Glycogenolysis
Glycogenolysis is simply the process in the degradation of glycogen for utilization as an energy source mainly in skeletal muscle and liver 1. Glycogen is an extensively branched glucose polymer that is used by humans as an energy reserve, stored mainly in the liver and the skeletal muscle that supplies glucose to the blood stream during fasting periods and to the muscle cells during muscle contraction 2. Glycogen exists from precursors of glucose, derived from recently ingested carbohydrates or gluconeogenic precursors, including lactate and alanine. Glycogen is highly concentrated in the liver, although skeletal muscles contain the most glycogen by weight. Glycogen is also present in lower levels in other tissues, such as kidney, heart, brain, adipose tissue, and red blood cells, but glycogen function in these tissues is mostly unknown 3. Glycogenolysis is the catabolism of glycogen by removal of a glucose monomer through cleavage with inorganic phosphate to produce glucose-1-phosphate (see Figure 1). The rate-limiting enzyme of glycogenolysis is glycogen phosphorylase and debranching enzyme. Glycogen phosphorylase takes care of breaking down linear chain bonds using an inorganic phosphate instead of water (see Figure 1), whereas debranching enzyme transfers glucose residues to the linear chain as to degrade branches. Both enzymes work in skeletal muscle and liver. Because skeletal muscle can utilize the phosphorylated form of glucose (glucose 1-phosphate) as fuel, it does not require glucose-6-phosphatase. The glucose 1-phosphate formed by glycogen phosphorylase is converted to glucose 6-phosphate, an intermediate in glycolysis, by the same mutase used in glycogen synthesis, as shown in Figure 1. Glycolysis is the metabolic pathway that converts glucose into pyruvate and a hydrogen ion, H+. The free energy released in this process is used to form the high-energy molecules ATP (adenosine triphosphate) and NADH (reduced nicotinamide adenine dinucleotide).
Glycogen degradation occurs in two distinct pathways depending on location. Concerning skeletal muscle, glycogen degradation leads to glucose-1-phosphate, and muscle uses it, per se, as fuel for contraction 1. Glycogenolysis in skeletal muscle occurs under conditions of physical activity.
On the other hand, the liver requires glucose-6-phosphatase to dephosphorylate glucose and export glucose outside the cell. In terms of lysosomal degradation, acid alpha-glucosidase is responsible for glycogenolysis 1. Deficiency in any of these enzymes manifests as unique disease states. Most cytosolic degradations alterations effectuate in glycogen storage diseases, including von Gierke disease, Cori disease, and Hers disease. A defect in lysosomal degradation can lead to Pompe disease. The impaired role of glycogenolysis in neural functioning appears via the presentation of Lafora disease. Detection of glycogenolysis alterations can be done mainly through DNA analysis and electron microscopy through the liver and skeletal muscle biopsies.
In skeletal muscle, glucose uptake gets mediated via GLUT1 (lying on the plasma membrane) and GLUT4 (lying inside intracellular storage vesicles) transporters. In the cytosol, glycogen phosphorylase becomes strongly activated by AMP for muscle tissue. For liver, glucagon activates glycogen phosphorylase via an increase in cyclic-AMP. Glycogen degradation in lysosomes undergoes mediation via acid alpha-glucosidase via autophagic vacuoles that engulf a portion of the cytoplasm and fuse with organelles to enclose the content.
Due to the vital role that glycogen breakdown has on normal physiology, including maintaining blood glucose levels and muscle contraction during activity, disruptions in glycogenolysis have exhibited pathophysiological conditions. Although researchers have investigated the role of glycogen, certain information is not well understood 1. For instance, the specific role that glycogen serves in muscle contraction or the method for glycogen transport into lysosomes is unclear 1.
Figure 1. Glycogenolysis
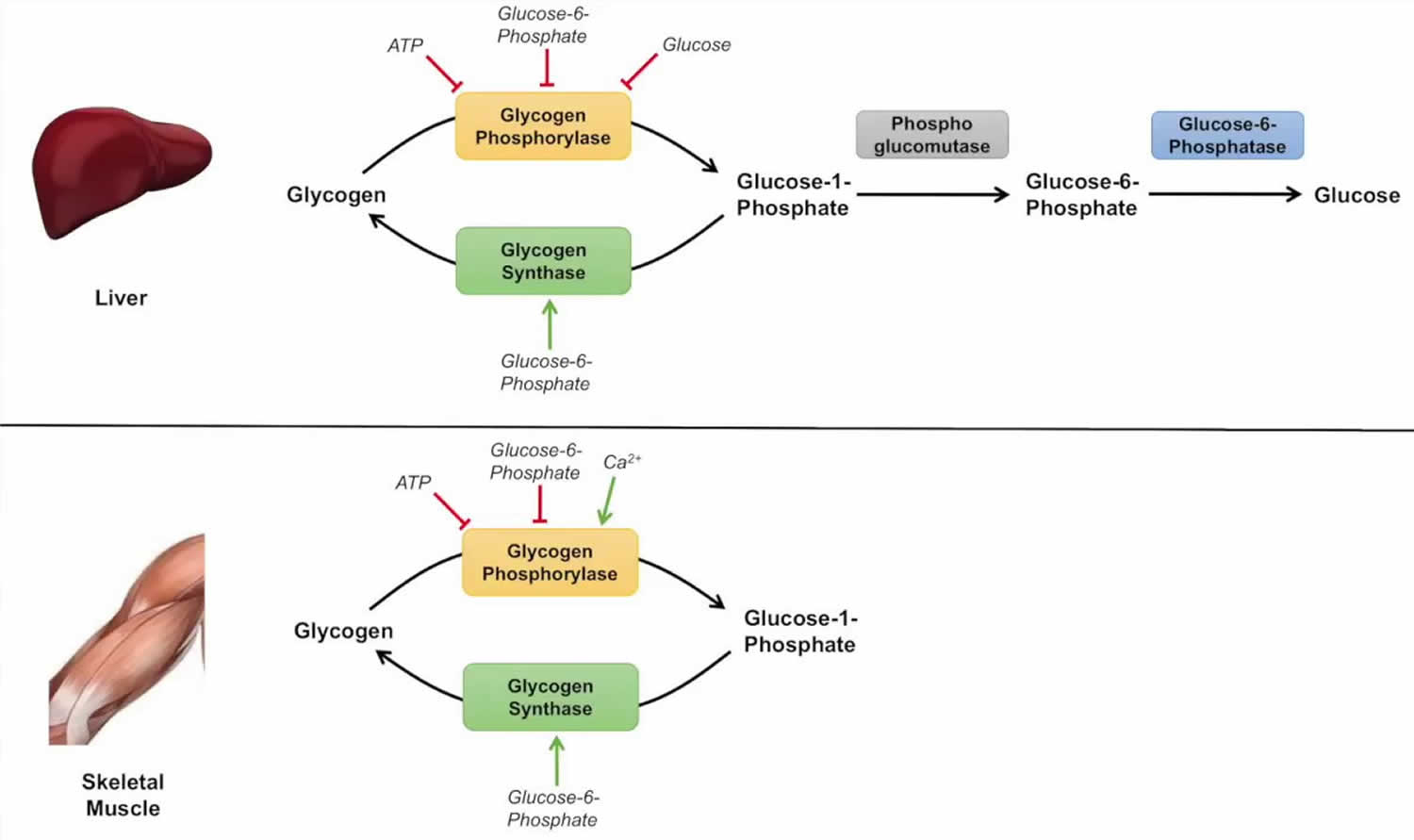
Glycogenolysis steps
First step
The overall reaction for the 1st step is:
Glycogen (n residues) + Pi <—–> Glycogen (n-1 residues)+ glucose-1-phosphate
Here, glycogen phosphorylase cleaves the bond at the 1 position by substitution of a phosphoryl group. It breaks down glucose polymer at α-1-4 linkages until 4 linked glucoses are left on the branch.
Second step
The 2nd step involves the debranching enzyme that moves the remaining glucose units to another non-reducing end. This results in more glucose units available to glycogen phosphorylase (step 1)
Third step
The 3rd and last stage converts glucose-1-phosphate to glucose-6-phosphate through the enzyme phosphoglucomutase.
Glycogenolysis pathway
Glycogenolysis can occur via two pathways. Whereas the first pathway revolves around cytosolic degradation via the synchronized action of glycogen phosphorylase and glycogen debranching enzyme, the second pathway revolves around lysosomal degradation via the enzyme alpha-glucosidase.
Corresponding to cytosolic degradation, glycogen phosphorylase, the rate-limiting enzyme of glycogenolysis, cleaves terminal glucose residue connected to a glycogen branch while substituting a phosphoryl group for the alpha 1-4 bond. Four residues before an alpha 1-6 bond, corresponding to a branch, glycogen debranching enzyme catalyzes the transfer of three of the four remaining glucose residues to the end of another glycogen chain, where they can again by degraded by glycogen phosphorylase. In other words, the breakage of alpha 1-4 glycosidic bonds present in linear chains is catalyzed by glycogen phosphorylase, and the addition of the phosphate group to position one results in the production of glucose-1-phosphate. The activity of glycogen phosphorylase is modulated allosterically and by phosphorylation. Glycogen production, conversely, inhibits glycogen degradation. Phosphoglucomutase is then in charge of converting glucose-1-phosphate to glucose-6-phosphate through an isomerization reaction that has no energy requirements. On the other hand, the debranching enzyme deals with alpha 1-6 bonds and transfers a branch to the end of the polymer so that glycogen phosphorylase can continue working with it. In most tissues, glucose-6-phosphate is internally utilized for glycolysis and energy production through conversion to pyruvate, acting as a critical metabolic intermediate for other pathways, including the citric acid cycle cycle, fatty acid synthesis, Cori cycle, and alanine cycle. Nevertheless, in gluconeogenic organs such as the liver, kidney, and intestines, glucose-6-phosphate needs to be dephosphorylated to glucose—with the aid of enzyme glucose-6-phosphatase— so that it can undergo transport from the endoplasmic reticulum to the interstitial space. Corresponding the lysosomal glycogen degradation, the primary enzyme involved in acid maltase. The hydrolysis of glycogen to glucose, catalyzed by acid alpha-glucosidase, has been hypothesized to serve a protective mechanism for the liver from high concentrations of glycogen. Of the total amount of glycogenolysis that happens in skeletal muscle, only 5% of glycogen degradation happens in lysosomes. For liver glycogenolysis, only 10% occurs in lysosomes 4.
Figure 2. Glycogenolysis pathway
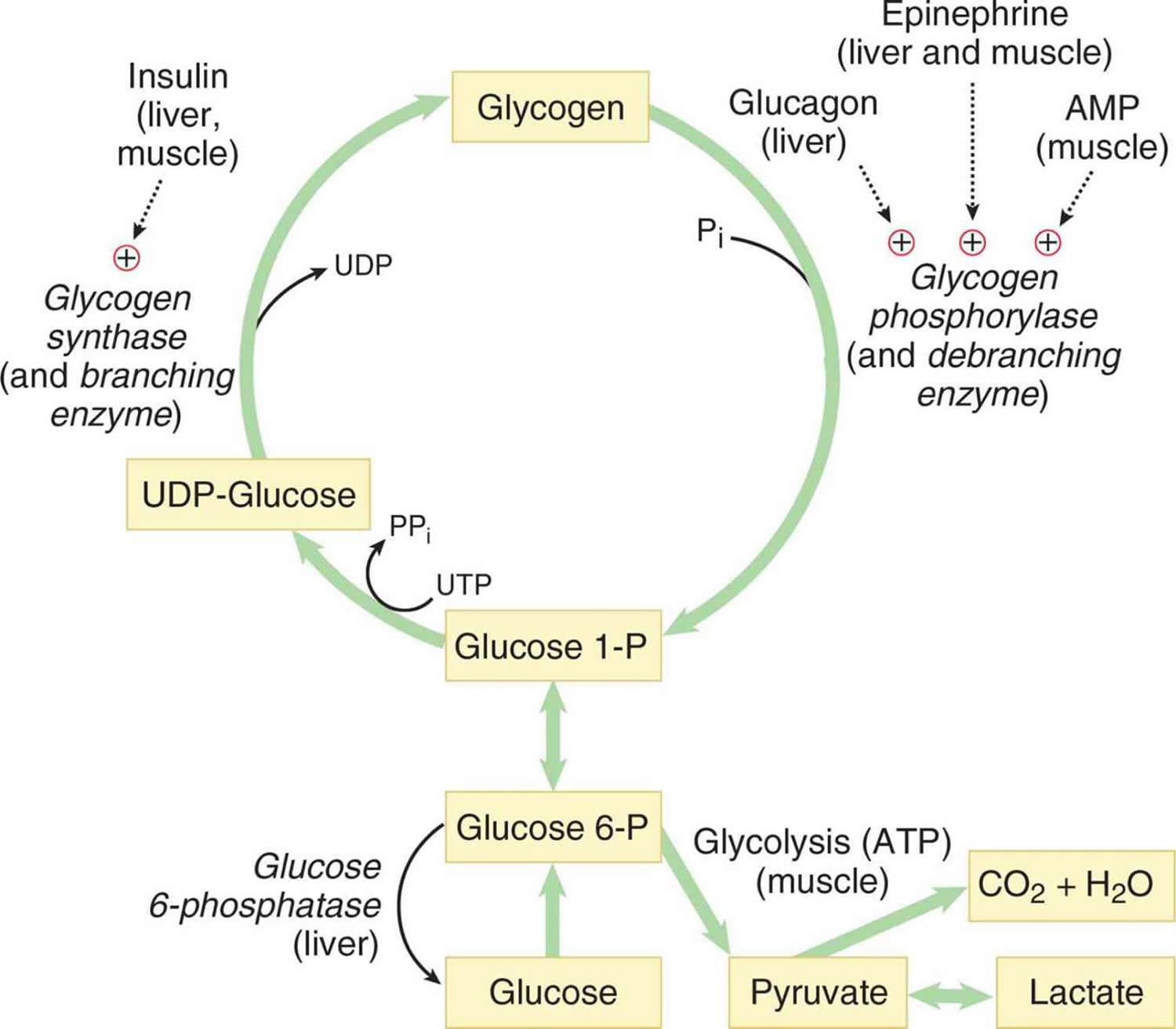
Abbreviations: UDP-Glucose = uracil diphosphate glucose
Glycogenolysis function
Glycogen degradation occurs during a fasting state or when there is a low ratio of insulin to glucagon. The primary energy reserves for the body are glycogen and lipids. Whereas lipids result in a higher number of ATP molecules after oxidation, glycogenolysis leads to a lower number of ATP molecules. However, two significant advantages exist for the metabolism of glycogen. The first advantage corresponds to the rapid mobilization of glycogen for metabolic requirements. Rapid mobilization is possible since glycogenolysis enzymes can adhere to the numerous branches of glycogen and begin simultaneous hydrolysis. The second advantage entails energy production under conditions of low lipid deposits, such as with anorexia 5.
Glycogen levels are quantitively more present in skeletal muscle than the liver. Nevertheless, glycogenolysis serves important roles in both tissues. In the liver, glycogen metabolism has a vital role during fasting conditions, leading to hepatic glucose production for maintaining healthy blood glucose levels and supporting the fuel needs of other tissues. The full manifestation of glycogen in skeletal muscle, on the other hand, indicates the critical function that glycogen plays in skeletal muscle in terms of rapid ATP generation. A close relationship exists between glycogen storage in skeletal muscle and fatigue resistance. The ability of a muscle to exercise during the first 30 minutes of activity, despite the abundance of other energy sources such as lipids, is severely compromised when glycogen levels become diminished in skeletal muscle. Glycogen depletion has shown to develop fatigue because the muscle is unable to provide adequate fuel to skeletal muscle for excitation and contraction. A probable reason revolves around the role of glycogen in calcium release from the sarcoplasmic reticulum 6.
Asides for providing energy, glycogenolysis can lead to precursors for participation in oxidative reactions of the pentose phosphate pathway, helping in the generation of NADPH, which is necessary for the synthesis of fatty acids and production of pentose phosphates, which are essential for the synthesis of RNA and DNA 5.
Glycogenolysis clinical significance
The importance of glycogenolysis is demonstrated through mutations in glycogen degradation leading to human genetic disorders and through the inability of skeletal muscle to cope with physical stress when glycogen scarcity exists.
Dysfunctions in glycogenolysis can lead to a variety of diseases, including glycogen storage diseases, lysosomal storage diseases, and Lafora progressive myoclonus epilepsy. Disruptions in glycogenolysis frequently effectuate in dysfunction of organs, including the liver, skeletal muscle, brain, and kidney. Depending on the affected enzyme in glycogenolysis, a particular spectrum of syndromes is possible.
A disruption in glycogenolysis can result in glycogen storage diseases such as von Gierke disease, the most common glycogen storage disease. Type 1 glycogen storage disease is due to a deficiency in glucose-6-phosphatase, responsible for dephosphorylating glucose-6-phosphate so that glucose can get transported outside the cell for the regulation of blood glucose levels and fuel usage in other tissues outside of the liver. The impaired ability to generate glucose from glycogenolysis results in severe hypoglycemia, hyperuricemia, and increased levels of lactic acid and triglycerides. Due to the deposition of fat, patients present with a rounded doll-like face. Without treatment, failure to thrive, hepatomegaly, abnormal swelling, and delayed motor development has been evident in patients with this disease. Long term complications can develop due to kidney glycogen accumulation leading to nephropathy, chronic kidney disease, and renal cancer. The main form of treatment for von Gierke disease patients is to maintain normal glucose levels while avoiding hypoglycemia by having frequent feeds 7.
While degradation of glycogen by phosphorylase and debranching enzyme can happen in the cytosol, glycogen is also degraded via a lysosomal pathway, leading to a lysosomal storage disease called Pompe disease (glycogen storage disease type 2). In Pompe disease, a mutation involving lysosomal alpha-glucosidase—also called acid maltase—developed. As a result, glycogen accumulates in the lysosome and its vesicles, leading to fatal outcomes, including cardiomyopathy and muscular hypotonia. The precise pathway in which glycogen in transported to lysosomes is still unknown but is hypothesized to be through macroautophagy, in which engulfment of cargo within double-membrane vesicles called autophagosomes fuse with the lysosome 3.
Glycogen storage disease type 3 also referred to as Cori disease, results due to a deficiency of glycogen debranching enzyme. As a result, this disease manifests with an accumulation of abnormal glycogen since glycogenolysis halts when glycogen phosphorylase encounters a branching point. The glycogen is then considered abnormal because it reflects very short outer chains. With Cori disease, patients present with ketotic hypoglycemia and hepatomegaly. In rare cases, it can lead to liver cirrhosis and hepatocellular carcinoma 6.
Glycogen storage disease type 5 (McArdle disease) develops due to a deficiency in skeletal muscle glycogen phosphorylase. In other words, with this disease, the liver is spared. Patients demonstrate exercise intolerance, muscle weakness, cramping, and pain. Creatine kinase levels become elevated, and myoglobinuria can be present. Typical of this disease is a phenomenon called second wind, where patients can resume exercise when resting briefly. Ingestion of sucrose before exercise can help alleviate symptoms since this becomes the source of energy during exercise before resorting to glycogen stores. When glycogen phosphorylase is deficient in the liver, a different disease develops—glycogen storage disease type 5. Hers disease exhibits normal creatine kinase and uric acid levels. Patients present with growth retardation and liver enlargement. Hyperlipidemia and ketotic hypoglycemia can be common 6.
In Lafora progressive myoclonus epilepsy, increased phosphorylation of glycogen is present in several tissues, leading to toxicity and cell death in neurons. Symptoms include ataxia, seizures, myoclonus, and dementia. The presence of more than normal phosphorylation levels in glycogen effectuates in longer chains and irregular branch points that render the polymer insoluble and degradation resistant. As a result, patients with this condition have a conglomerate of inclusion bodies called Lafora bodies 6.
- Patino SC, Mohiuddin SS. Biochemistry, Glycogenesis. [Updated 2019 Nov 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK549820[↩][↩][↩][↩][↩]
- Adeva-Andany MM, González-Lucán M, Donapetry-García C, Fernández-Fernández C, Ameneiros-Rodríguez E. Glycogen metabolism in humans. BBA Clin. 2016;5:85–100. Published 2016 Feb 27. doi:10.1016/j.bbacli.2016.02.001 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4802397[↩]
- Roach PJ, Depaoli-Roach AA, Hurley TD, Tagliabracci VS. Glycogen and its metabolism: some new developments and old themes. Biochem. J. 2012 Feb 01;441(3):763-87.[↩][↩]
- Prats C, Graham TE, Shearer J. The dynamic life of the glycogen granule. J. Biol. Chem. 2018 May 11;293(19):7089-7098.[↩]
- Bezborodkina NN, Chestnova AY, Vorobev ML, Kudryavtsev BN. Spatial Structure of Glycogen Molecules in Cells. Biochemistry Mosc. 2018 May;83(5):467-482.[↩][↩]
- Ellingwood SS, Cheng A. Biochemical and clinical aspects of glycogen storage diseases. J. Endocrinol. 2018 Sep;238(3):R131-R141.[↩][↩][↩][↩]
- Kanungo S, Wells K, Tribett T, El-Gharbawy A. Glycogen metabolism and glycogen storage disorders. Ann Transl Med. 2018 Dec;6(24):474.[↩]





{kind=link}