Epithelioid sarcoma
Epithelioid sarcoma is a rare soft tissue sarcoma (high-grade malignant tumor of the soft tissue) that mimics granulomatous disease, carcinoma, and synovial sarcoma 1. Epithelioid sarcoma, a rare and highly malignant soft tissue tumor, shows a high tendency for local recurrence, regional lymph node involvement, and distant metastases 1. Epithelioid sarcoma most often starts in tissues under the skin of a finger, hands, forearms, feet, or lower legs, though it can start in other areas of the body. Teens and young adults are often affected 2. Typically, epithelioid sarcoma starts as a painless, slow-growing soft tissue swelling in the distal extremity of young adult males 3. It usually starts out as a single growth, but multiple growths may occur by the time a person seeks medical help. Fifty-four percent of cases arise in the distal upper extremities 4. Pain or tenderness is rarely a prominent symptom, unless large nerves are affected by the tumor. epithelioid sarcoma is commonly seen in young patients; the peak incidence is in the third decade of life, and it is rare in children and the elderly.
Sometimes epithelioid sarcoma appears as ulcers that don’t heal, looking like open wounds over the growths. The classic form (distal-type) of epithelioid sarcoma mainly occurs in teenagers and young adults. A rarer form, called large-cell (proximal-type) epithelioid sarcoma, tends to be more aggressive and mainly affects adults 5. Epithelioid sarcoma is locally invasive and frequently metastasizes to regional lymph nodes and distant sites, most commonly to the lungs. Complete surgical resection is curative in low-stage disease; however, a risk of recurrence and late metastasis remains 6. So if epithelioid sarcoma is suspected, it’s best to seek care at a comprehensive cancer center that sees more cases of soft tissue sarcoma to ensure proper diagnosis and treatment, and to help prevent the cancer from recurring.
Recurrence and metastasis are reported in 77 % and 45 % of patients with epithelioid sarcoma, respectively. The most common sites of metastasis are the lungs (51 %), regional lymph nodes (34 %), scalp (22 %), and bone (13 %) 4. Epithelioid sarcoma is highly malignant; 32 % of patients die as a result of the disease 7. Male sex, proximal location, large tumor size (greater than 5 cm), deep location, high mitotic index, hemorrhage, necrosis, vascular invasion, and inadequate initial excision causing local recurrence have been shown to be poor prognostic factors 8.
Epithelioid sarcomas are rare, representing less than 1% of soft tissue sarcomas 6 and have a predilection for men (up to 2:1 male to female ratio) 9. Most reported tumors occur in young males ranging in age from 10 to 45 years 6. The extremes of ages include ages 4 to 90, with a median age of 27 years 9.
Epithelioid sarcomas are tumors of purportedly mesenchymal origin that show ultrastructural and immunophenotypic evidence of epithelial differentiation 10. The mixed differentiation of epithelioid sarcoma can make the differential diagnosis challenging from a histopathologic perspective. The differential diagnosis is narrowed by the somewhat unique epithelioid sarcoma immunophenotype expressing cytokeratin, epithelial membrane antigen, and CD34. Epithelioid sarcoma is one of only a few tumors that characteristically lacks integrase interactor-1 (INI-1) or SMARCB1 (SWI/SNF Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily B, Member 1) expression 3.
After definitive tissue-based diagnosis, treatment depends on the presence or absence of metastatic disease. Metastatic disease should be ruled out based on a thorough clinical and radiological examination, particularly focused on regional lymph nodes and pulmonary examination. Ultrasound scanning of regional lymph nodes and sentinel lymph node biopsy is recommended to improve metastatic workup accuracy 6.
Surgical resection of primary tumors with no metastases can achieve a cure if the primary tumor is amenable to complete resection with no residual tumor 10. Radiation therapy is often added to mitigate local recurrence; however, the use of radiation therapy is relatively undefined 11. Surgery with curative intent still carries a risk of recurrence and late discovery of metastases. Inoperable tumors, incomplete resections, and metastatic disease can be offered palliative measures such as radiation therapy 11.
Figure 1. Epithelioid sarcoma
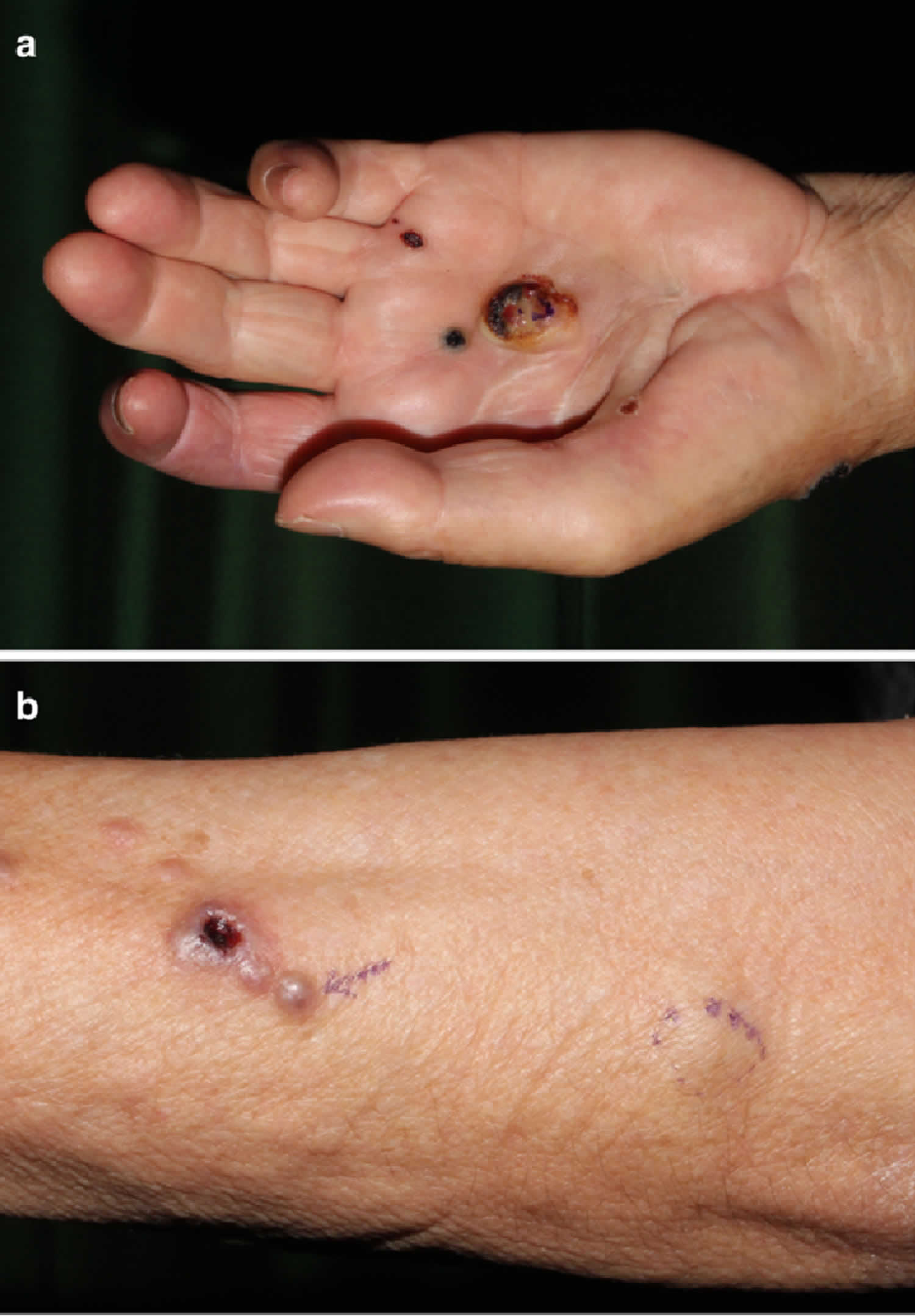
Footnote: (a) A 2-cm ulcerated nodule on the left hand. (b) Multiple lesions on the patient’s left forearm
[Source 12 ]Proximal type epithelioid sarcoma
A new subtype, a proximal variant of epithelioid sarcoma, has been proposed 13. This subtype exhibits more aggressive clinical behavior, and its histopathological appearance is distinct. Clinically, the proximal subtype differs from the classical type by its multinodular growth pattern, its more frequent occurrence in older patients, its more proximal/axial distribution, its more deep-seated location, and its more aggressive clinical behavior from the outset. Histologically, it is characterized by predominantly large cells, epithelioid cytomorphology, marked cytological atypia, the frequent manifestation of rhabdoid features, and the absence of a granuloma-like pattern in most patients. On the basis of our clinical and histological findings, we considered the epithelioid sarcoma in our patient to be of the conventional distal type.
Epithelioid sarcoma cause
Epithelioid sarcomas are malignant tumors with mixed differentiation toward both mesenchymal and epithelial cell types. Up to 90% of epithelioid sarcomas show loss of integrase interactor-1 (INI-1) expression 14. INI-1 is part of the SWI/SNF chromatin remodeling complex expressed in all normal nucleated cells. This chromatin remodeling complex is essential to biological function by altering nucleosomes to allow for transcription of DNA. INI-1, also commonly known as SMARCB1, is on chromosome 22 at the 22q11 band. A spectrum of gene deletions and rearrangements including translocation events involving the 22q11 locus are putatively responsible for tumorigenesis in INI-1 deficient epithelioid sarcomas 15. Intriguingly, an association with prior trauma at the site of the tumor has been noted in up to 27% of occurrences 10. Prior trauma to the distal extremity, however, is logically an all too common of an occurrence for further speculation as to an inciting event.
Epithelioid sarcoma pathophysiology
Epithelioid sarcomas have no definitive cell of origin but are considered to be true mesenchymal cells that show epithelial differentiation. This is based on ultrastructural studies that show the malignant cells to resemble mesenchymal cells closely, often with myofibroblastic characteristics 10. The proximal-type variant of epithelioid sarcoma, which often microscopically has rhabdoid morphology, ultrastructurally shows perinuclear whorls of intermediate filaments consistent with at least some degree of rhabdomyocyte differentiation 16. The immunohistochemical staining profile, discussed in more detail below, also supports bi-differentiation toward mesenchymal and epithelial lines. The tumor has a unique immunophenotype, showing strong expression of the epithelial markers cytokeratin and epithelial membrane antigen (EMA) as well as positive labeling with the mesenchymal markers CD34 and vimentin.
Epithelioid sarcoma symptoms
Epithelioid sarcoma most often presents as a distal upper-extremity multinodular swelling that is slow-growing, painless, and sometimes ulcerates 6. The lesion can be centered in the dermis, subcutis, or deep fascia. It characteristically invades along fascial planes and tracks along tendons and aponeuroses 6. A proximal type of epithelioid sarcoma predominantly affects adults, tends to occur more often in the perineal, pubic, genital and truncal regions; although it is defined by its histologic features rather than its location 6.
Masses can be large, up to 20 cm, especially in the proximal variant, but are often less than 5 centimeters in greatest dimension 10. Epithelioid sarcoma aggressively invades locally and has a propensity for regional lymph node metastasis. Distant metastatic spread, especially to the lungs and scalp, has been well documented 6. The clinical behavior of the tumor has probably best been described by Chase and Enzinger as an “indolent, relentless clinical course” due to the aggressive nature of local recurrences and metastatic potential 9.
Epithelioid sarcoma diagnosis
Epithelioid sarcoma can be difficult to diagnose because it may be confused with other conditions that have similar signs. For example, an ulcer on the skin that isn’t healing could be mistaken for a skin infection.
Tests and procedures used in diagnosis of epithelioid include:
- Imaging. Magnetic resonance imaging (MRI) is typically the method of imaging because of the level of detail it provides. Sometimes other imaging tests, such as computed tomography (CT) or positron emission tomography (PET), may be used.
- Biopsy. The doctor performs a biopsy using a long, thin needle to remove a sample of the suspected sarcoma or a larger lump for testing in a lab. Sometimes a biopsy sample is removed during surgery. A pathologist analyzes the sample to determine whether it’s cancer, and if so, the type and whether it’s aggressive.
Epithelioid sarcoma is characterized by inactivation of the SMARCB1 gene, which is present in both conventional and proximal types of epithelioid sarcoma 17. This abnormality leads to increased dependence on EZH2 and tumor formation 18.
Sentinel node biopsy
Since epithelioid sarcoma can metastasize to the lymph nodes, some have proposed sentinel lymph node biopsy and regional lymph node dissection 19. Other tumors where sentinel node biopsy has shown to be beneficial include melanoma and breast cancer, but outcome literature for sarcomas is lacking. Further research into this topic is needed.
Histopathology
Microscopically, the conventional or usual type of epithelioid sarcoma has a nodular or lobular architecture with central areas of necrosis. Epithelioid cells (polygonal with relatively abundant cytoplasm) with eosinophilic cytoplasm surround the necrosis, imparting a necrotizing granulomatous pattern to the low-power diagnosis. The nuclei are generally not too atypical, and mitotic figures are conspicuously present 10. Tumor cells at the periphery often take on a spindled or sarcomatous appearance. Various histologic patterns further complicate the histologic diagnosis as some tumors take on a more angiomatoid or fibromatoid appearance 20. These features can lead the pathologist toward vascular neoplasms and more benign processes like fibromatosis, or even reactive lesions such as nodular fasciitis.
The proximal-type variant of epithelioid sarcoma lacks the granulomatous pattern of necrosis. The tumor consists of cellular nodules with more cytologic pleomorphism, nuclear atypia, and even signet-ring-like vacuolation 10. Rhabdoid morphology with prominent nucleoli is characteristic of this lesion. Although the proximal type is characteristically found near the trunk, it is defined by its histologic features and not its location 6. Furthermore, the often proximal location of this variant leads to more diagnostic difficulty because of the overlapping features with metastatic carcinoma that would be more common to occur at proximal sites such as the groin and pelvic area 16.
Immunohistochemical examination
Immunohistochemical examination is essential to making a diagnosis of epithelioid sarcoma. Characteristically, epithelioid sarcoma shows expression of vimentin and low-molecular-weight cytokeratins such as those recognized by the monoclonal antibody CAM5.2 21 and 85–96 % of cases are EMA-positive 8. The staining pattern suggests that epithelioid sarcoma is a neoplasm of mesenchymal origin with epithelial dedifferentiation. Vimentin is rarely negative. CD34, expressed in approximately 50 % of epithelioid sarcoma lesions, is used as an additional diagnostic marker in vimentin-negative cases. The INI1 gene is a member of the adenosine triphosphate-dependent SWI/SWF chromatin-remodeling complex and thus a candidate as a tumor suppressor gene. Loss of the INI1 gene is seen in some patients with malignant rhabdoid tumors; its loss is 93 % sensitive for a diagnosis of epithelioid sarcoma 22.
ERG, a member of the erythroblast transformation-specific family of transcription factors, is a marker of endothelial differentiation 23. Kohashi et al. 24, who analyzed the expression of ERG in INI1-deficient tumors, including epithelioid sarcoma, reported that ERG was expressed in 13 of 24 conventional distal type lesions and 5 of 20 patients with proximal-type epithelioid sarcoma. All of their patients with malignant rhabdoid tumors had negative test results for ERG, suggesting that ERG may be a useful marker to distinguish epithelioid sarcoma from malignant rhabdoid tumors with loss of INI1.
Cancer antigen 125 (CA-125) is a high-molecular-weight glycoprotein commonly used for the identification of epithelial ovarian carcinomas; it has also been detected in various neoplasms, including carcinomas of the breast, lung, and colon, and in patients with lymphoma. Kato et al. 25 documented that 10 (91 %) of 11 patients with epithelioid sarcoma had positive test results for CA-125; 10 manifested the classical type and the other had the proximal type of the disease. In other soft tissue tumors, including synovial sarcomas, clear cell sarcomas, leiomyosarcomas, rhabdomyosarcomas, liposarcomas, malignant peripheral nerve sheath tumors, malignant fibrous histiocytomas, desmoid tumors, cutaneous squamous cell carcinomas, and rheumatoid nodules, CA-125 was negative. Other authors 21 have suggested that CA-125 immunoreactivity in the presence of an elevated serum CA-125 level could be a useful tumor marker for the diagnosis of epithelioid sarcoma and for monitoring the clinical course of patients with epithelioid sarcoma.
The most diagnostically useful finding is lack of INI-1 expression in tumor cells. Also helpful in the differential diagnosis, expanded upon in the differential diagnosis section below, is the typically negative immunolabeling of the tumor cells for S100, desmin, and CD31 26.
Immunohistochemical profile summary 26.:
Positive labeling
- Cytokeratin
- Epithelial membrane antigen
- Vimentin
- CD 34 (approximately 50% of cases)
Negative labeling
- INI-1/SMARCB1
- S100
- Desmin
- Factor VIII-related antigen
- Cytokeratin 7
Epithelioid sarcoma treatment
Surgery is the most common treatment for epithelioid sarcoma, but other treatments may be used in addition to surgery.
- Surgery. Surgery generally involves removing the cancer and a margin of healthy tissue surrounding it to help prevent recurrence. In severe cases, depending on the size, depth and location of the cancer, there’s a rare possibility that part of the affected limb would need to be amputated, but surgeons try their best to avoid doing this.
- Radiation therapy. Radiation therapy uses high-energy beams, such as X-rays or protons, to kill cancer cells. Radiation therapy is sometimes used before surgery to shrink the tumor and reduce the risk of the cancer returning, and after surgery to help reduce the risk of recurrence. Radiation therapy is used in some institutions for primary and recurrent cases 27 for limb salvage, with favorable results compared to amputation, but it has not shown to improve overall survivorship 28. The late effects (scarring, stiffness, and neuropathy) can be particularly debilitating in the hand 29.
- Chemotherapy. Chemotherapy uses drugs to kill cancer cells. Chemotherapy appears to be less effective in treating epithelioid sarcoma compared with other cancers, though it’s sometimes used in addition to surgery or when the cancer has spread. Doxorubicin has been used for multifocal, large (>5cm), or metastatic disease. It has not been shown to improve survivorship 28, but there are not large published trials.
- Clinical trials. Clinical trials are studies of new treatments. These studies provide a chance to try the latest treatment options, but the risk of side effects may not be known. Targeted cancer therapies — drugs or other substances that target certain molecules to block cancer growth — show promise in the treatment of epithelioid sarcoma. Ask your doctor about the availability of clinical trials, including the potential risks and benefits.
Patients should be carefully evaluated for the presence of involved lymph nodes; suspicious lymph nodes are biopsied. Surgical removal of primary and recurrent tumor(s) is the most effective treatment 30. Because of the propensity of epithelioid sarcoma to have occult metastasis to the lymph nodes, sentinel lymph node biopsy is recommended for epithelioid sarcoma of the extremities or buttocks in the absence of clinically (by imaging or physical examination) enlarged lymph nodes 31.
In a review of 30 pediatric patients with epithelioid sarcoma (median age at presentation, 12 years), responses to chemotherapy were reported in 40% of patients using sarcoma-based regimens, and 60% of patients were alive at 5 years after initial diagnosis 32. A single-institution retrospective review of 20 patients, which included children and adults (median age, 27.3 years), found no difference in the probability of recurrence between patients who received chemotherapy and those who did not receive chemotherapy and suggested that radiation therapy may be useful 30.
Epithelioid sarcoma prognosis
Prognosis, as with most malignancies, is primarily determined by the clinical stage of the disease. Epithelioid sarcoma prognosis is most closely associated with tumor size, vascular invasion, resectability, and metastases 9. Large tumor size and early metastases are independently associated with poor outcomes 33. The proximal type is generally described as a more aggressive tumor; however, at least one study 6 demonstrated no significant difference in overall survival between the proximal and usual types of epithelioid sarcoma.
Epithelioid sarcoma survival rate
Five year survival and ten year survival rate for patients with epithelioid sarcoma are approximately 50-70% and 42-55% respectively 34. Gender, site, age of diagnosis, tumor size and microscopic pathology have been shown to affect prognosis 35. Gender has been shown in multiple studies to be an important factor in prognosis with female patients showing a more favorable outcome. Proximal lesions have been shown to have worse outcomes compared to distal lesions 35. Tumors that presented at an earlier age had better outcome. Tumors more than 2 cm in diameter and tumors with necrosis and vascular invasion have been correlated with worse outcome 35. Mitotic index is a prognostic factor as well 36.
In a German Cooperative Weichteilsarkom Studiengruppe retrospective analysis of 67 children, adolescents, and young adults (median age, 14 years) with epithelioid sarcoma, 53 patients presented with localized disease and 14 patients presented with metastatic disease 37. Fifty-eight of 67 patients were treated with primary resections. Resections were microscopically complete in 35 patients, microscopically incomplete in 12 patients, and macroscopically incomplete in 20 patients. Forty-nine patients received chemotherapy, and 33 patients received radiation therapy. Complete remission was achieved in 45 of 53 patients (85%) with localized disease. Twenty-seven patients relapsed after a median time of 0.9 years (range, 0.1–2.3 years). Patients with localized disease had a 5-year event-free survival rate of 35% and an overall survival rate of 48%. Patients with metastatic disease had a 5-year event-free survival rate of 7% and an overall survial rate of 9%. Smaller tumor size, lower Intergroup Rhabdomyosarcoma Study group, less tumor invasiveness, negative nodal status, and microscopically complete resection correlated with a favorable prognosis in patients with localized disease.
In a phase I trial of the EZH2 inhibitor tazemetostat, two patients with INI1-negative epithelioid sarcoma had prolonged stable disease for more than 20 months after starting therapy 38.
- Enzinger FM. Epitheloid sarcoma. A sarcoma simulating a granuloma or a carcinoma. Cancer. 1970 Nov;26(5):1029-41.[↩][↩]
- What Is a Soft Tissue Sarcoma? https://www.cancer.org/cancer/soft-tissue-sarcoma/about/soft-tissue-sarcoma.html[↩]
- Needs T, Fillman EP. Cancer, Epithelioid Sarcoma. [Updated 2019 Jun 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK532911[↩][↩]
- Chase DR, Enzinger FM. Epithelioid sarcoma: diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241–63.[↩][↩]
- Soft tissue sarcoma. https://www.mayoclinic.org/diseases-conditions/epithelioid-sarcoma/cdc-20392420[↩]
- de Visscher SA, van Ginkel RJ, Wobbes T, Veth RP, Ten Heuvel SE, Suurmeijer AJ, Hoekstra HJ. Epithelioid sarcoma: Still an only surgically curable disease. Cancer. 2006 Aug 01;107(3):606-12.[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Enzinger FM, Weiss SW. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, editors. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby-Year Book; 2001. p. 1521–38.[↩]
- Coates SJ, Ogunrinade O, Lee HJ, Desman GJ. Epidermotropic metastatic epithelioid sarcoma: a potential diagnostic pitfall. Cutan Pathol. 2014;41(8):672–6.[↩][↩]
- Chase DR, Enzinger FM. Epithelioid sarcoma. Diagnosis, prognostic indicators, and treatment. Am. J. Surg. Pathol. 1985 Apr;9(4):241-63.[↩][↩][↩][↩]
- Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006 May;13(3):114-21.[↩][↩][↩][↩][↩][↩][↩]
- Noujaim J, Thway K, Bajwa Z, Bajwa A, Maki RG, Jones RL, Keller C. Epithelioid Sarcoma: Opportunities for Biology-Driven Targeted Therapy. Front Oncol. 2015;5:186.[↩][↩]
- Nishibaba, R., Higashi, Y., Goto, Y. et al. Epithelioid sarcoma with multiple lesions on the left arm: a case report. J Med Case Reports 10, 295 (2016). https://doi.org/10.1186/s13256-016-1088-z[↩]
- Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features: clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am J Surg Pathol. 1997;21:130–46.[↩]
- Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am. J. Surg. Pathol. 2011 Oct;35(10):e47-63.[↩]
- Modena P, Lualdi E, Facchinetti F, Galli L, Teixeira MR, Pilotti S, Sozzi G. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005 May 15;65(10):4012-9.[↩]
- Guillou L, Wadden C, Coindre JM, Krausz T, Fletcher CD. “Proximal-type” epithelioid sarcoma, a distinctive aggressive neoplasm showing rhabdoid features. Clinicopathologic, immunohistochemical, and ultrastructural study of a series. Am. J. Surg. Pathol. 1997 Feb;21(2):130-46.[↩][↩]
- Hornick JL, Dal Cin P, Fletcher CD: Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol 33 (4): 542-50, 2009.[↩]
- Knutson SK, Warholic NM, Wigle TJ, et al.: Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110 (19): 7922-7, 2013.[↩]
- Blazer, DG et al.: Is there a role for sentinel lymph node biopsy in the management of sarcoma?. Surgical Oncology; 12; 201-206; 2003[↩]
- Miettinen M, Wang Z, Sarlomo-Rikala M, Abdullaev Z, Pack SD, Fetsch JF. ERG expression in epithelioid sarcoma: a diagnostic pitfall. Am. J. Surg. Pathol. 2013 Oct;37(10):1580-5.[↩]
- Lynch MC, Graber EM, Johnson TS, Clarke LE. Epithelioid sarcoma resembling benign fibrous histiocytoma. Cutis. 2015;95(2):83–6.[↩][↩]
- Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal-type epithelioid sarcoma. Am J Surg Pathol. 2009;33(4):542–50.[↩]
- Martens JH. Acute myeloid leukemia: a central role for the ETS factor ERG. Int J Biochem Cell Biol. 2011;43(10):1413–6.[↩]
- Kohashi K, Yamada Y, Hotokebuchi Y, Yamamoto H, Taguchi T, Iwamoto Y, et al. ERG and SALL4 expressions in SMARCB1/INI1-deficient tumors: A useful tool for distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol. 2015;46(2):225–30.[↩]
- Kato H, Hatori M, Kokubun S, Watanabe M, Smith RA, Hotta T, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34(3):149–54.[↩]
- Stockman DL, Hornick JL, Deavers MT, Lev DC, Lazar AJ, Wang WL. ERG and FLI1 protein expression in epithelioid sarcoma. Mod. Pathol. 2014 Apr;27(4):496-501.[↩][↩]
- Callister, MD et al.: Epithelioid Sarcoma: results of conservative surgery and radiotherapy. International Journal of Radiation Oncology-Biology-Physics. 51(2):384-391, Oct 2001[↩]
- Wolf, PS. et al.: Epithelioid Sarcoma: the University of Washington experience. CTOS annual meeting presentation 2005[↩][↩]
- McPhee, M., McGrath, B.E., Zhang, P.,Driscoll D., Gibbs, J. and Peimer, C: Soft Tissue Sarcomas of the Hand. J of Hand Surgery, 24A: 1001-1007, 1999[↩]
- Guzzetta AA, Montgomery EA, Lyu H, et al.: Epithelioid sarcoma: one institution’s experience with a rare sarcoma. J Surg Res 177 (1): 116-22, 2012.[↩][↩]
- Hawkins DS, Spunt SL, Skapek SX, et al.: Children’s Oncology Group’s 2013 blueprint for research: Soft tissue sarcomas. Pediatr Blood Cancer 60 (6): 1001-8, 2013.[↩]
- Casanova M, Ferrari A, Collini P, et al.: Epithelioid sarcoma in children and adolescents: a report from the Italian Soft Tissue Sarcoma Committee. Cancer 106 (3): 708-17, 2006.[↩]
- Hasegawa T, Matsuno Y, Shimoda T, Umeda T, Yokoyama R, Hirohashi S. Proximal-type epithelioid sarcoma: a clinicopathologic study of 20 cases. Mod. Pathol. 2001 Jul;14(7):655-63.[↩]
- Gary D. et al: Epithelioid sarcoma, An analysis of Fifty-One cases, Journal of bone and Joint Surgery (Am), 70-A: 862-870, 1988[↩]
- Chase D.R. and Enzinger, F.M: Epithelioid sarcoma: Diagnosis, Prognostic indicators and treatment. The American J of Surgical Pathology, 9(4): 241-263, 1985[↩][↩][↩]
- Chbani et al: Epithelioid Sarcoma, A Clinicopathologic and Immunohistochemical Analysis of 106 Cases From the French Sarcoma Group, American Journal of Clinical Pathology;131:222-227; 2009[↩]
- Sparber-Sauer M, Koscielniak E, Vokuhl C, et al.: Epithelioid sarcoma in children, adolescents, and young adults: Localized, primary metastatic and relapsed disease. Treatment results of five Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr Blood Cancer 66 (9): e27879, 2019.[↩]
- Italiano A, Soria JC, Toulmonde M, et al.: Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol 19 (5): 649-659, 2018.[↩]





{kind=link}