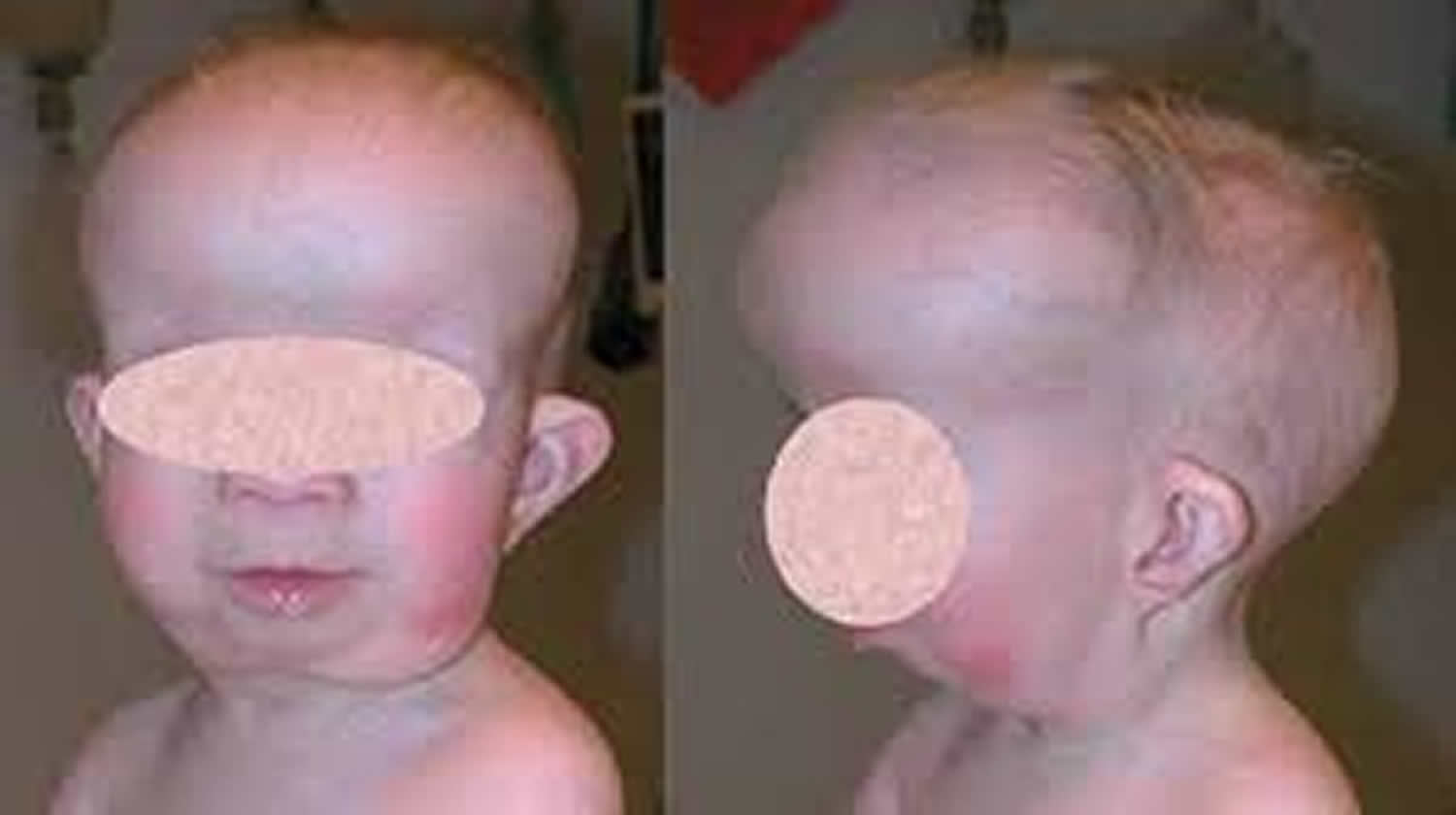
Gorlin syndrome
Gorlin syndrome also known as basal cell nevus syndrome, Gorlin-Goltz syndrome or nevoid basal cell carcinoma syndrome, is a rare inherited condition that affects many areas of the body and increases the risk of developing various cancerous and noncancerous tumors. In people with Gorlin syndrome, the type of cancer diagnosed most often is basal cell carcinoma (BCC), which is the most common form of skin cancer. Individuals with Gorlin syndrome typically begin to develop basal cell carcinomas during adolescence or early adulthood. These cancers occur most often on the face, chest, and back. The number of basal cell carcinomas that develop during a person’s lifetime varies among affected individuals. Some people with Gorlin syndrome never develop any basal cell carcinomas, while others may develop thousands of these cancers. Individuals with lighter skin are more likely to develop basal cell carcinomas than are people with darker skin. The number of carcinomas may be reduced with ongoing treatment.
Gorlin syndrome affects an estimated 1 in 31,000 people 1. While more than 1 million new cases of basal cell carcinoma are diagnosed each year in the United States, fewer than 1 percent of these skin cancers are related to Gorlin syndrome 1.
Most people with Gorlin syndrome also develop noncancerous (benign) tumors of the jaw, called keratocystic odontogenic tumors. These tumors usually first appear during adolescence, and new tumors form until about age 30. Keratocystic odontogenic tumors rarely develop later in adulthood. If untreated, these tumors may cause painful facial swelling and tooth displacement.
Individuals with Gorlin syndrome have a higher risk than the general population of developing other tumors. A small proportion of affected individuals develop a brain tumor called medulloblastoma during childhood. A type of benign tumor called a fibroma can occur in the heart or in a woman’s ovaries. Heart (cardiac) fibromas often do not cause any symptoms, but they may obstruct blood flow or cause irregular heartbeats (arrhythmia). Ovarian fibromas are not thought to affect a woman’s ability to have children (fertility).
Other features of Gorlin syndrome include small depressions (pits) in the skin of the palms of the hands and soles of the feet; an unusually large head size (macrocephaly) with a prominent forehead; and skeletal abnormalities involving the spine, ribs, or skull. These signs and symptoms are typically apparent from birth or become evident in early childhood.
Figure 1. Gorlin syndrome
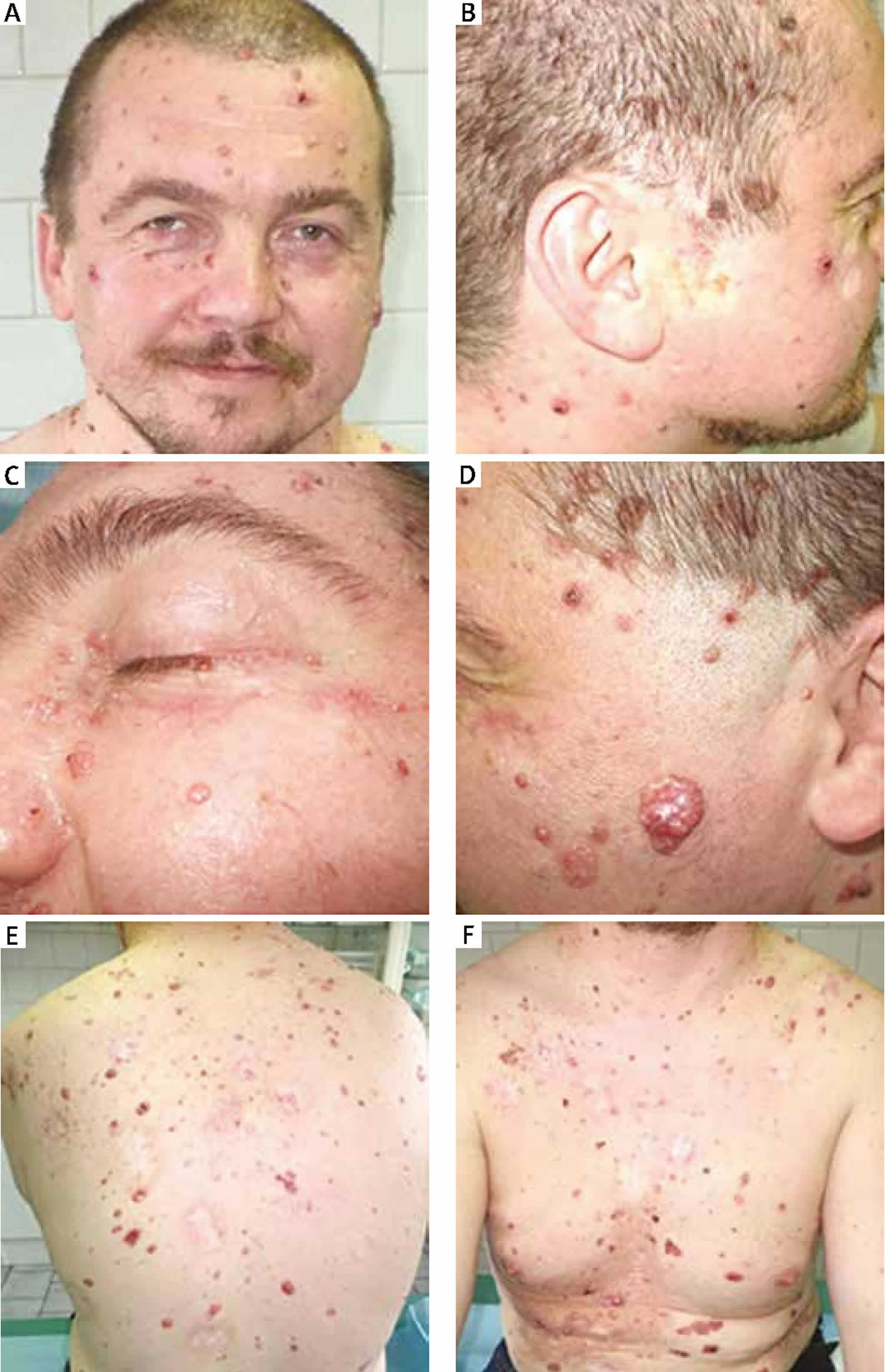
Footnote: Photograph of the patient showing multiple basal cell carcinomas of the face and trunk.
[Source 2 ]Basal cell nevus syndrome causes
Mutations in the PTCH1 gene (patched gene) on chromosome 9q22.3-q31 are the main cause Gorlin syndrome. PTCH1 gene provides instructions for making a protein called patched-1, which functions as a receptor. Receptor proteins have specific sites into which certain other proteins, called ligands, fit like keys into locks. Together, ligands and their receptors trigger signals that affect cell development and function. A protein called Sonic Hedgehog is the ligand for the patched-1 receptor. Patched-1 blocks cell growth and division (proliferation) until Sonic Hedgehog is attached.
The PTCH1 gene is a tumor suppressor gene, which means it stops cells from proliferating too rapidly or in an uncontrolled way. Mutations in this gene prevent the production of patched-1 or lead to the production of an abnormal version of the receptor. An altered or missing patched-1 receptor cannot effectively suppress cell growth and division. As a result, cells proliferate uncontrollably to form the tumors that are characteristic of Gorlin syndrome. It is less clear how PTCH1 gene mutations cause the other signs and symptoms related to this condition.
The characteristic features of Gorlin syndrome can also be associated with a chromosomal change called a 9q22.3 microdeletion, in which a small piece of chromosome 9 is deleted in each cell. This deletion includes the segment of chromosome 9 that contains the PTCH1 gene, and as a result, people with a 9q22.3 microdeletion are missing one copy of this gene. Loss of this gene underlies the signs and symptoms of Gorlin syndrome in people with 9q22.3 microdeletions. Affected individuals also have features that are not typically associated with Gorlin syndrome, including delayed development, intellectual disability, overgrowth of the body (macrosomia), and other physical abnormalities. Researchers believe that these other signs and symptoms may result from the loss of additional genes in the deleted region of chromosome 9.
Mutations in the PTCH1 gene account for 50 to 85 percent of cases of Gorlin syndrome, while mutations in other genes, some known and some unidentified, are responsible for the remaining cases.
Gorlin syndrome inheritance pattern
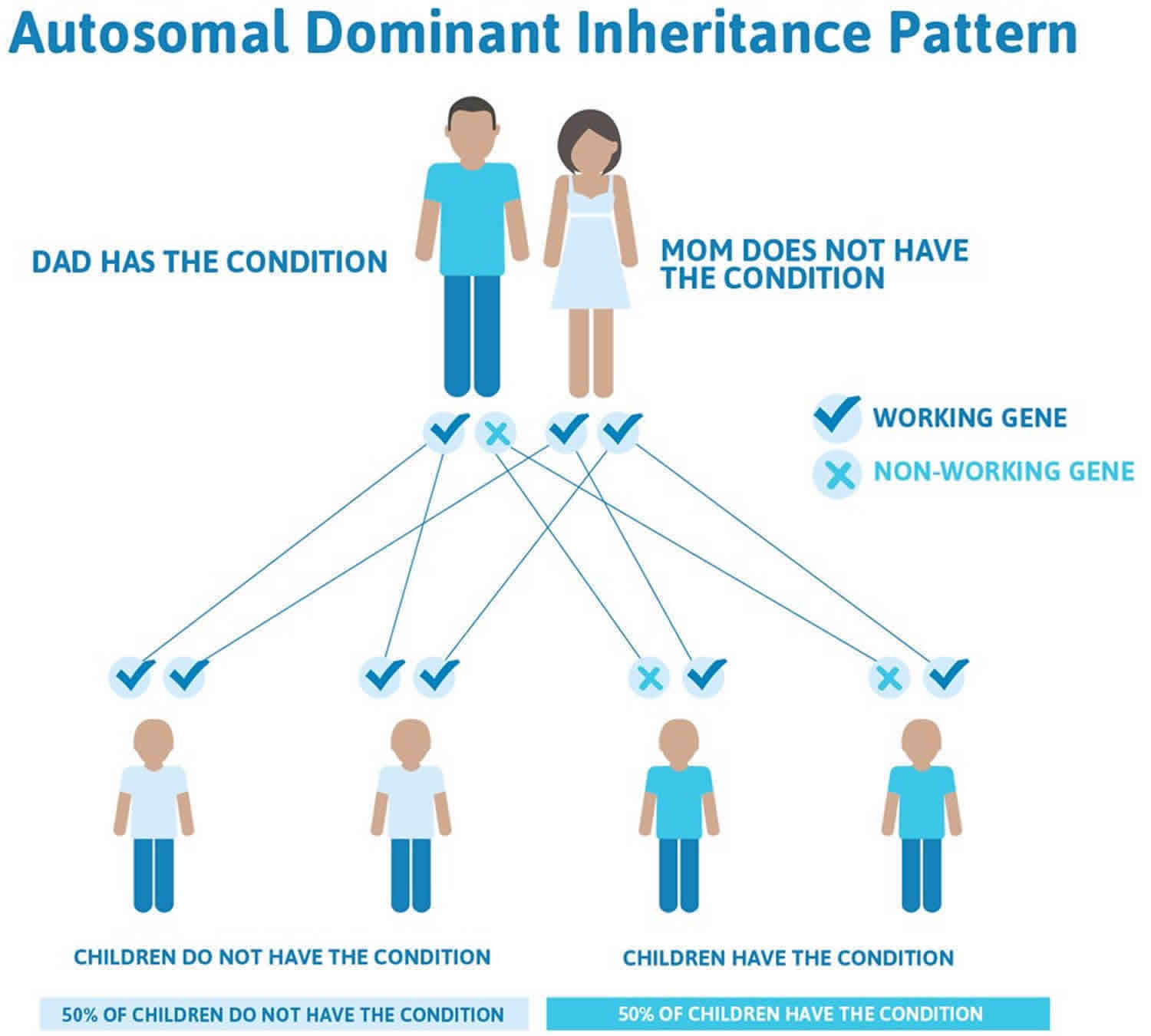
Gorlin syndrome is inherited in an autosomal dominant pattern, which means one copy of the altered gene in each cell is sufficient to cause the condition. In most cases, an affected person inherits the mutation from one affected parent. Other cases result from new mutations in the gene and occur in people with no history of the disorder in their family.
Having one mutated copy of the PTCH1 gene in each cell is enough to cause the features of Gorlin syndrome that are present early in life, including macrocephaly and skeletal abnormalities. For basal cell carcinomas and other tumors to develop, a mutation in the second copy of the PTCH1 gene must also occur in certain cells during the person’s lifetime. Most people who are born with one PTCH1 gene mutation eventually acquire a second mutation in some cells and consequently develop various types of tumors.
Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
There are cases of autosomal dominant gene changes, or mutations, where no one in the family has it before and it appears to be a new thing in the family. This is called a de novo mutation. For the individual with the condition, the chance of their children inheriting it will be 50%. However, other family members are generally not likely to be at increased risk.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. Gorlin syndrome autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Basal cell nevus syndrome symptoms
More than 100 features that are variable within and among families have been associated with nevoid basal cell carcinoma syndrome 3. Findings are presented here in the usual order of manifestation.
Macrocephaly (large head). The first feature likely to be observed is relative macrocephaly. A large proportion of babies with nevoid basal cell carcinoma syndrome require delivery by cesarean section because of large head size. After birth, the head growth pattern often resembles that of arrested hydrocephalus, but hydrocephaly requiring treatment is rare. Head circumference increases above the 97th centile until age ten to 18 months and then maintains its centile.
Other congenital malformations, found in approximately 5%, include cleft lip/palate (5%), polydactyly, and severe eye anomalies. Eye findings include strabismus, cataract, orbital cyst, microphthalmia, and pigmentary changes of the retinal epithelium 4.
Gross motor delay. There is often some delay in motor milestones; most individuals catch up by about age five years. No published psychometric evidence for global delay exists.
Medulloblastoma. Approximately 5% of all individuals with nevoid basal cell carcinoma syndrome develop the childhood brain malignancy medulloblastoma now often called primitive neuroectodermal tumor 5. The tumor tends to be of desmoplastic histology 6 and to have a favorable prognosis. Peak incidence of medulloblastoma in nevoid basal cell carcinoma syndrome is at approximately age one to two years, compared to age seven years in its sporadic form 6.
More recently, nonsense and missense variants and multiexon deletion of SUFU were identified in three families with classic nevoid basal cell carcinoma syndrome features; one individual in each family had medulloblastoma 7. SUFU-related nevoid basal cell carcinoma syndrome is associated with a high risk for medulloblastoma of up to 33% (3/9) and a high meningioma risk post radiation. The risk for medulloblastoma in PTCH1-related nevoid basal cell carcinoma syndrome was less than 2% 7.
Facies. Approximately 60% of individuals with a PTCH1 pathogenic variant have a recognizable appearance with frontal bossing, coarse facial features, and facial milia. Facial features are likely more subtle in individuals with an SUFU pathogenic variant.
Skeletal features. Congenital bone anomalies are present at birth but will not be evident clinically in a newborn. The shoulders slope downward. Most individuals have skeletal anomalies identified on radiographs (e.g., bifid ribs, wedge-shaped vertebrae). Severe skeletal defects resulting from multiple rib/vertebral anomalies have been reported but are uncommon, as is open spina bifida.
Ectopic calcification, particularly in the falx, is present in more than 90% of individuals by age 20 years 8. Sella calcification, when present, is visible on lateral x-rays of the skull.
Jaw keratocysts. Approximately 90% of individuals with PTCH1-related nevoid basal cell carcinoma syndrome develop multiple jaw keratocysts. They can occur as early as age five years, but the peak occurrence is in the teenage years. Jaw keratocysts usually present as painless swellings. Untreated, they can lead to major tooth disruption and fracture of the jaw. Jaw cysts rarely occur after age 30 years. Jaw cysts have not been reported in individuals with SUFU-related nevoid basal cell carcinoma syndrome 7.
A rare malignant transformation of a keratocyst called ameloblastoma has been reported in individuals with nevoid basal cell carcinoma syndrome at least six times 9.
Basal cell carcinomas (BCCs). Brownish/pink/orange basal cell nevi may occur in early childhood and may lie quiescent without evidence of aggressive behavior. The histologic appearance is that of a typical basal cell carcinoma which, when excised, can be the first, unexpected finding of nevoid basal cell carcinoma syndrome in simplex cases (i.e., affected individuals with no known family history of nevoid basal cell carcinoma syndrome), especially children. Active basal cell carcinomas may grow from existing basal cell nevi that may be numerous, or typical basal cell carcinomas may appear from virtually blemish-free skin. basal cell carcinomas may also crust, bleed, and ulcerate, or may present as a localized infection.
Basal cell carcinomas can occur in early childhood, but in general do not present until the late teens or early adulthood. They occur more frequently with age, although 10% of individuals with nevoid basal cell carcinoma syndrome never develop a basal cell carcinoma. Individuals with type 1 skin (white skin that burns, but never tans, e.g., Celtic skin) and individuals with excessive ultraviolet light exposure seem especially prone to developing large numbers of basal cell carcinomas. Clinically some affected individuals appear to be particularly radiosensitive, with new basal cell carcinomas appearing in the field of radiation following radiotherapy.
Other skin manifestations include meibomian cysts in the eyelids, sebaceous cysts, and dermoid cysts. Skin tags (especially around the neck) often have the histologic appearance of basal cell carcinomas but do not act aggressively.
Other tumors. Cardiac and ovarian fibromas occur, respectively, in approximately 2% and 20% of females 10. Cardiac fibromas are usually present at birth or soon after. They can be asymptomatic or can cause arrhythmia or obstruction of cardiac flow. Rhabdomyomas may occur at other sites as well as in the heart 11.
Ovarian fibromas occur with both SUFU and PTCH1-related nevoid basal cell carcinoma syndrome and may be more common in individuals with SUFU-related nevoid basal cell carcinoma syndrome 12. They are usually an incidental finding on ultrasound examination or at cesarean section. They may cause torsion of the ovary, but are not thought to affect fertility. They can become large and calcified; however, malignant transformation is uncommon.
The risk for other malignant tumors is not clearly increased, although lymphoma 13 and meningioma have been reported 14.
Basal cell nevus syndrome diagnosis
A diagnosis of basal cell nevus syndrome can be made if there are 2 major or 1 major and 2 minor criteria 15.
Major criteria
- Multiple (>2) basal cell carcinomas at any age or one basal cell carcinoma less than 20 years or >10 basal cell nevi
- Histologically proven odontogenic keratocyst or a polyostotic bone cyst
- Palmar or plantar pits (3 or more)
- Ectopic calcification: lamellar or early (<20 years) calcification of the falx cerebri. Falx calcification is nearly always present and is visible on anteroposterior (AP) x-rays of the skull after age 20 years.
- Family history of basal cell nevus syndrome
Minor criteria
- Congenital skeletal defects: bifid, fused, splayed, or missing rib, or bifid, wedged, or fused vertebra. Vertebral/rib anomalies observed on chest x-ray and/or spinal x-ray
- Large head or macrocephaly with occipitofrontal circumference >97th percentile, with frontal bossing
- Cardiac or ovarian fibroma (benign tumor in heart or ovary)
- Childhood medulloblastoma also called primitive neuroectodermal tumor (a malignant brain tumor that usually arises in young children); this is usually associated with SUFU gene mutations
- Lympho-mesenteric cysts (abdominal cysts full of lymph fluid) or pleural cysts
- Congenital malformation: cleft lip and/or cleft palate, polydactyly (extra fingers or toes), congenital eye defect such as cataract, microphthalmos (small eye) or coloboma (iris tumor)
Basaloid follicular hamartoma has also been linked to a mutation in the patched gene but is a benign tumor. There are solitary and multiple hereditary variants.
Notes regarding radiographs
- To verify a clinical diagnosis of nevoid basal cell carcinoma syndrome, AP and lateral x-rays of the skull, an orthopantogram, chest x-ray, and spinal x-ray are usually necessary.
- Clinicians should avoid using x-rays in childhood if the diagnosis is obvious without them or if a known pathogenic variant exists in the family.
- If radiographs have already been taken (i.e., before the diagnosis of nevoid basal cell carcinoma syndrome is being considered) it is preferable to obtain and review the original radiographs rather than repeat them because individuals with nevoid basal cell carcinoma syndrome are susceptible to x-irradiation.
- Even when present, bifid ribs, bifid vertebrae, and falx calcification are often not mentioned in formal reports of radiographic findings, as these can also be normal variations in the general population.
- X-ray findings may be helpful in suggesting or confirming the diagnosis in young children with cardiac fibromas, cleft lip/palate, polydactyly, or macrocephaly 16.
Establishing the diagnosis
The diagnosis of nevoid basal cell carcinoma syndrome is established in a proband with the following findings:
- Two major diagnostic criteria and one minor diagnostic criterion or one major and three minor diagnostic criteria 17. A similar series of diagnostic criteria was proposed by Kimonis et al 18. No study has been able to assess which combination of diagnostic criteria represents the best trade-off between sensitivity and specificity.
- Identification of a heterozygous germline PTCH1 or SUFU pathogenic variant on molecular genetic testing. This finding establishes the diagnosis if clinical features are inconclusive.
- Note: (1) Occasional variants in PTCH2 have been found in individuals with nevoid basal cell carcinoma syndrome but these may not be conclusive 19. Likewise, SUFU pathogenic variants may not always cause typical nevoid basal cell carcinoma syndrome. (2) Identification of an identical PTCH1 pathogenic variant in two or more separate tumors but not present (or present at a lower-than-normal ratio) in lymphocyte DNA confirms the presence of mosaicism 20.
Basal cell nevus syndrome treatment
The first sign of basal cell nevus syndrome may be the development of a medulloblastoma in a child aged 2 to 5 years, but luckily this is uncommon. Only a few children with medulloblastoma also have basal cell nevus syndrome. If detected early enough, the tumor may be treated by surgery and chemotherapy.
Patients with basal cell nevus syndrome often require surgery to remove jaw cysts in their 20s. Often, it is not until they are in their 30s or 40s that the basal cell carcinomas begin to appear so the diagnosis of the syndrome is often delayed.
All patients with basal cell nevus syndrome should see a dermatologist for regular skin examinations so that basal cell carcinomas can be treated when they are small. This may require surgery or one of the many other treatments available for these tumors including cryotherapy, photodynamic therapy, fluorouracil cream and imiquimod cream. They should not receive treatment with irradiation as this is liable to provoke the development of more tumors.
Some patients may require long term treatment with oral retinoids such as isotretinoin or acitretin. Advanced basal cell carcinomas may sometimes be treated with vismodegib.
Affected individuals should avoid UV exposure and cover up exposed skin by wearing long sleeves, high collars, and hats; complete sunblock should be used.
Avoid unnecessary radiation exposure from the environment, investigative radiology, or radiotherapy treatment.
Sun protection is vital to reduce the number of skin cancers developing but even complete protection will not prevent all basal cell carcinomas in patients with basal cell nevus syndrome.
Surveillance
Head circumference should be followed throughout childhood and plotted on appropriate growth charts. Rapid enlargement should prompt evaluation for possible hydrocephalus.
Awareness of the risk of medulloblastoma in the first years of life is important and may justify developmental assessment and physical examination every six months. No evidence for the efficacy of regular neuroimaging exists; frequent computed tomography scans should be avoided because of risks associated with radiation sensitivity. A consensus meeting has suggested annual head MRI scans until age eight years in affected children 21, but this would require general anesthesia for many children and is probably not now justified in PTCH1-related Gorlin syndrome (basal cell nevus syndrome) with only a 2% risk 22. However, it may well be justified in infants with SUFU pathogenic variants [Smith et al 2014]; this has been supported by a consensus statement recommendation to “consider brain MRI every four months through age three years, then brain MRI every six months until the age five years” 22.
A baseline heart ultrasound examination in infants has been advocated by Foulkes et al 22.
Ovarian ultrasound in women at age 18 has been advocated by Foulkes et al 22.
No other tumors occur at a frequency that warrants surveillance above that offered to members of the general population.
Orthopantogram is indicated every 12-18 months in individuals older than age eight years to identify jaw keratocysts 22.
Skin should be examined at least annually; some physicians recommend skin examination by a professional every three to four months.
Agents and circumstances to avoid
Use of radiotherapy can lead to the development of thousands of basal cell carcinomas in the radiation field 23 and therefore should be avoided if there are alternative treatments, especially in childhood. If the treating team believes that no other treatment modality is possible, radiotherapy should be used through as few skin ports as possible.
Diagnostic x-rays should be used sparingly.
Individuals with Gorlin syndrome (basal cell nevus syndrome) should be advised to avoid direct sun exposure as much as possible. Excessive sun exposure increases the likelihood of developing basal cell carcinomas.
Pregnancy management
Since individuals with Gorlin syndrome (basal cell nevus syndrome) have a large head circumference, a woman who is carrying an affected fetus should be assessed for the need for either early induction of labor or cesarean section delivery due to cephalopelvic disproportion.
Basal cell nevus syndrome life expectancy
Life expectancy in nevoid basal cell carcinoma syndrome is not significantly different from average 24. The major problem is with the cosmetic effect of treatment of multiple skin tumors and usually, to a lesser extent, treatment of jaw keratocysts. A poor cosmetic outcome can lead to social difficulties, including difficulty maintaining employment.
- Gorlin syndrome. https://ghr.nlm.nih.gov/condition/gorlin-syndrome[↩][↩]
- Witmanowski, Henryk et al. “Basal cell nevus syndrome (Gorlin-Goltz syndrome): genetic predisposition, clinical picture and treatment.” Postepy dermatologii i alergologii (2017).[↩]
- Farndon PA. Gorlin (naevoid basal cell carcinoma) syndrome. In: Eeles R, Easton DF, Ponder BAJ, Eng C, eds. Genetic Predisposition to Cancer. London, UK: Hodder Arnold; 2004:193-213.[↩]
- Ragge NK, Salt A, Collin JR, Michalski A, Farndon PA. Gorlin syndrome: the PTCH gene links ocular developmental defects and tumour formation. Br J Ophthalmol. 2005;89:988–91.[↩]
- Cowan R, Hoban P, Kelsey A, Birch JM, Gattamaneni R, Evans DG. The gene for the naevoid basal cell carcinoma syndrome acts as a tumour-suppressor gene in medulloblastoma. Br J Cancer. 1997;76:141–5.[↩]
- Amlashi SF, Riffaud L, Brassier G, Morandi X. Nevoid basal cell carcinoma syndrome: relation with desmoplastic medulloblastoma in infancy. A population-based study and review of the literature. Cancer. 2003;98:618–24.[↩][↩]
- Smith MJ, Beetz C, Williams SG, Bhaskar SS, O’Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, Cheesman E, Kelsey A, McCabe MG, Newman WG, Evans DG. Germline mutations in SUFU cause Gorlin syndrome-associated childhood medulloblastoma and redefine the risk associated with PTCH1 mutations. J Clin Oncol. 2014;32:4155–61.[↩][↩][↩]
- Kimonis VE, Mehta SG, Digiovanna JJ, Bale SJ, Pastakia B. Radiological features in 82 patients with nevoid basal cell carcinoma (NBCC or Gorlin) syndrome. Genet Med. 2004;6:495–502.[↩]
- Ponti G, Pollio A, Mignogna MD, Pellacani G, Pastorino L, Bianchi-Scarrà G, Di Gregorio C, Magnoni C, Azzoni P, Greco M, Seidenari S. Unicysticameloblastoma associated with the novel K729M PTCH1 mutation in a patient with nevoid basal cell carcinoma (Gorlin) syndrome. Cancer Genet. 2012;205:177–81.[↩]
- Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome. Genet Med. 2004;6:530–9.[↩]
- Watson J, Depasquale K, Ghaderi M, Zwillenberg S. Nevoid basal cell carcinoma syndrome and fetal rhabdomyoma: a case study. Ear Nose Throat J. 2004;83:716–8.[↩]
- Evans DG, Oudit D, Smith MJ, Rutkowski D, Allan E, Newman WG, Lear JT. First evidence of genotype-phenotype correlations in Gorlin syndrome. J Med Genet. 2017;54:530–6.[↩]
- Pereira CM, Lopes AP, Meneghini AJ, Silva AF, Botelho Tde L. Oral diffuse B-cell non-Hodgkin’s lymphoma associated to Gorlin-Goltz syndrome: a case report with one year follow-up. Indian J Pathol Microbiol. 2011;54:388–90.[↩]
- Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer. 2012;11:565–70.[↩]
- Evans DG, Farndon PA. Nevoid Basal Cell Carcinoma Syndrome. 2002 Jun 20 [Updated 2018 Mar 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1151[↩]
- Debeer P, Devriendt K. Early recognition of basal cell naevus syndrome. Eur J Pediatr. 2005;164:123–5.[↩]
- Evans DG, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complications of the naevoid basal cell carcinoma syndrome: results of a population based study. J Med Genet. 1993;30:460–4.[↩]
- Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308.[↩]
- Fujii K, Ohashi H, Suzuki M, Hatsuse H, Shiohama T, Uchikawa H, Miyashita T. Frameshift mutation in the PTCH2 gene can cause nevoid basal cell carcinoma syndrome. Fam Cancer. 2013;12:611–4.[↩]
- Evans DG, Ramsden RT, Shenton A, Gokhale C, Bowers NL, Huson SM, Wallace A. Mosaicism in NF2 an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including MLPA. J Med Genet. 2007;44:424–8.[↩]
- Bree AF, Shah MR, et al. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155A:2091–7.[↩]
- Foulkes WD, Kamihara J, Evans DGR, Brugières L, Bourdeaut F, Molenaar JJ, Walsh MF, Brodeur GM, Diller L. Cancer Surveillance in Gorlin Syndrome and Rhabdoid Tumor Predisposition Syndrome. Clin Cancer Res. 2017;23:e62–e67.[↩][↩][↩][↩][↩]
- Evans DG, Birch JM, Orton CI. Brain tumours and the occurrence of severe invasive basal cell carcinoma in first degree relatives with Gorlin syndrome. Br J Neurosurg. 1991a;5:643–6.[↩]
- Wilding A, Ingham SL, Lalloo F, Clancy T, Huson SM, Moran A, Evans DG. Life expectancy in hereditary cancer predisposing diseases: an observational study. J Med Genet. 2012;49:264–9.[↩]





{kind=link}