Griscelli syndrome
Griscelli syndrome also called Griscelli-Pruniéras syndrome, is a rare hereditary cutaneous disease characterized by a silvery-gray sheen of the hair and hypopigmentation of the skin, which can be associated to primary neurological impairment (type 1), immunologic impairment (type 2) or be isolated (type 3). Griscelli syndrome type 2 appears to be the most common of the three known types, while Griscelli syndrome type 3 is the least common. Griscelli syndrome is a form of partial albinism. Griscelli syndrome usually presents in infancy or early childhood, in most cases between the ages of 4 months and 7 years. Untreated, most children with Griscelli syndrome die in early childhood 1.
Griscelli syndrome is a rare condition, with a prevalence of less than 1 per million. Most cases are reported in Turkish and Mediterranean populations. To date, approximately 150 cases have been reported, predominantly in Turkish and Mediterranean populations.
Griscelli syndrome is an autosomal recessive condition meaning that two defective genes are inherited, one from each parent.
Griscelli syndrome is caused by genetic mutations leading to defective transport of melanosomes. Melanosomes are pigment granules containing melanin that are found within melanocytes. Normally, the melanosomes are moved from the centre of the melanocyte to its border so that melanin can be transferred to surrounding keratinocytes (skin cells). When transportation fails, melanosomes accumulate within the melanocyte and the skin appears pale.
Figure 1. Griscelli syndrome
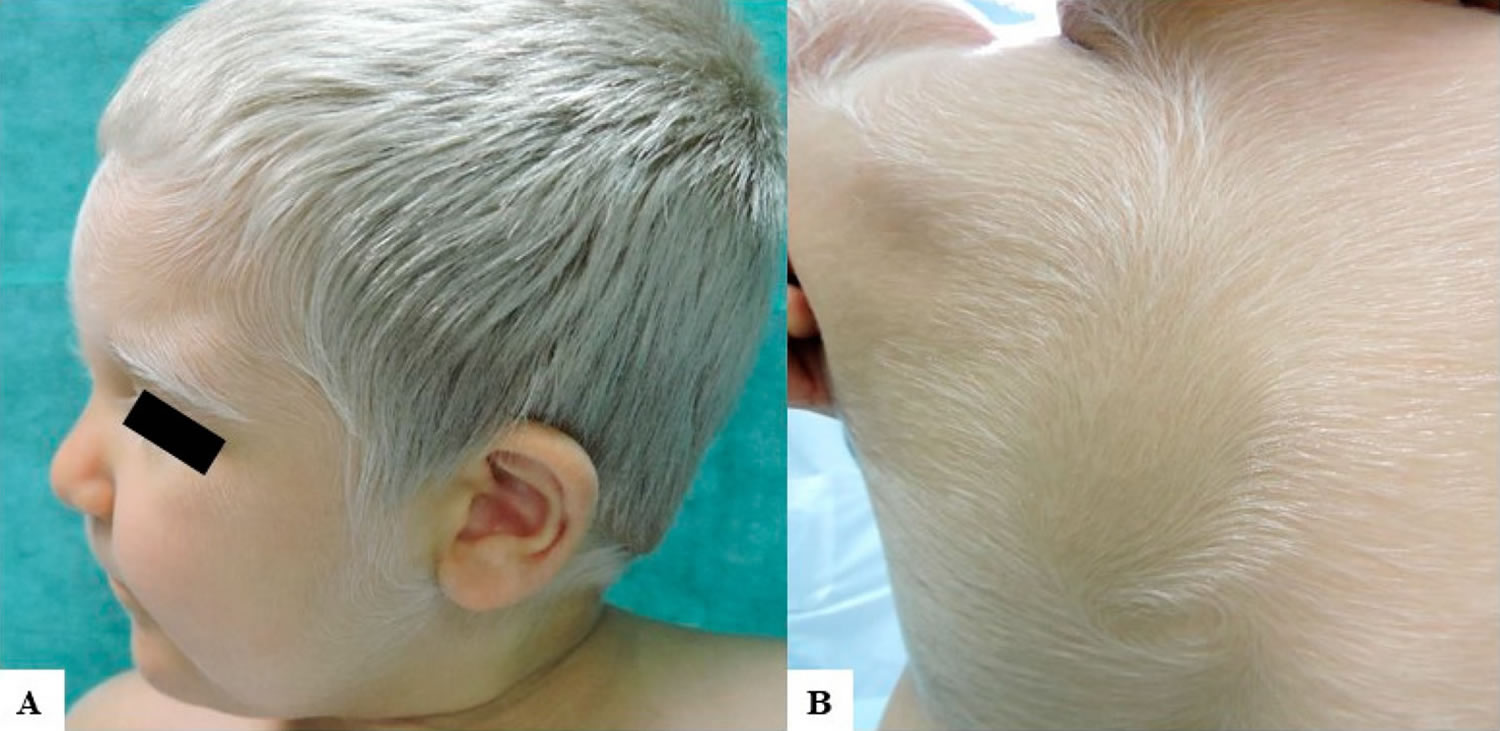
Footnote: (A) A 2-year-old Caucasian child with congenital silvery gray scalp hair, eyebrows, and eyelashes and fair skin. He had also brown irises and very fair skin with mild tanning capacities after sun exposure; (B) Diffuse silvery hypertrichosis: unmedullated, short, and soft hypopigmented hairs on the back of the patient.
[Source 2 ]Griscelli syndrome type 1
Griscelli syndrome type 1 involves severe problems with brain function in addition to the distinctive skin and hair coloring. Affected individuals typically have neurological dysfunction with delayed development, intellectual disability, seizures, weak muscle tone (hypotonia), and eye and vision abnormalities. Another condition called Elejalde disease has many of the same signs and symptoms, and some researchers have proposed that Griscelli syndrome type 1 and Elejalde disease are actually the same disorder.
Griscelli syndrome type 1 is due to a defect in the MYO5A gene (myosin VA, myoxin). Neurological problems are often present at birth or develop in infancy. These include changes in muscle tone or paralysis, seizures, and developmental delay.
Additional findings in type 1 and 2 Griscelli syndrome include:
- Generalized lymphadenopathy (swollen lymph nodes)
- Enlargement of the liver and spleen
- Hepatitis (liver inflammation)
- Eye defects including partial ocular albinism
- Pancytopaenia (reduced numbers of red cells, white cells and platelets)
Griscelli syndrome type 2
People with Griscelli syndrome type 2 have immunological deficiency in addition to having hypopigmented skin and hair. Affected individuals are prone to recurrent infections. They also develop an immune condition called hemophagocytic lymphohistiocytosis (HLH), in which the immune system produces too many activated immune cells called T-lymphocytes and macrophages (histiocytes). Overactivity of these cells can damage organs and tissues throughout the body, causing life-threatening complications if the condition is untreated. People with Griscelli syndrome type 2 do not have the neurological abnormalities of type 1.
Griscelli syndrome type 2 is due to a defect in the RAB27A gene (a member of the RAS oncogene family). There is uncontrolled T lymphocyte and macrophage activation leading to hemophagocytic lymphohistiocytosis (HLH). Hemophagocytic lymphohistiocytosis is a blood disorder where activated macrophages (a type of blood cell) phagoctyose (engulf) other blood cells. Reduced numbers of circulating blood cells leads to anaemia, bleeding tendency and increased risk of infection.
Additional findings in type 1 and 2 Griscelli syndrome include:
- Generalized lymphadenopathy (swollen lymph nodes)
- Enlargement of the liver and spleen
- Hepatitis (liver inflammation)
- Eye defects including partial ocular albinism
- Pancytopaenia (reduced numbers of red cells, white cells and platelets)
Griscelli syndrome type 3
Unusually light skin and hair coloring without systemic problems are the only features of Griscelli syndrome type 3. People with Griscelli syndrome type 3 of the disorder do not have neurological abnormalities or immune system problems.
Griscelli syndrome type 3 is caused by mutation in either the MYO5A or MLPH (melanophilin) gene.
What causes Griscelli syndrome?
The three types of Griscelli syndrome are caused by mutations in different genes: Type 1 results from mutations in the MYO5A gene, type 2 is caused by mutations in the RAB27A gene, and type 3 results from mutations in the MLPH gene.
The proteins produced from these genes are found in pigment-producing cells called melanocytes. Within these cells, the proteins work together to transport structures called melanosomes. These structures produce a pigment called melanin, which is the substance that gives skin, hair, and eyes their color (pigmentation). Melanosomes are formed near the center of melanocytes, but they must be transported to the outer edge of these cells and then transferred into other types of cells to provide normal pigmentation.
Mutations in any of the three genes, MYO5A, RAB27A, or MLPH, impair the normal transport of melanosomes within melanocytes. As a result, these structures clump near the center of melanocytes, trapping melanin within these cells and preventing normal pigmentation of skin and hair. The clumps of pigment, which can be seen in hair shafts when viewed under a microscope, are a hallmark feature of the condition.
In addition to their roles in melanosome transport, the MYO5A and RAB27A genes have functions elsewhere in the body. Specifically, the protein produced from the MYO5A gene transports materials within nerve cells (neurons) that appear to be critical for cell function. The protein produced from the RAB27A gene is found in immune system cells, where it is involved in the release of certain compounds that kill foreign invaders (such as viruses and bacteria). Mutations in these genes impair these critical cell activities, leading to the neurological problems and immune system abnormalities found in Griscelli syndrome types 1 and 2, respectively.
Griscelli syndrome inheritance pattern
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
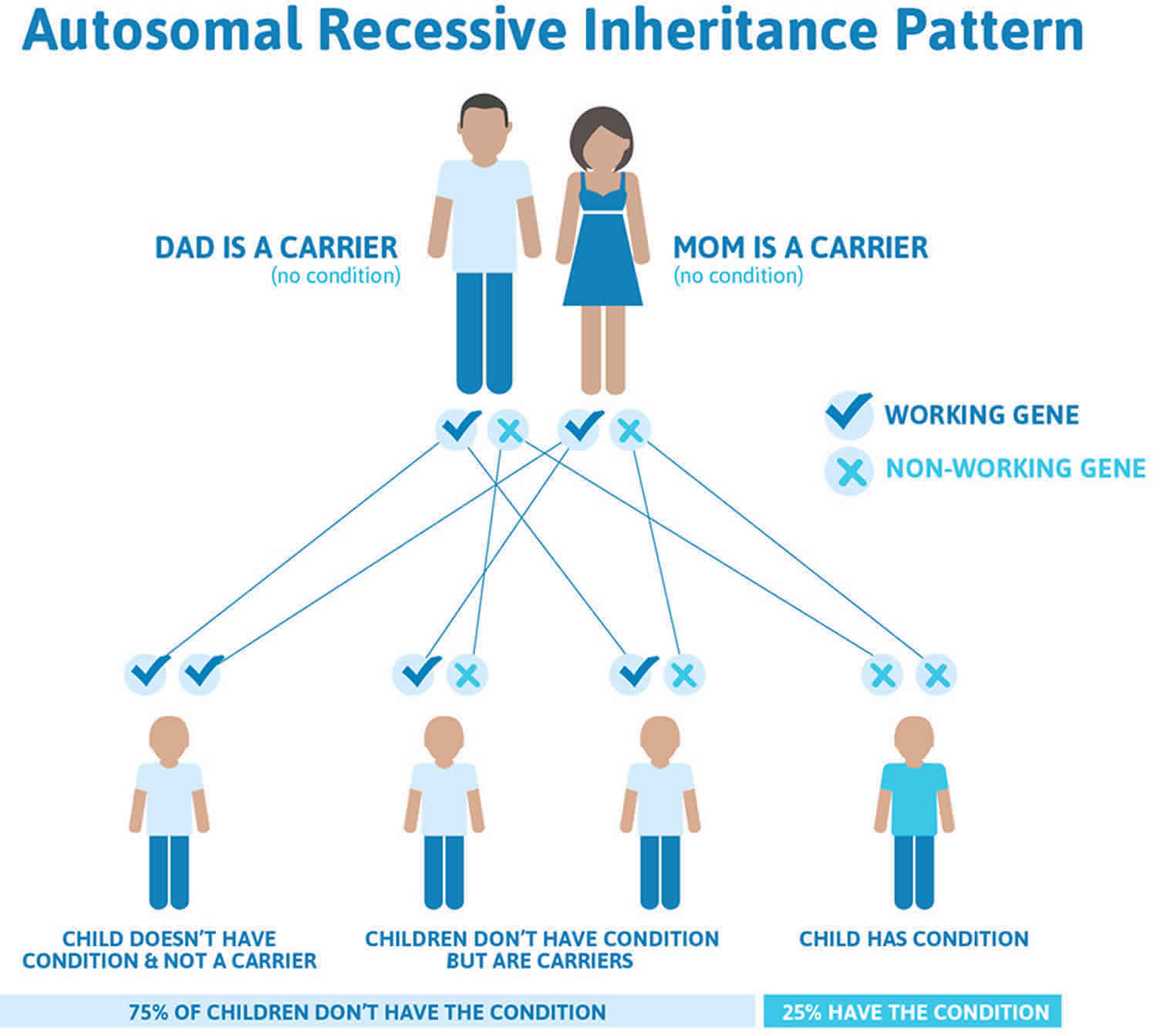
Figure 2 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 2. Griscelli syndrome autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Griscelli syndrome symptoms
Griscelli syndrome occurs in infancy to childhood. In addition to the silvery-gray sheen of the hair and the light-colored skin, Griscelli syndrome type 1 patients present with delayed motor development, intellectual disability and hypotonia. Griscelli syndrome type 2 patients have the same hypopigmentation features but in association with immune pathology. Patients exhibit a lymphocyte cytotoxic defect resulting in an uncontrolled T-lymphocyte and macrophage-activation syndrome, also known as hemophagocytic syndrome (HLH), in which activated T cells and macrophages infiltrate the lymph nodes and other organs (including the brain), producing hemophagocytosis. Patients with Griscelli syndrome type 2 can present neurological symptoms due to brain infiltration by the activated hematopoietic cells. In Griscelli syndrome type 3 patients, hypopigmentation of the skin and hair is the only feature.
Griscelli syndrome diagnosis
The diagnosis of the three types of Griscelli syndrome can be established by the clinical signs and light microscopic examination, evidencing large clumps of pigment in hair shafts and the accumulation of mature melanosomes in melanocytes. A decrease in T and NK lymphocyte degranulation and cytotoxicity characterize Griscelli syndrome type 2. No immunological or cytotoxic defects have been observed in Griscelli syndrome type 1 or 3. Thus, based on the patient’s clinical and biological features, sequencing of the corresponding causative gene allows confirmation of the type of Griscelli syndrome.
Griscelli syndrome should be considered in a child with silver hair, eyebrows and eyelashes, particularly if there are neurological or immune defects.
Microscopic analysis of hair shafts shows clumps of pigment. Histological examination of a skin biopsy may reveal prominent melanosomes within epidermal and hair follicle melanocytes.
In suspected Griscelli syndrome, chromosomal analysis may be performed to look for the specific gene defect.
Antenatal diagnosis
Antenatal diagnosis of Griscelli syndrome type 1 and 2 can be performed through chorionic villus sampling (CVS) by the sequencing of the MYO5A or RAB27A gene, respectively.
Griscelli syndrome treatment
Treatment for Griscelli syndrome type 1 is only symptomatic. In Griscelli syndrome type 2, the hemophagocytic syndrome is often fatal and the only cure is hematopoietic stem cell transplantation. Currently there is no specific management for Griscelli syndrome type 3.
Griscelli syndrome life expectancy
The prognosis for long-term survival of patients with Griscelli syndrome is relatively poor. In the form caused by the RAB27A defect, Griscelli syndrome type 2 is usually rapidly fatal within 1-4 years without treatment with hematopoietic stem cell transplantation (bone marrow transplantation) at onset of an accelerated phase. Some patients die after transplantation, and some patients have had lasting remissions. The mean patient age at the time of death is 5 years.
The prognosis of Griscelli syndrome type 1 is good. Griscelli syndrome type 3 should be better considered as a pigmentation phenotype rather than a pathology with a prognosis similar to the control population.





{kind=link}