
What is Friedreich’s ataxia
Friedreich’s ataxia is a rare inherited disease that damages your nervous system and causes movement problems 1. The damage affects your spinal cord and the nerves that control muscle movement in your arms and legs. Symptoms usually begin between the ages of 5 and 15. The main symptom is ataxia, which means trouble coordinating movements. People with Friedreich ataxia develop impaired muscle coordination (ataxia) that worsens over time. Other features of Friedreich’s ataxia include the gradual loss of strength and sensation in the arms and legs; muscle stiffness (spasticity); and impaired speech, hearing, and vision. Individuals with Friedreich ataxia often have a form of heart disease called hypertrophic cardiomyopathy, which enlarges and weakens the heart muscle and can be life-threatening. Some affected individuals develop diabetes or an abnormal curvature of the spine (scoliosis).
Friedreich’s ataxia is named after Nikolaus Friedreich, a German doctor who described the condition in the 1860s.
In Friedreich ataxia nerve fibers in the spinal cord and peripheral nerves degenerate, becoming thinner. Peripheral nerves carry information from the brain to the body and from the body back to the brain, such as a message that the feet are cold or a signal to the muscles to generate movement. The cerebellum, part of the brain that coordinates balance and movement, also degenerates to a lesser extent. This damage results in awkward, unsteady movements and impaired sensory functions. The disorder also causes problems in the heart (in as many as one-third of affected individuals) and spine, and some people with the condition will also develop diabetes. The disorder does not affect thinking and reasoning abilities (cognitive functions).
Friedreich ataxia is caused by a defect (mutation) in a gene labeled FXN, which carries the genetic code for a protein called frataxin. Individuals who inherit two defective copies of the FXN gene, one from each parent, will develop the disease. Although rare, Friedreich ataxia is the most common form of hereditary ataxia in the United States, affecting about 1 in every 50,000 people. Both male and female children can inherit the disorder.
Most people with Friedreich ataxia begin to experience the signs and symptoms of the disorder between ages 5 and 15. Poor coordination and balance are often the first noticeable features. Affected individuals typically require the use of a wheelchair about 10 years after signs and symptoms appear.
About 25 percent of people with Friedreich ataxia have an atypical form in which signs and symptoms begin after age 25. Affected individuals who develop Friedreich ataxia between ages 26 and 39 are considered to have late-onset Friedreich ataxia (LOFA). When the signs and symptoms begin after age 40 the condition is called very late-onset Friedreich ataxia (VLOFA). Late-onset Friedreich ataxia (LOFA) and very late-onset Friedreich ataxia (VLOFA) usually progress more slowly than typical Friedreich ataxia.
Friedreich’s ataxia is estimated to affect 1 in 40,000 people in the United States. This condition is found in people with European, Middle Eastern, or North African ancestry. It is rarely identified in other ethnic groups.
Friedreich’s ataxia symptoms include:
- Difficulty walking
- Muscle weakness
- Speech problems
- Involuntary eye movements
- Scoliosis (curving of the spine to one side)
- Heart palpitations, from the heart disease which can happen along with Friedreich’s ataxia
Complications of Friedreich’s ataxia may include:
- Diabetes
- Heart failure or heart disease
- Loss of ability to move around
People with Friedreich’s ataxia usually need a wheelchair 15 to 20 years after symptoms first appear. In severe cases, people become incapacitated. There is no cure. You can treat symptoms with medicines, braces, surgery, and physical therapy.
Alcohol can exacerbate ataxia and should be consumed in moderation. Illicit drugs may well affect neuronal well-being and may exacerbate Friedreich’s ataxia and thus should be avoided. Environments that place an ambulant individual at risk for falls (e.g., rough surfaces) should be avoided.
- Medications that are toxic or potentially toxic to persons with Friedreich’s ataxia comprise a spectrum of risk ranging from definite high risk to negligible risk. For an up-to-date list please go here: https://www.ncbi.nlm.nih.gov/books/NBK1281/bin/cmt_and_medications.pdf
There is currently no effective cure or treatment for Friedreich ataxia. However, many of the symptoms and accompanying complications can be treated to help individuals maintain optimal functioning as long as possible. Diabetes and heart problems can be treated with medications. Orthopedic problems such as foot deformities and scoliosis can be treated with braces or surgery. Rehabilitative therapy (physical, occupational, and vocational) can help individuals become as functionally independent as possible. Hearing impairments can be helped with hearing aids.
Support and information for families is also available through a number of private organizations. These groups can offer ways to network and communicate with others affected by Friedreich’s ataxia. They can also provide access to patient registries, clinical trials information, and other useful resources.
- Friedreich’s Ataxia Research Alliance: http://www.curefa.org/index.php
- National Ataxia Foundation: https://ataxia.org/
- Muscular Dystrophy Association: https://www.mda.org/
- Ataxia UK: https://www.ataxia.org.uk/
Can Friedreich ataxia be cured or treated?
As with many degenerative diseases of the nervous system, there is currently no cure or effective treatment for Friedreich ataxia. However, many of the symptoms and accompanying complications can be treated to help individuals maintain optimal functioning as long as possible. A multi-specialty team approach is essential to the treatment of the individual with Friedreich ataxia. Doctors can prescribe treatments for diabetes, if present; some of the heart problems can be treated with medication as well. Orthopedic problems such as foot deformities and scoliosis can be corrected with braces or surgery. Physical therapy may prolong use of the arms and legs. Swallowing and speech issues should be followed closely. Hearing impairment can be helped with hearing aids.
Can Friedreich ataxia affect individuals of any ethnicity?
Some genetic disorders are more likely to occur among people who trace their ancestry to a particular geographic area. People in an ethnic group often share certain versions of their genes, which have been passed down from common ancestors. If one of these shared genes contains a disease-causing mutation, a particular genetic disorder may be more frequently seen in the group 2. Although rare, Friedreich ataxia is the most common form of hereditary ataxia, affecting about 1 in every 50,000 people in the United States 1. This condition is typically found in people with European, Middle Eastern, or North African ancestry and it is rarely identified in other ethnic groups. There have been no reports of individuals with Friedreich ataxia in Southeast Asia, in sub-Saharan Africa, or among Native Americans 3. About 1 in 100 Americans are Friedreich ataxia carriers, but in some ethnic groups the frequency is higher. For example, about one in 70 people of Acadian (Cajun) ancestry are carriers 4.
How can I find a genetics professional in my area?
To find a medical professional who specializes in genetics, you can ask your doctor for a referral or you can search for one yourself. Online directories are provided by the American College of Medical Genetics (https://www.acmg.net/) and the National Society of Genetic Counselors (https://www.findageneticcounselor.com/).
Friedreich’s ataxia prognosis
Generally, within 10 to 20 years after the appearance of the first symptoms, the person is confined to a wheelchair. In later stages of the disease, individuals may become completely incapacitated. Friedreich ataxia can shorten life expectancy, and heart disease is the most common cause of death. Many individuals with Friedreich ataxia die in early adulthood, but some people with less severe symptoms live into their 60s or older.
Friedreich’s ataxia life expectancy
The rate of progression of Friedreich’s ataxia is variable. The average time from symptom onset to wheelchair dependence is ten years 5. A number of studies have found that progression is more rapid in those with earlier disease onset 6.
In a large study conducted in the early 1980s, the average age at death was 37 years 7. In a more recent study, the mean and median age of death was 36.5 years and 30 years, respectively 8. Survival into the sixth and seventh decades has been documented. The most common cause of death was cardiac (38/61), with the remainder (17/61) being non-cardiac (most commonly pneumonia) or unknown cause (6/61) 8.
Friedreich’s ataxia symptoms
Symptoms typically begin between the ages of 5 and 15 years, although they sometimes appear in adulthood. Approximately 15 percent of people with Friedreich ataxia have onset after age 25. The first neurological symptom to appear is usually difficulty walking and poor balance (gait ataxia, often described as appearing dizzy or even drunk). Another early sign of the disease is slowness and slurring of speech (dysarthria). With time speech becomes hesitant and jerky (often referred to as “scanning of speech”). The difficulty coordinating movement (ataxia) can affect all of the muscles. It gradually worsens and slowly spreads to the arms and the trunk (torso). As the muscle weakness progresses most affected individuals develop increased muscle tone (spasticity). Up to two-thirds of people with Friedreich ataxia also develop scoliosis (a curving of the spine to one side) that often requires surgical intervention for treatment. Most affected individuals also develop difficulty swallowing, due to difficulty coordinating the muscles of the tongue and throat.
In addition to the movement impairments, there is often a loss of sensation in the arms and legs, which may spread to other parts of the body. Other features include loss of normal reflexes, especially in the knees and ankles, and muscle weakness. Many individuals with later stages of Friedreich ataxia also develop hearing and vision loss.
Other symptoms that may occur include heart palpitations and shortness of breath. These symptoms are the result of various forms of heart disease that often accompany Friedreich ataxia, such as enlargement of the heart (hypertrophic cardiomyopathy), formation of fiber-like material in the muscles of the heart (myocardial fibrosis), and heart failure. Heart rhythm abnormalities such as a fast heart rate (tachycardia) and impaired conduction of cardiac impulses within the heart (heart block) are also common.
About 50 percent of people with Friedreich’s ataxia develop carbohydrate intolerance and 30 percent develop diabetes. Most individuals with the disease tire very easily and find that they require more rest and take a longer time to recover from common illnesses such as colds and flu.
Typical Friedreich Ataxia
Neurologic manifestations
Individuals with typical Friedreich ataxia develop progressive ataxia with onset from early childhood through to early adulthood, starting with poor balance when walking, followed by slurred speech and upper-limb ataxia. The mean age at onset of symptoms is ten to 15 years 9; onset can be as early as age two years and as late as the eighth decade. Gait ataxia, caused by a combination of spinocerebellar degeneration and loss of joint-position sense (proprioception), is the earliest symptom in the vast majority. The poor balance is accentuated when visual input is eliminated, such as in darkness or when the eyes are closed (Romberg sign). Ankle and knee jerks are generally absent, and plantar responses are up-going.
Within five years of symptom onset, most individuals with Friedreich’s ataxia exhibit “scanning” dysarthria, lower-extremity weakness, and diminished or absent joint-position and vibration sense distally ‒ neurologic manifestations that result from progressive degeneration of the dorsal root ganglia, posterior columns, corticospinal tracts, dorsal spinocerebellar tracts of the spinal cord, and cerebellum. Involvement of peripheral sensory and motor neurons results in a mixed axonal peripheral neuropathy.
Muscle weakness is often present and is most prominent in hip extensors and abductors; as disease advances, distal limb muscle weakness and wasting become evident.
Spasticity in the lower limbs is common and can be significant, affecting foot plantar flexors and inverters to a greater extent than dorsiflexors and everters. Thus, in the late stages of disease, equinovarus deformity is commonly seen 10 and may result in contractures ‒ more commonly in non-ambulatory affected individuals 11 ‒ and significant morbidity. Pes cavus is common (55%) but generally causes little problem for affected individuals. Restless leg syndrome is common in individuals with Friedreich ataxia, affecting 32%-50% of individuals in two studies 12.
Scoliosis is present in approximately two thirds of individuals with Friedreich’s ataxia when assessed clinically and 100% when assessed radiographically. A study found that 49 of 77 individuals with Friedreich’s ataxia had scoliosis; ten were treated with a brace and 16 required spinal surgery 13.
Autonomic disturbance becomes more common with disease progression. The most common manifestations are cold, cyanosed feet; bradycardia is less common.
Electrodiagnostic findings
Nerve conduction studies generally show a motor nerve conduction velocity of greater than 40 m/s with reduced or absent sensory nerve action potential with an absent H reflex.
Central motor conduction time is abnormal after transcranial magnetic stimulation 14.
Speech
Dysarthria, present in the majority of individuals with Friedreich’s ataxia, is generally of three types: mild dysarthria, increased velopharyngeal involvement manifest as hypernasality, and increased laryngeal dysfunction manifest as increased strained-strangled vocal quality 15. Dysarthria becomes worse as the disease progresses with the main changes seen over time being in speaking rate and utterance duration 16.
Mild dysphonia characterized by hoarseness (combined roughness and breathiness), increased strain, and altered pitch variability is also seen 17.
Swallowing
Dysphagia (difficulty swallowing) is common in Friedreich’s ataxia with 92% of individuals reporting issues with swallowing 18. Dysphagia in Friedreich’s ataxia relates to oropharyngeal incoordination, weakness, and spasticity.
Hypertrophic cardiomyopathy, defined as increased thickness of the interventricular septum, is present in about two thirds of individuals with Friedreich’s ataxia 5. Echocardiographic evaluation may reveal left ventricular hypertrophy that is more commonly asymmetric than concentric 19. When more subtle cardiac involvement is sought by methods such as tissue Doppler echocardiography, an even larger percentage of individuals have detectable abnormalities 20. Between 12% and 20% of individuals have reduced ejection fraction 21 and longitudinal strain is commonly reduced 22.
Later in the disease course, the cardiomyopathy may become dilated. Progressive systolic dysfunction is common 23 and reduction in left ventricular wall thickness is often seen as the disease progresses 24. A longitudinal study identified two groups; a “low risk” group (approximately 80%) with normal ejection fraction that declined slowly and remained in the normal range and a “high risk” group (approximately 20%) in whom ejection fraction declined into the abnormal range and was associated with high mortality 25. Those in the “high risk” group had longer GAA expansions on the shorter allele. The degree of neurologic impairment did not predict whether an affected individual would have stable or rapid progression of cardiomyopathy.
Electrocardiography (ECG) is abnormal in the vast majority, with T wave inversion, left axis deviation, and repolarization abnormalities being most commonly seen 26.
Symptoms related to cardiomyopathy usually occur in the later stages of the disease 26 but in rare instances may precede ataxia 27. Quercia et al 28 established the diagnosis of Friedreich’s ataxia in a young child evaluated for sudden death. Subjective symptoms of exertional dyspnea (40%), palpitations (11%), and anginal pain may be present in moderately advanced disease. Arrhythmias (especially atrial fibrillation) and congestive heart failure frequently occur in the later stages of the disease and are the most common cause of mortality 29. Coronary artery disease may occur and should be considered if there is angina and/ or sudden deterioration in cardiac function 30.
Urinary issues
Bladder symptoms including urinary frequency and urgency were reported by 41% of individuals in one study 5. A study of 158 individuals with Friedreich’s ataxia revealed lower urinary tract symptoms in 82% with impact on quality of life in 22% of those 31. Of 28 who underwent urodynamic studies, all had normal serum creatinine and four had upper urinary tract dilatation.
Sleep-disordered breathing
Sleep-disordered breathing and sleep apnea are more prevalent in those with Friedreich’s ataxia than in the healthy population. There is a minimum prevalence of 21% of obstructive sleep apnea compared to an incidence of about 5% in the general population 32.
Diabetes mellitus occurs in up to 30% of individuals with Friedreich’s ataxia 33. Impaired glucose tolerance is seen in up to an additional 49% 34. Non-diabetic individuals with Friedreich’s ataxia demonstrate high insulin responsiveness to oral glucose testing and low insulin sensitivity 35.
Ophthalmic manifestations
Optic nerve atrophy, often asymptomatic, occurs in approximately 25% of individuals with Friedreich’s ataxia. Reduced visual acuity was found in 13% in one study 36. Study of the anterior and posterior visual pathways in Friedreich’s ataxia by visual field testing and optical coherence tomography, pattern visual evoked potentials, and diffusion-weighted imaging revealed that all individuals studied had optic nerve abnormalities, but only 5/26 (19%) had related symptoms 37. Progressive diminution of contrast acuity is typical with disease progression 38.
Abnormal extraocular movements include irregular ocular pursuit, dysmetric saccades, saccadic latency, square wave jerks, ocular flutter, and marked reduction in vestibulo-ocular reflex gain and increased latency 39. Horizontal and vertical gaze palsy does not occur.
Hearing loss
Sensorineural hearing loss occurs in 13% of individuals with Friedreich’s ataxia 40. Auditory neuropathy may occur and difficulty hearing in background noise is common 41.
Cognitive skills
While cognition is generally not impaired in Friedreich’s ataxia, motor and mental reaction times can be significantly slowed 42. Motor planning is markedly impaired 43. The intelligence profile of individuals with Friedreich’s ataxia is characterized by concrete thinking and poor capacity in concept formation and visuospatial reasoning with reduced speed of information processing 44. Problems with attention and working memory have also been demonstrated 45. Motor overflow is also more prevalent in Friedreich’s ataxia than in controls 46. Those with earlier onset and larger FXN intron 1 GAA repeats tend to have more severe cognitive difficulties than those with later onset and smaller GAA repeats 47. Impairment of inhibition and cognitive flexibility was identified in individuals with Friedreich’s ataxia on the Haylings Sentence Completion Task 48.
Bone mineral density
A study of 28 individuals with Friedreich’s ataxia identified that six (21.4%) had reduced bone mineral density for age in at least one site assessed 49. There was a negative correlation between disease severity and femoral neck bone density. Females were more likely to have clinical fractures than males but no association was found between bone mineral density and fracture occurrence. In fact, all fractures occurred in those with a Z-score better than -2.
Other. Inflammatory bowel disease and growth hormone deficiency are more common in individuals with Friedreich’s ataxia than the general community 50.
Pregnancy
A study of 65 pregnancies in 31 women with Friedreich’s ataxia found no increase in the rate of spontaneous miscarriage, preeclampsia, prematurity, or cesarean section delivery 51. Worsening, improving, or unchanged symptoms during pregnancy were each reported by approximately one third of women with Friedreich’s ataxia.
Neuroimaging
MRI is often normal in the early stages of Friedreich’s ataxia. With advanced disease, atrophy of the cervical spinal cord and cerebellum may be observed 52. Atrophy of the superior cerebellar peduncle, the main outflow tract of the dentate nucleus, may also be seen 53. Cervical spinal cord size correlates with disease severity as measured by the Friedreich Ataxia Rating Scale 54.
A voxel-based morphometry study showed a symmetric volume loss in the dorsal medulla, infero-medial portions of the cerebellar hemispheres, rostral vermis, and dentate region 55. No volume loss in cerebral hemispheres was observed. Lower fractional anisotropy, higher mean diffusivity, and increased radial diffusivity compared to controls have been found in the dentatorubral, dentatothalamic, and thalamocortical tracts in individuals with Friedreich’s ataxia 56.
Atypical Presentations
Approximately 25% of individuals homozygous for full-penetrance GAA expansions in FXN have atypical findings 36 that include the following.
Late-onset Friedreich’s ataxia (LOFA) and very late-onset Friedreich’s ataxia (VLOFA). In approximately 15% of individuals with Friedreich’s ataxia, onset is later than age 25 years. In individuals with late-onset Friedreich’s ataxia (LOFA), the age of onset is 26-39 years; and, in very late-onset Friedreich’s ataxia (VLOFA), onset is after age 40 years 52. The oldest reported age of onset among individuals homozygous for the GAA expansion is 80 years 57.
A study of 44 individuals with LOFA and 30 individuals with VLOFA found that dysarthria, absent tendon reflexes, extensor plantar reflexes, weakness, amyotrophy, ganglionopathy, cerebellar atrophy, scoliosis, cardiomyopathy, and functional disability were milder, and GAA expansion on the smaller allele shorter, than in individuals with typical-onset Friedreich’s ataxia 58. Another study of 18 individuals with LOFA reported similar findings [59.
- Friedreich’s ataxia with retained reflexes (FARR) accounts for approximately 12% of individuals who are homozygous for the GAA expansion 60. Some individuals with FARR show brisk tendon reflexes that can be accompanied by clonus. Tendon reflexes may be retained for more than ten years after the onset of the disease. FARR usually has a later age of onset and lower incidence of secondary skeletal involvement and cardiomyopathy 60.
- Friedreich’s ataxia in Acadians. Montermini et al 61 showed that Acadians with Friedreich’s ataxia have a later age of onset (on average 3.0 years later than those with typical Friedreich’s ataxia) and of wheelchair confinement, and a much lower incidence of cardiomyopathy (48% vs 82%).
Spastic paraparesis without ataxia. Individuals who have biallelic full-penetrance GAA expansions may rarely present with spastic gait disturbance without gait or limb ataxia. These individuals usually have hyperreflexia and a later age of onset (on average 5.8 years later than those with typical Friedreich’s ataxia); they develop ataxia with time 62.
Other rare presentations of Friedreich’s ataxia
- Chorea and pure sensory ataxia 63
- Apparently isolated cardiomyopathy, with ataxia only becoming evident some time later 27.
What causes Friedreich’s ataxia?
Mutations in the FXN gene cause Friedreich ataxia. This gene provides instructions for making a protein called frataxin. Although its role is not fully understood, frataxin is important for the normal function of mitochondria, the energy-producing centers within cells. One region of the FXN gene contains a segment of DNA known as a GAA trinucleotide repeat (or a triplet repeat expansion). This segment is made up of a series of three DNA building blocks (one guanine and two adenines) that appear multiple times in a row. Normally, this segment is repeated 5 to 33 times within the FXN gene.
In people with Friedreich ataxia, the GAA segment is repeated 66 to more than 1,000 times. The length of the GAA trinucleotide repeat appears to be related to the age at which the symptoms of Friedreich ataxia appear, how severe they are, and how quickly they progress. People with GAA segments repeated fewer than 300 times tend to have a later appearance of symptoms (after age 25) than those with larger GAA trinucleotide repeats. The abnormally long GAA trinucleotide repeat disrupts the production of frataxin, which severely reduces the amount of this protein in cells. Certain nerve and muscle cells cannot function properly with a shortage of frataxin, leading to the characteristic signs and symptoms of Friedreich ataxia.
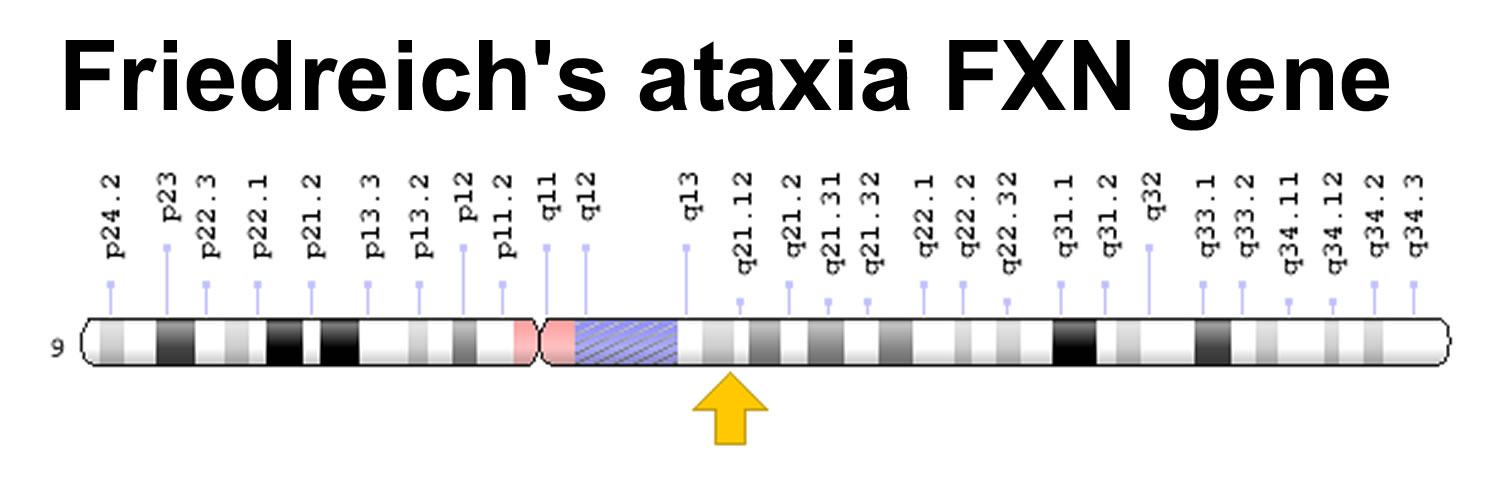
Friedreich’s ataxia gene
The FXN gene is located at 9q21.11, which is the long (q) arm of chromosome 9 at position 21.11. The FXN gene provides instructions for making a protein called frataxin. This protein is found in cells throughout the body, with the highest levels in the heart, spinal cord, liver, pancreas, and muscles used for voluntary movement (skeletal muscles). Within cells, frataxin is found in energy-producing structures called mitochondria. Although its function is not fully understood, frataxin appears to help assemble clusters of iron and sulfur molecules that are critical for the function of many proteins, including those needed for energy production.
One region of the FXN gene contains a segment of DNA known as a GAA trinucleotide repeat. This segment is made up of a series of three DNA building blocks (one guanine and two adenines) that appear multiple times in a row. In most people, the number of GAA repeats in the FXN gene is fewer than 12 (referred to as short normal). Sometimes, however, the GAA segment is repeated 12 to 33 times (referred to as long normal).
Friedreich ataxia is the only known genetic disorder that requires inheriting two copies of the abnormal FXN gene to cause the disease. Almost all people with Friedreich’s ataxia (98 percent) have two copies of this mutant form of FXN, but it is not found in all cases of the disease. About two percent of affected individuals have other defects in the FXN gene that are responsible for causing the disease.
The triplet repeat expansion greatly disrupts the normal production of frataxin. Frataxin is found in the energy-producing parts of the cell called mitochondria. Research suggests that without a normal level of frataxin, certain cells in the body (especially peripheral nerve, spinal cord, brain and heart muscle cells) produce energy less effectively and have been hypothesized to have a buildup of toxic byproducts leading to what is called “oxidative stress.” Lack of normal levels of frataxin also may lead to increased levels of iron in the mitochondria. When the excess iron reacts with oxygen, free radicals can be produced. Although free radicals are essential molecules in the body metabolism, they can also destroy cells and harm the body.
Figure 1. Friedreich’s ataxia gene (FXN gene)
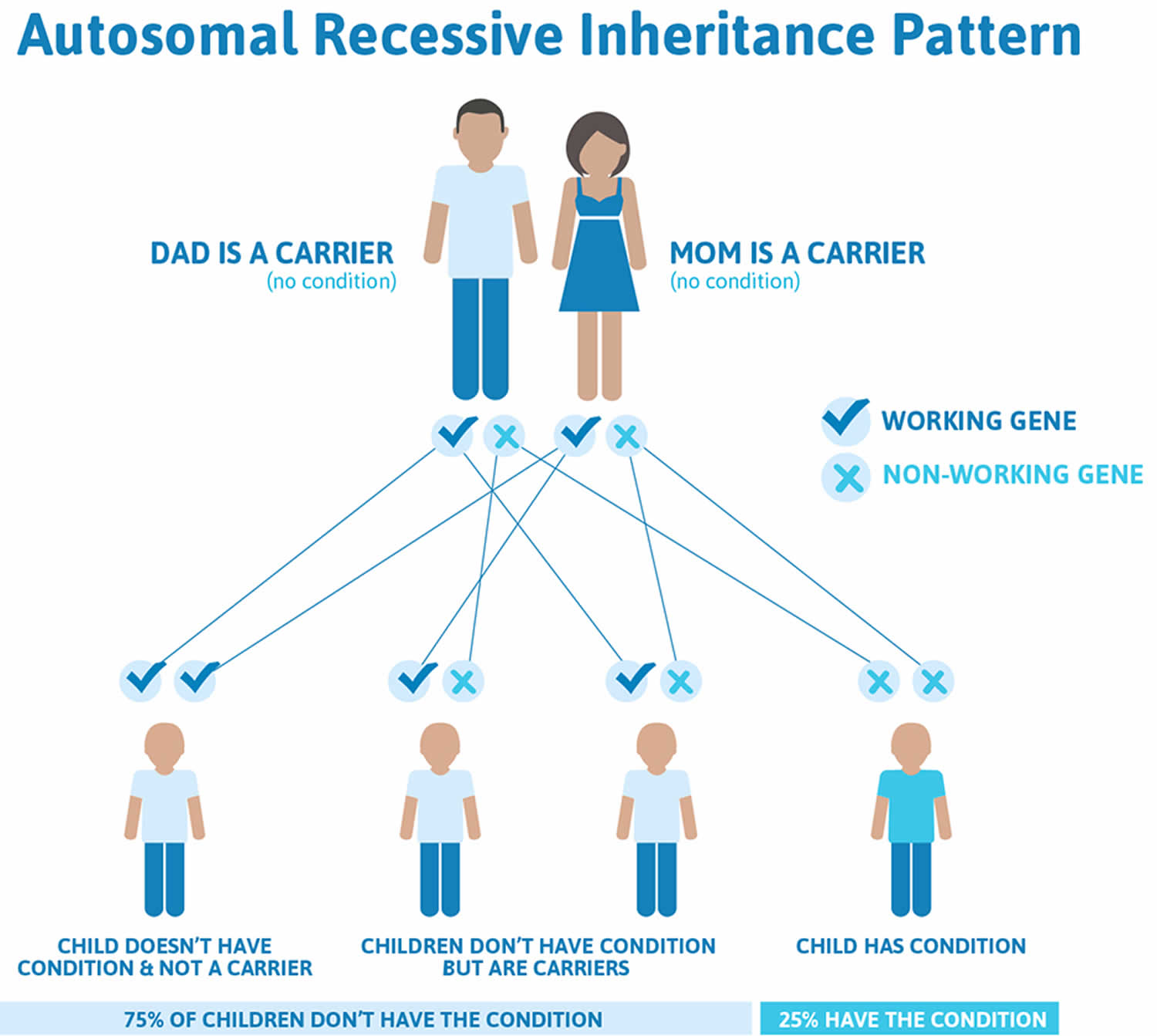
Friedreich’s ataxia inheritance pattern
Friedreich’s ataxia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
Figure 2. Friedreich’s ataxia autosomal recessive inheritance pattern
Friedreich’s ataxia diagnosis
A diagnosis of Friedreich ataxia requires a careful clinical examination, which includes a medical history and a thorough physical exam, in particular looking for balance difficulty, loss of joint sensation (proprioception), absence of reflexes, and signs of neurological problems. Genetic testing now provides a conclusive diagnosis. Other tests that may aid in the diagnosis or management of the disorder include:
- Electromyogram (EMG), which measures the electrical activity of muscle cells,
- Nerve conduction studies, which measure the speed with which nerves transmit impulses,
- Electrocardiogram (also called EKG or ECG), which gives a graphic presentation of the electrical activity or beat pattern of the heart,
- Echocardiogram, which records the position and motion of the heart muscle,
- Blood tests to check for elevated glucose levels and vitamin E levels, and
- Magnetic resonance imaging (MRI) or computed tomography (CT) scans, tests which provide brain and spinal cord images that are useful for ruling out other neurological conditions.
Friedreich’s ataxia family members
Genetic testing is essential for proper clinical diagnosis, and can aid in prenatal diagnosis and determining a person carrier status. Genetic counselors can help explain how Friedreich ataxia is inherited.
A primary care physician can screen people for complications such as heart disease, diabetes, and scoliosis, and can refer individuals to specialists such as cardiologists, physical therapists, and speech therapists to help deal with some of the other associated problems.
Support and information for families is also available through a number of private organizations. These groups can offer ways to network and communicate with others affected by Friedreich’s ataxia. They can also provide access to patient registries, clinical trials information, and other useful resources.
- Friedreich’s Ataxia Research Alliance: http://www.curefa.org/index.php
- National Ataxia Foundation: https://ataxia.org/
- Muscular Dystrophy Association: https://www.mda.org/
- Ataxia UK: https://www.ataxia.org.uk/
Friedreich’s ataxia treatment
Treatment for Friedreich ataxia includes:
- Counseling
- Speech therapy
- Physical therapy
- Walking aids or wheelchairs
Orthopedic devices (braces) may be needed for scoliosis and foot problems. Treating heart disease and diabetes help people live longer and improve their quality of life.
There is little objective evidence regarding management of Friedreich’s ataxia. A multidisciplinary approach is essential for maximal benefit because Friedreich’s ataxia affects multiple organ systems:
- Prostheses, walking aids, wheelchairs, and physical therapy as prescribed by a physiatrist (rehabilitation medicine specialist) to maintain an active lifestyle
- In-patient rehabilitation, which has been shown to improve physical function as measured by the Functional Independence Measure 65
- Occupational therapy assessment to ensure a safe home and work environment
- To manage spasticity: physical therapy including stretching programs, standing frame and splints, pharmacologic agents such as baclofen and botulinum toxin. Intrathecal baclofen can be beneficial where oral administration is unsuccessful or side effects are excessive. Orthopedic interventions, both operative and non-operative, for scoliosis and foot deformities may be necessary.
- Speech therapy to maximize communication skills
- Management of dysphagia that may include dietary modification and, in the late stages of disease, use of nasogastric or gastrostomy feeding
- Treatment of cardiac disease to reduce morbidity and mortality, including antiarrhythmic agents, anticardiac failure medication, anticoagulants, and pacemaker/implantable cardioverter defibrillator insertion. Cardiac transplantation is more controversial but has been used particularly when there is severe cardiac disease in the setting of mild neurologic symptoms.
- Antispasmodic agents for bladder dysfunction, with some individuals requiring botulinum toxin for the bladder and some requiring intermittent or permanent catheterization
- Treatment of sleep apnea by continuous positive airway pressure
- Treatment of diabetes mellitus with diet and, if necessary, oral hypoglycemic agents or insulin
- Hearing aids, microphone, and receiver as needed.
- Psychological (counseling and/or pharmacologic) support for affected individuals and family
Prevention of Secondary Manifestations
Measures include the following:
- Active management of spasticity with physiotherapy and botulinum toxin to prevent permanent contracture and the need for surgery
- Treatment of scoliosis with physiotherapy, botulinum toxin, and surgery to prevent cardiopulmonary complications that can result from severe scoliosis
- Treatment of diabetes mellitus to prevent complications that can arise from untreated/inadequately treated diabetes
- Treatment of cardiac complications of Friedreich’s ataxia to prevent arrhythmias that can result in mortality
- Treatment of sleep apnea to prevent neurologic and cardiopulmonary complications that can result from untreated sleep apnea
Surveillance
Published clinical management guidelines provide detailed discussion of recommended surveillance 66.
The following are appropriate.
- If ECG and echocardiogram performed at the time of initial diagnosis are normal, annual repeat testing
- Annual fasting blood sugar to monitor for diabetes mellitus
- Hearing assessment every two to three years or more often if symptoms are present. This should include testing of hearing in background noise, as it is more often abnormal than the common audiogram assessed in a quiet environment
- Sleep study to investigate for obstructive sleep apnea if concerns are raised by clinical history or a screening test such as the Epworth Sleepiness Scale
- Friedreich ataxia. https://ghr.nlm.nih.gov/condition/friedreich-ataxia[↩][↩]
- Friedreich Ataxia Fact Sheet. https://www.ninds.nih.gov/Disorders/Patient-Caregiver-Education/Fact-Sheets/Friedreichs-Ataxia-Fact-Sheet[↩]
- Bidichandani SI, Delatycki MB. Friedreich Ataxia. 1998 Dec 18 [Updated 2017 Jun 1]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1281/[↩]
- Friedreich’s Ataxia (FA). https://www.mda.org/disease/friedreichs-ataxia[↩]
- Delatycki MB, Knight M, Koenig M, Cossee M, Williamson R, Forrest SM. G130V, a common FRDA point mutation, appears to have arisen from a common founder. Hum Genet. 1999a;105:343–6.[↩][↩][↩]
- Patel M, Isaacs CJ, Seyer L, Brigatti K, Gelbard S, Strawser C, Foerster D, Shinnick J, Schadt K, Yiu EM, Delatycki MB, Perlman S, Wilmot GR, Zesiewicz T, Mathews K, Gomez CM, Yoon G, Subramony SH, Brocht A, Farmer J, Lynch DR. Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann Clin Transl Neurol. 2016;3:684–94.[↩]
- Harding AE. Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. https://www.ncbi.nlm.nih.gov/pubmed/7272714[↩]
- Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD, Wilmot GR, Ravina B, Koeppen AH, Lynch DR. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307:46–9. https://www.ncbi.nlm.nih.gov/pubmed/21652007[↩][↩]
- Delatycki MB, Paris DB, Gardner RJ, Nicholson GA, Nassif N, Storey E, MacMillan JC, Collins V, Williamson R, Forrest SM. Clinical and genetic study of Friedreich ataxia in an Australian population. Am J Med Genet. 1999b;87:168–74.[↩]
- Delatycki MB, Holian A, Corben L, Rawicki HB, Blackburn C, Hoare B, Toy M, Churchyard A. Surgery for equinovarus deformity in Friedreich’s ataxia improves mobility and independence. Clin Orthop Relat Res. 2005;(430):138–41.[↩]
- Milne SC, Corben LA, Yiu E, Delatycki MB, Georgiou-Karistianis N. Gastrocnemius and soleus spasticity and muscle length in Friedreich’s ataxia. J Clin Neurosci. 2016;29:29–34.[↩]
- Frauscher B, Hering S, Högl B, Gschliesser V, Ulmer H, Poewe W, Boesch SM. Restless legs syndrome in Friedreich ataxia: a polysomnographic study. Mov Disord. 2011;26:302–6.[↩]
- Milbrandt TA, Kunes JR, Karol LA. Friedreich’s ataxia and scoliosis: the experience at two institutions. J Pediatr Orthop. 2008;28:234–8.[↩]
- Brighina F, Scalia S, Gennuso M, Lupo I, Matta F, Piccoli T, Fierro B. Hypo-excitability of cortical areas in patients affected by Friedreich ataxia: a TMS study. J Neurol Sci. 2005;235:19–22.[↩]
- Folker J, Murdoch B, Cahill L, Delatycki M, Corben L, Vogel A. Dysarthria in Friedreich’s ataxia: a perceptual analysis. Folia Phoniatr Logop. 2010;62:97–103.[↩]
- Rosen KM, Folker JE, Vogel AP, Corben LA, Murdoch BE, Delatycki MB. Longitudinal change in dysarthria associated with Friedreich ataxia: a potential clinical endpoint. J Neurol. 2012;259:2471–7.[↩]
- Vogel AP, Wardrop MI, Folker JE, Synofzik M, Corben LA, Delatycki MB, Awan SN. Voice in Friedreich ataxia. J Voice. 2017;31:243.e9–243.e19.[↩]
- Vogel AP, Brown SE, Folker JE, Corben LA, Delatycki MB. Dysphagia and swallowing-related quality of life in Friedreich ataxia. J Neurol. 2014;261:392–9.[↩]
- Koc F, Akpinar O, Yerdelen D, Demir M, Sarica Y, Kanadasi M. The evaluation of left ventricular systolic and diastolic functions in patients with Friedreich ataxia. A pulse tissue Doppler study. Int Heart J. 2005;46:443–52.[↩]
- Mottram PM, Delatycki MB, Donelan L, Gelman JS, Corben L, Peverill RE. Early changes in left ventricular long-axis function in Friedreich ataxia: relation with the FXN gene mutation and cardiac structural change. J Am Soc Echocardiogr. 2011;24:782–9.[↩]
- Regner SR, Lagedrost SJ, Plappert T, Paulsen EK, Friedman LS, Snyder ML, Perlman SL, Mathews KD, Wilmot GR, Schadt KA, Sutton MS, Lynch DR. Analysis of echocardiograms in a large heterogeneous cohort of patients with friedreich ataxia. Am J Cardiol. 2012a;109:401–5.[↩]
- St John Sutton M, Ky B, Regner SR, Schadt K, Plappert T, He J, D’Souza B, Lynch DR. Longitudinal strain in friedreich ataxia: a potential marker for early left ventricular dysfunction. Echocardiography. 2014;31:50–7.[↩]
- Kipps A, Alexander M, Colan SD, Gauvreau K, Smoot L, Crawford L, Darras BT, Blume ED. The longitudinal course of cardiomyopathy in Friedreich’s ataxia during childhood. Pediatr Cardiol. 2009;30:306–10.[↩]
- Rajagopalan B, Francis JM, Cooke F, Korlipara LV, Blamire AM, Schapira AH, Madan J, Neubauer S, Cooper JM. Analysis of the factors influencing the cardiac phenotype in Friedreich’s ataxia. Mov Disord. 2010;25:846–52.[↩]
- Pousset F, Legrand L, Monin ML, Ewenczyk C, Charles P, Komajda M, Brice A, Pandolfo M, Isnard R, Tezenas du Montcel S, Durr A. A 22-year follow-up study of long-term cardiac outcome and predictors of survival in Friedreich ataxia. JAMA Neurol. 2015;72:1334–41.[↩]
- Dutka DP, Donnelly JE, Nihoyannopoulos P, Oakley CM, Nunez DJ. Marked variation in the cardiomyopathy associated with Friedreich’s ataxia. Heart. 1999;81:141–7.[↩][↩]
- Leonard H, Forsyth R. Friedreich’s ataxia presenting after cardiac transplantation. Arch Dis Child. 2001;84:167–8.[↩][↩]
- Quercia N, Somers GR, Halliday W, Kantor PF, Banwell B, Yoon G. Friedreich ataxia presenting as sudden cardiac death in childhood: clinical, genetic and pathological correlation, with implications for genetic testing and counselling. Neuromuscul Disord. 2010;20:340–2.[↩]
- Tsou AY, Paulsen EK, Lagedrost SJ, Perlman SL, Mathews KD, Wilmot GR, Ravina B, Koeppen AH, Lynch DR. Mortality in Friedreich ataxia. J Neurol Sci. 2011;307:46–9.[↩]
- Giugliano GR, Sethi PS. Friedreich’s ataxia as a cause of premature coronary artery disease. Tex Heart Inst J. 2007;34:214–7.[↩]
- Musegante AF, Almeida PN, Monteiro RT, Barroso U Jr. Urinary symptoms and urodynamics findings in patients with Friedreich’s ataxia. Int Braz J Urol. 2013;39:867–74.[↩]
- Corben LA, Ho M, Copland J, Tai G, Delatycki MB. Increased prevalence of sleep-disordered breathing in Friedreich ataxia. Neurology. 2013;81:46–51.[↩]
- Cnop M, Mulder H, Igoillo-Esteve M. Diabetes in Friedreich ataxia. J Neurochem. 2013;126 Suppl 1:94–102.[↩]
- Cnop M, Igoillo-Esteve M, Rai M, Begu A, Serroukh Y, Depondt C, Musuaya AE, Marhfour I, Ladrière L, Moles Lopez X, Lefkaditis D, Moore F, Brion JP, Cooper JM, Schapira AH, Clark A, Koeppen AH, Marchetti P, Pandolfo M, Eizirik DL, Féry F. Central role and mechanisms of β-cell dysfunction and death in friedreich ataxia-associated diabetes. Ann Neurol. 2012;72:971–82.[↩]
- Isaacs CJ, Brigatti KW, Kucheruk O, Ratcliffe S, Sciascia T, McCormack SE, Willi SM, Lynch DR. Effects of genetic severity on glucose homeostasis in Friedreich ataxia. Muscle Nerve. 2016;54:887–94.[↩]
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–75.[↩][↩]
- Fortuna F, Barboni P, Liguori R, Valentino ML, Savini G, Gellera C, Mariotti C, Rizzo G, Tonon C, Manners D, Lodi R, Sadun AA, Carelli V. Visual system involvement in patients with Friedreich’s ataxia. Brain. 2009;132:116–23.[↩]
- Seyer LA, Galetta K, Wilson J, Sakai R, Perlman S, Mathews K, Wilmot GR, Gomez CM, Ravina B, Zesiewicz T, Bushara KO, Subramony SH, Ashizawa T, Delatycki MB, Brocht A, Balcer LJ, Lynch DR. Analysis of the visual system in Friedreich ataxia. J Neurol. 2013;260:2362–9.[↩]
- Fahey MC, Cremer PD, Aw ST, Millist L, Todd MJ, White OB, Halmagyi M, Corben LA, Collins V, Churchyard AJ, Tan K, Kowal L, Delatycki MB. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain. 2008;131:1035–45.[↩]
- Dürr A, Cossee M, Agid Y, Campuzano V, Mignard C, Penet C, Mandel JL, Brice A, Koenig M. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med. 1996;335:1169–75[↩]
- Rance G, Fava R, Baldock H, Chong A, Barker E, Corben L, Delatycki MB. Speech perception ability in individuals with Friedreich ataxia. Brain. 2008;131:2002–12.[↩]
- Corben LA, Georgiou-Karistianis N, Fahey MC, Storey E, Churchyard A, Horne M, Bradshaw JL, Delatycki MB. Towards an understanding of cognitive function in Friedreich Ataxia. Brain Res Bull. 2006;70:197–202.[↩]
- Corben LA, Akhlaghi H, Georgiou-Karistianis N, Bradshaw JL, Egan GF, Storey E, Churchyard AJ, Delatycki MB. Impaired inhibition of prepotent motor tendencies in Friedreich ataxia demonstrated by the Simon interference task. Brain Cogn. 2011;76:140–5.[↩]
- Mantovan MC, Martinuzzi A, Squarzanti F, Bolla A, Silvestri I, Liessi G, Macchi C, Ruzza G, Trevisan CP, Angelini C. Exploring mental status in Friedreich’s ataxia: a combined neuropsychological, behavioral and neuroimaging study. Eur J Neurol. 2006;13:827–35.[↩]
- Klopper F, Delatycki MB, Corben LA, Bradshaw JL, Rance G, Georgiou-Karistianis N. The test of everyday attention reveals significant sustained volitional attention and working memory deficits in friedreich ataxia. J Int Neuropsychol Soc. 2011;17:196–200.[↩]
- Low SC, Corben LA, Delatycki MB, Ternes AM, Addamo PK, Georgiou-Karistianis N. Excessive motor overflow reveals abnormal inter-hemispheric connectivity in Friedreich ataxia. J Neurol. 2013;260:1757–64.[↩]
- Nachbauer W, Bodner T, Boesch S, Karner E, Eigentler A, Neier L, Benke T, Delazer M. Friedreich ataxia: executive control is related to disease onset and GAA repeat length. Cerebellum. 2014;13:9–16.[↩]
- Corben LA, Klopper F, Stagnitti M, Georgiou-Karistianis N, Bradshaw JL, Rance G, Delatycki MB. Measuring inhibition and cognitive flexibility in Friedreich ataxia. Cerebellum. 2017. Epub ahead of print.[↩]
- Eigentler A, Nachbauer W, Donnemiller E, Poewe W, Gasser RW, Boesch S. Low bone mineral density in Friedreich ataxia. Cerebellum. 2014;13:549–57.[↩]
- Shinnick JE, Schadt K, Strawser C, Wilcox N, Perlman SL, Wilmot GR, Gomez CM, Mathews KD, Yoon G, Zesiewicz T, Hoyle C, Subramony SH, Yiu EM, Delatycki MB, Brocht AF, Farmer JM, Lynch DR. Comorbid medical conditions in Friedreich ataxia: association with inflammatory bowel disease and growth hormone deficiency. J Child Neurol. 2016;31:1161–5[↩]
- Friedman LS, Paulsen EK, Schadt KA, Brigatti KW, Driscoll DA, Farmer JM, Lynch DR. Pregnancy with Friedreich ataxia: a retrospective review of medical risks and psychosocial implications. Am J Obstet Gynecol. 2010;203:224.e1–5.[↩]
- Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH. Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol. 2005;62:1865–9.[↩][↩]
- Akhlaghi H, Corben L, Georgiou-Karistianis N, Bradshaw J, Storey E, Delatycki MB, Egan GF. Superior cerebellar peduncle atrophy in Friedreich’s ataxia correlates with disease symptoms. Cerebellum. 2011;10:81–7.[↩]
- Chevis CF, da Silva CB, D’Abreu A, Lopes-Cendes I, Cendes F, Bergo FP, França MC Jr. Spinal cord atrophy correlates with disability in Friedreich’s ataxia. Cerebellum. 2013;12:43–7.[↩]
- Della Nave R, Ginestroni A, Giannelli M, Tessa C, Salvatore E, Salvi F, Dotti MT, De Michele G, Piacentini S, Mascalchi M. Brain structural damage in Friedreich’s ataxia. J Neurol Neurosurg Psychiatry. 2008;79:82–5.[↩]
- Akhlaghi H, Yu J, Corben L, Georgiou-Karistianis N, Bradshaw JL, Storey E, Delatycki MB, Egan GF. Cognitive deficits in Friedreich ataxia correlate with micro-structural changes in dentatorubral tract. Cerebellum. 2014;13:187–98[↩]
- Alvarez V, Arnold P, Kuntzer T. Very late-onset Friedreich ataxia: later than life expectancy? J Neurol. 2013;260:1408–9.[↩]
- Lecocq C, Charles P, Azulay JP, Meissner W, Rai M, N’Guyen K, Péréon Y, Fabre N, Robin E, Courtois S, Guyant-Maréchal L, Zagnoli F, Rudolf G, Renaud M, Sévin-Allouet M, Lesne F, Alaerts N, Goizet C, Calvas P, Eusebio A, Guissart C, Derkinderen P, Tison F, Brice A, Koenig M, Pandolfo M, Tranchant C, Dürr A, Anheim M. Delayed-onset Friedreich’s ataxia revisited. Mov Disord. 2016;31:62–9.[↩]
- Martinez AR, Moro A, Abrahao A, Faber I, Borges CR, Rezende TJ, Martins CR Jr, Moscovich M, Munhoz RP, Segal SL, Arruda WO, Saraiva-Pereira ML, Karuta S, Pedroso JL, D’Abreu A, Jardim LB, Lopes-Cendes Í, Barsottini OG, Teive HA, França MC Jr. Nonneurological involvement in late-onset Friedreich ataxia (LOFA): exploring the phenotypes. Cerebellum. 2017;16:253–6.[↩]
- Coppola G, De Michele G, Cavalcanti F, Pianese L, Perretti A, Santoro L, Vita G, Toscano A, Amboni M, Grimaldi G, Salvatore E, Caruso G, Filla A. Why do some Friedreich’s ataxia patients retain tendon reflexes? A clinical, neurophysiological and molecular study. J Neurol. 1999;246:353–7[↩][↩]
- Montermini L, Richter A, Morgan K, Justice CM, Julien D, Castellotti B, Mercier J, Poirier J, Capozzoli F, Bouchard JP, Lemieux B, Mathieu J, Vanasse M, Seni MH, Graham G, Andermann F, Andermann E, Melancon SB, Keats BJ, Di Donato S, Pandolfo M. Phenotypic variability in Friedreich ataxia: role of the associated GAA triplet repeat expansion. Ann Neurol. 1997b;41:675–82.[↩]
- Badhwar A, Jansen A, Andermann F, Pandolfo M, Andermann E. Striking intrafamilial phenotypic variability and spastic paraplegia in the presence of similar homozygous expansions of the FRDA1 gene. Mov Disord. 2004;19:1424–31[↩]
- Zhu D, Burke C, Leslie A, Nicholson GA. Friedreich’s ataxia with chorea and myoclonus caused by a compound heterozygosity for a novel deletion and the trinucleotide GAA expansion. Mov Disord. 2002;17:585–9.[↩]
- FXN gene. https://ghr.nlm.nih.gov/gene/FXN[↩]
- Milne SC, Campagna EJ, Corben LA, Delatycki MB, Teo K, Churchyard AJ, Haines TP. Retrospective study of the effects of inpatient rehabilitation on improving and maintaining functional independence in people with Friedreich ataxia. Arch Phys Med Rehabil. 2012;93:1860–3[↩]
- http://www.curefa.org/pdf/research/ClinicalManagementGuidelinesForFA(1).pdf[↩]




{kind=link}