the less severe form) have a mutation in both of their copies of the HBB gene. People who have only one HBB gene mutation (carriers) typically are said to have thalassemia minor (or trait) and usually do not have symptoms, but may have some symptoms of anemia 3. Very rarely, the inheritance of beta-thalassemia may be dominant. In this case, a person has only one mutated HBB gene, but has signs and symptoms of beta-thalassemia major or beta-thalassemia intermedia 5.
Some mutations in the HBB gene prevent the production of any beta-globin. The absence of beta-globin is referred to as beta-zero (β0) thalassemia. Other HBB gene mutations allow some beta-globin to be produced but in reduced amounts. A reduced amount of beta-globin is called beta-plus (β+) thalassemia. Having either β0 or β+ thalassemia does not necessarily predict disease severity, however; people with both types have been diagnosed with thalassemia major and thalassemia intermedia.
A lack of beta-globin leads to a reduced amount of functional hemoglobin. Without sufficient hemoglobin, red blood cells do not develop normally, causing a shortage of mature red blood cells (ineffective erythropoiesis). The low number of mature red blood cells leads to anemia and other associated health problems in people with beta thalassemia.
Cooley’s anemia inheritance pattern
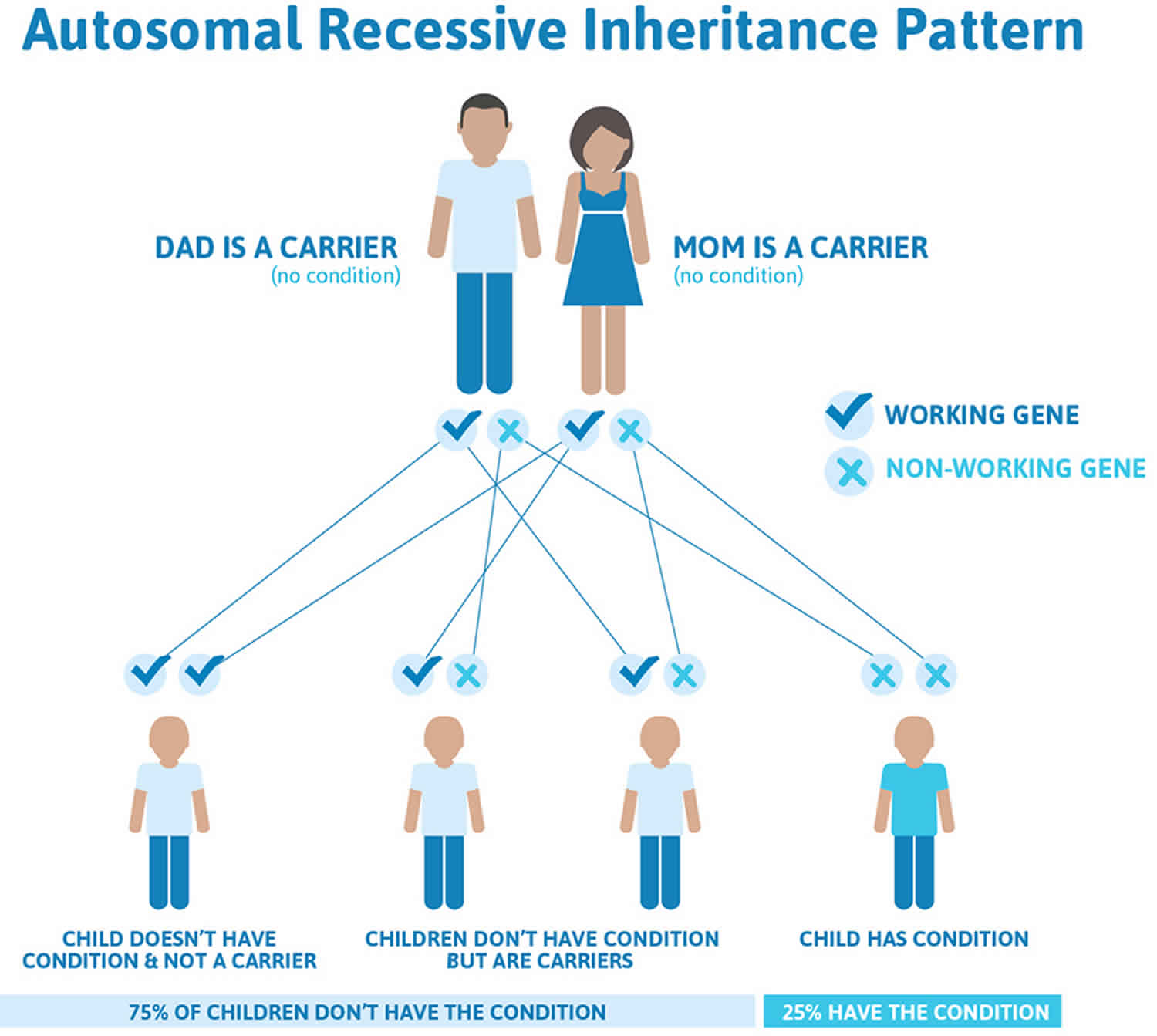
Cooley’s anemia (thalassemia major) and beta-thalassemia intermedia are usually inherited in an autosomal recessive manner, which means both copies of the HBB gene in each cell have mutations. The parents of a person with an autosomal recessive condition each carry one copy of the mutated gene and are referred to as carriers. When two carriers have children, each child has a 25% (1 in 4) chance to be affected, a 50% (1 in 2) chance to be a carrier like each parent, and a 25% (1 in 4) chance to be unaffected and not a carrier. Sometimes, people with only one HBB gene mutation in each cell (carriers) do have mild anemia. These people are said to have ‘beta-thalassemia minor’ or ‘beta-thalassemia trait’ 6.
In a small percentage of families, the condition is inherited in an autosomal dominant manner. In these cases, one mutated copy of the gene in each cell is enough to cause the signs and symptoms of beta-thalassemia 6.
Figure 3. Cooley’s anemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Cooley’s anemia symptoms
Patients with Cooley’s anemia (beta-thalassaemia major) are healthy until the age of around 6 months due to the transition from fetal hemoglobin (HbF) to adult hemoglobin (HbA) and then become very pale due to profound anemia 2. Fetal hemoglobin (HbF) is the primary hemoglobin until six months of age and consists of two alpha chains and two gamma chains. Adult hemoglobin is primarily hemoglobin A (HbA), consisting of two alpha chains and two beta chains. Without treatment, a child grows slowly (failure to thrive), presents with bone deformations, shortness of breath, irritability, and hepatosplenomegaly due to extramedullary hematopoiesis. Other features include recurrent fever, feeding problems, diarrhea and abdominal distention. Patients who are either untreated or received poor transfusion (especially in developing countries where there is no availability of well-developed transfusion programs) develop a wide range of symptoms such as retarded growth, brown discoloration of the skin, jaundice, leg ulcers, knock knee (genu valgum), and skeletal deformities such as long bone abnormalities, craniofacial deformities like frontal bossing, prominent cheekbones, depressed nasal bridge, and maxillary hypertrophy resulting from bone marrow expansion giving the chipmunk appearance 7. In addition to that, gallstone formation usually occurs due to chronic hemolysis 8.
Cooley’s anemia complications
Patients should be closely monitoring for potential complications owing to ineffective erythropoiesis, bone marrow overstimulation, and blood transfusion. The complications of the transfusion are of two types.
- Acute (e.g., severe allergic reactions, septic shock, hemolysis), and
- Chronic (e.g., diseases associated with iron overload (i.e., cardiomyopathy, endocrinopathy, and hepatotoxicity), alloimmunization, and transfusion-related infections.
Transfusion-related complications:
- Iron overload: It is the primary chronic complication associated with long-term red blood cell transfusion in beta-thalassemia patients. Transfused red blood cells contain the iron load. Iron accumulates over time, and when the body’s excess iron eliminating function is overwhelmed, iron starts depositing in various organs, commonly heart, pancreas, liver, and brain. Cardiac failure secondary to cardiac iron overload is the leading cause of death in transfusion-dependent thalassemia patients. Usually, the patient dies at a young age (<20) if not promptly addressed. Iron overload in the liver results in liver fibrosis, cirrhosis, and eventually hepatocellular carcinoma. Furthermore, deposition of iron leads to diabetes, hypogonadism, hypothyroidism, and hypoparathyroidism secondary to pituitary gland disruption. Also, it can manifest as bronze skin, growth, and developmental delay. Therefore, iron chelation therapy is essential in preventing these complications.
- Alloimmunizations: It is the production of antibodies against specific red blood cell antigens or alloantigen (e.g., ABO, Rh, C, c, E, e, D, and Kell antigens) resulting from chronic transfusion. As per international consensus recommendation, donated blood should be screened and crossmatched, and thalassemia patients should receive antigen-compatible blood to avoid alloimmunization.
- Transfusion-related infections: Viral, bacterial, and parasitic infections are common transfusion-related infections. Donated blood should be screened for hepatitis B virus (HBV), hepatitis C virus (HCV), and HIV as per World Health Organization (WHO) and Thalassemia International Federation guidelines. Screening for endemic infections is also recommended in some countries.
- Thrombosis or hypercoagulable state: Hypercoagulability is seen in patients with Cooley’s anemia (beta-thalassaemia major) and beta-thalassemia intermedia who are either poorly transfused, non-transfused, and splenectomized individuals.
- Others include: Transfusion-related acute lung injury (TRALI), febrile non-hemolytic transfusion reaction, and delayed hemolytic transfusion reactions 8.
Cooley’s anemia diagnosis
In infants and children under 2 years of age, the diagnosis of Cooley’s anemia (beta-thalassaemia major) is suspected based on clinical features of jaundice, severe hypochromic microcytic anemia, and hepatosplenomegaly.
Lab findings
Complete blood count (CBC) will show microcytic anemia with decreased hemoglobin level of <7g/dL, mean corpuscular volume (MCV) ranges from 50 to 70 fl, and mean corpuscular hemoglobin (MCH) level ranges from 12 to 20 pg.
Morphological changes of red blood cells like microcytosis, hypochromia, poikilocytosis, anisocytosis, and nucleated red blood cells can be seen on the peripheral blood smear. Additionally, target cells, teardrop cells, and basophilic stippling (course type) can also be visualized on the peripheral blood smear. The number of erythroblasts depends on the degree of anemia and after splenectomy. Moreover, either hemoglobin electrophoresis (a lab procedure that differentiates the types of hemoglobin present) or high-performance liquid chromatography (HPLC) is warranted for definitive diagnosis. It analyzes hemoglobin both quantitatively and qualitatively. Cooley’s anemia (beta-thalassaemia major) demonstrates a complete absence of beta-globin chain (beta0/beta0), therefore the HbF is 92 to 95%, HbA2 is 5 to 8%, and HbA 0%. Beta-thalassemia major typically shows markedly elevated HbF (30-to-greater than 95%) with normal to mildly elevated HbA2. The distinction between beta-thalassemia major and intermedia is a clinical one and does not have diagnostic laboratory findings 8.
Elevated HbA2 is also caused by hyperthyroidism, vitamin B12 and folic acid deficiency, and anti-retroviral therapy (ART). Apart from that, prenatal screening and genetic counseling are additional valuable tools in the detection and prevention of beta-thalassemia and other autosomal recessive diseases.
Radiographic features
Due to active bone marrow hyperplasia, wide medullary spaces, and trabeculae mimic, a classic “chicken wire” pattern is seen on maxillofacial radiographs. On x-rays of long bones, osteopenia, and cortical thinning secondary to expanded bone marrow spaces can be seen, which often presents with pathological fractures. Similarly, the classic “hair on end” appearance seen on a cranial vault radiograph. Additionally, vertebral bodies have a ground-glass appearance. MRI of the brain, heart, and liver are also done to determine iron deposition in various tissues in cases of suspected iron overload 7.
Cooley’s anemia treatment
Blood transfusion has been the mainstay treatment of Cooley’s anemia (beta-thalassaemia major). Usually, the patient dies before reaching puberty if he/she does not receive periodic blood transfusions. It provides normal red blood cells that compensate for anemia, halts ineffective erythropoiesis, which results in decreased oxidative stress, thus reducing bone deformities, improved symptoms, and increased survival. There is general consensus between the Thalassemia International Federation and other national guidelines (the United Kingdom and the United States) about thalassemia management despite few differences in terms of iron overload and iron chelation therapy strategies 2.
The degree of anemia and/or clinical findings should be considered when the frequency of blood transfusions is decided and leuko-reduced red blood cells are preferred. To accelerate growth and minimize symptoms, the pretransfusion goal hemoglobin (Hb) level is 9-10g/dL or if clinical complications are present irrespective of hemoglobin (Hb) level, and the post-transfusion is between 14-15g/dL. A higher pretransfusion level of 10-12g/dL is set in cases of a worsening of extramedullary hematopoiesis or cardiac abnormalities. However, care should be taken during regular blood transfusion because of the high risk of iron overload, infections, transfusion reactions, and allogeneic antibody formation.
Clinical features and serum ferritin are the reliable tools in close monitoring of iron overload in routinely transfused Cooley’s anemia (beta-thalassaemia major) patients. Iron chelation therapy is the treatment of choice for iron overload. Deferoxamine was the first to be used via subcutaneous (SC) or intravenously (IV) route over 8 to 24 hrs, 5 to 7 nights/week. Compliance was the main issue with deferoxamine. Currently, oral iron chelation therapy such as deferasirox or deferiprone is widely used either alone or in combination and is well tolerated. Due to rapid cell turnover, other nutritional deficiencies such as folate and Vitamin B12, and calcium need to be addressed as well.
No added benefit of hydroxyurea has been established in patients receiving adequate blood to achieve the pretransfusion goal. Unlike sickle cell disease (in which it reduces sickling by increasing HbF level), the role of hydroxyurea is unclear in Hb <9 g/dL.
The only definitive treatment at present is allogeneic hematopoietic stem cell transplant (Allo-HSCT) in transfusion dependant patients with >90% success rate if it takes place before the iron overload related damage occurs.
Luspatercept
Luspatercept is an erythroid maturation agent that has been approved for anemia in adult Cooley’s anemia (beta-thalassaemia major) patients who require regular blood transfusions. Recently an open-label phase 3 study (BELIEVE) of adult thalassemia patients, luspatercept has yielded promising results in terms of transfusion independence.
Gene therapy is also in the pipeline to be considered a potential cure for beta-thalassemia and sickle cell disease. Several clinical trials are currently underway to establish its efficacy and safety. However, preliminary results of one of the recent ongoing trials have been promising. A total of 3 beta-thalassemia patients (n=3) have been transduced with autologous CD34 hematopoietic stem cells using GLOBE or TNS9.3.55 as lentivirus and have demonstrated minimal and reversible adverse effects. However, to further validate its efficacy and safety, more data is warranted.
Surgical treatment
Cooley’s anemia (beta-thalassaemia major) patients can develop hypersplenism and massive splenomegaly, therefore, they require splenectomy as well as vaccination against encapsulated bacteria due to the high risk of infections following splenectomy.
Follow up
In patients with Cooley’s anemia (beta-thalassaemia major), follow up is mandatory to monitor transfusion therapy response and iron chelation therapy side effects. It is recommended a physical exam is performed every month, assessment of liver function tests every two months, serum ferritin every three months, growth and development every six months in pediatric patients, ophthalmologic, and audiology examinations. In patients 10 years of age and older, a complete cardiac evaluation, thyroid, parathyroid endocrine pancreas, adrenal and pituitary function evaluations, liver ultrasound, serum alpha-fetoprotein (AFP) for early detection of hepatocarcinoma, and bone densitometry for osteoporosis are recommended to be performed annually 9. MRI of the liver and heart to detect iron overload and repeated according to the severity of iron overload. In patients with Gilbert syndrome genotype, is recommended regular gallbladder echography for early detection of cholelithiasis 10.
Cooley’s anemia prognosis
Prior to 2000, Cooley’s anemia (beta-thalassaemia major) had a poor prognosis. The median survival was approximately 17 years in the US between 1965 to 1975. In the United Kingdom, approximately 50% of Cooley’s anemia (beta-thalassaemia major) patients died before age 35 years 11. The median age was 12 years for the cohort born in Italy in the 1960s. But now, life expectancy has dramatically improved due to a multidisciplinary approach among health care providers. Furthermore, the introduction of iron chelation therapy (oral and injectable), awareness among patients and health care providers, management of complications, and utilization of standard techniques in the screening of blood products has contributed to improved outcomes 12. The life span of Cooley’s anemia (beta-thalassaemia major) patients in Palestine has increased from 7 to 8 years in 1996 to 19 to 20 years in 2015. Similarly, in the UK, the mortality rate significantly dropped to 4.7/1000 in the years 2000 to 2003 from 12.7/1000 in the years 1990 to 1999 8.
- Choudhry VP. Thalassemia Minor and Major: Current Management. Indian J Pediatr. 2017 Aug;84(8):607-611. doi: 10.1007/s12098-017-2325-1. Epub 2017 Apr 24.[↩]
- Khan I, Shaikh H. Cooley Anemia. [Updated 2020 Jun 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK557522[↩][↩][↩][↩]
- Origa R. Beta-Thalassemia. 2000 Sep 28 [Updated 2018 Jan 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1426[↩][↩]
- Thom CS, Dickson CF, Gell DA, Weiss MJ. Hemoglobin Variants: Biochemical Properties and Clinical Correlates. Cold Spring Harbor Perspectives in Medicine. 2013;3(3):a011858. doi:10.1101/cshperspect.a011858. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3579210/[↩]
- Khera R, Singh T. Dominant β-thalassemia – A rare entity!. Indian J Pathol Microbiol 2012;55:422-3[↩]
- Beta thalassemia. https://ghr.nlm.nih.gov/condition/beta-thalassemia[↩][↩]
- Origa R. β-Thalassemia. Genet Med. 2017 Jun;19(6):609-619. doi: 10.1038/gim.2016.173. Epub 2016 Nov 3.[↩][↩]
- Needs T, Gonzalez-Mosquera LF, Lynch DT. Beta Thalassemia. [Updated 2020 May 23]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531481[↩][↩][↩][↩]
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010 Feb;12(2):61-76. doi: 10.1097/GIM.0b013e3181cd68ed [↩]
- Wood JC, Origa R, Agus A, Matta G, Coates TD, Galanello R. Onset of cardiac iron loading in pediatric patients with thalassemia major. Haematologica. 2008 Jun;93(6):917-20.[↩]
- Modell B, Khan M, Darlison M. Survival in beta-thalassaemia major in the UK: data from the UK Thalassaemia Register. Lancet. 2000 Jun 10;355(9220):2051-2.[↩]
- Rund D. Thalassemia 2016: Modern medicine battles an ancient disease. Am. J. Hematol. 2016 Jan;91(1):15-21.[↩]





{kind=link}