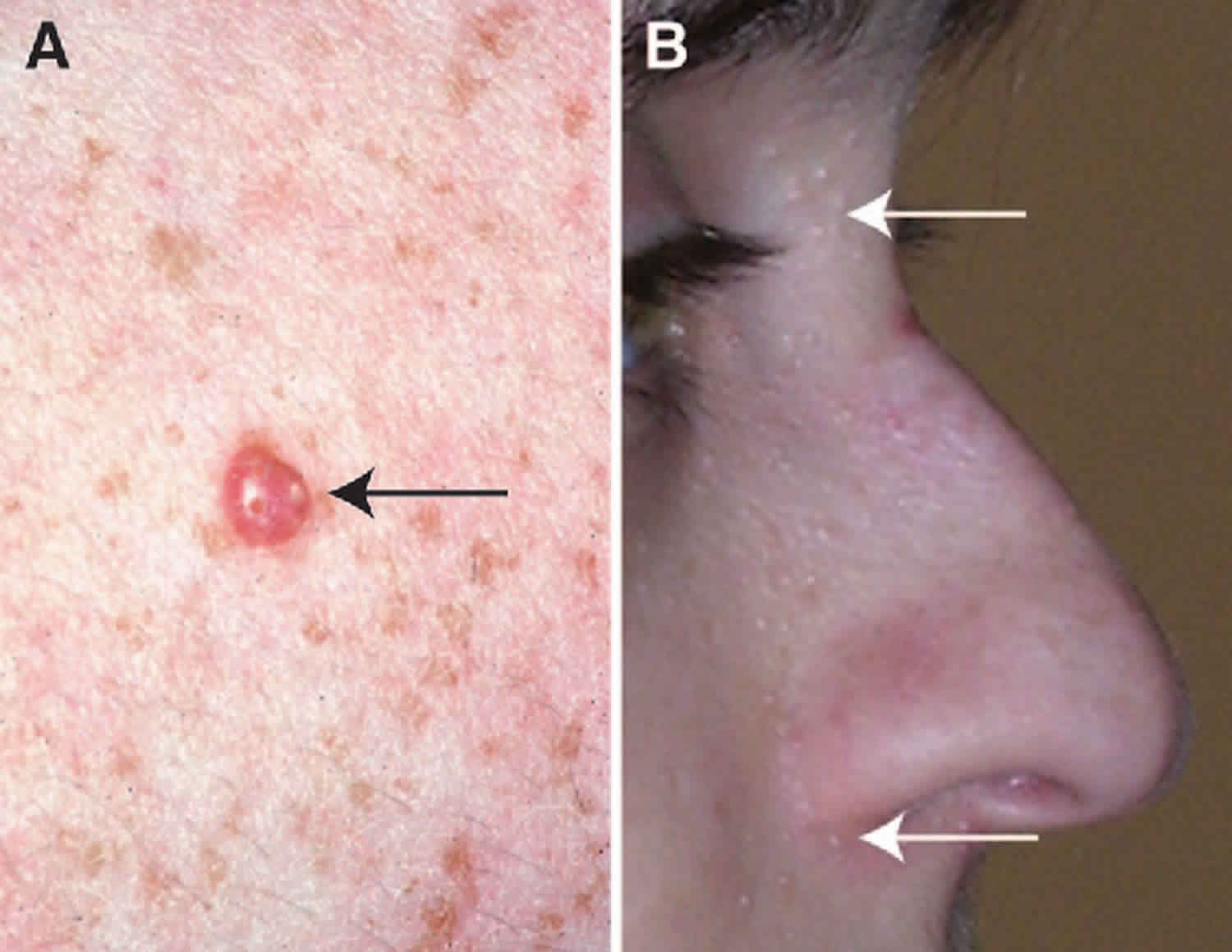
What is hamartoma
Hamartoma is a benign (not cancer) growth made up of an abnormal mixture of cells and tissues normally found in the area of the body where the growth occurs. A lesion first described by German pathologist Eugen Albrecht in 1904, hamartomas are generally benign tumors that may occur in the lungs, skin, heart, breast and other regions of the body 1. The word hamartoma derives from “hamartia,” the Greek word for ‘erroneous,” or “faulty” 2. The cellular make-up of a hamartoma is that of an abnormal mixture of tissue components that are common in the organ of origin, though organ architecture is usually not preserved within the lesion.
The literature describes several examples of hamartomas, including the following 3:
- Hemangioma and other vascular tumors that are not true neoplasms
- Peutz-Jeghers polyp of the bowel, juvenile or retention
- Polyp of the large bowel
- Bronchial hamartoma
- Melanocytic nevi
- Neurofibromatosis in von Recklinghausen disease
- Neuroepithelial cells in tuberous sclerosis
- Hamartomas of the hypothalamus and tuber cinereum
- A variety of bony hamartomas
A hamartoma resembles a neoplasm, but in most cases, it does not show any tendency to evolve into one. However, cases of neoplastic evolution have occurred with these lesions. Examples are chondrosarcomas arising in osteochondromas and neurofibrosarcomas arising in patients with von Recklinghausen disease.
In addition, neoplasms can be associated with hamartomas without directly arising from them. Examples are fibromas of the ovary and malignant ovarian tumors arising in patients with Peutz-Jeghers syndrome. Hamartomas should be distinguished from choristomas; the former are composed of tissues that are normally present at a given site, whereas the latter are malformations of tissues that are not normally found at that site.
Hamartomas may not cause any problems and are usually identified incidentally. Uncomplicated hamartomas have no tendency to grow, except as determined by the normal growth controls of the body. However, this does not mean that hamartomas are harmless. Morbidity can arise by means of a variety of mechanisms, including the following:
- Obstruction
- Pressure, direct or indirect
- Infection
- Infarction
- Hemorrhage and iron deficiency anemia
- Fracture
- Misdiagnosis of neoplasm
- Neoplastic transformation
Hamartoma breast
Breast hamartoma is an uncommon benign breast tumor that accounts for approximately 4.8% of all benign breast masses 4, that contains lobular breast tissue involving various fibrous, fibrocystic and adipose tissues 5. The pathogenesis is still poorly understood and breast hamartoma is not a well-known disorder, so its diagnosis is underestimated by clinicians and pathologists. Breast hamartomas are underdiagnosed because of the categorization of hamartomas as fibroadenomas by pathologists. Pathologic examinations can show variability from one case to another. Thus, the true incidence may be higher than the literature indicates. With increasing social awareness and widespread breast cancer screening, hamartomas are being routinely diagnosed with greater frequency 4. The pathogenesis of breast hamartomas remains unclear and its diagnosis is underestimated by clinicians and pathologists. In the literature, breast hamartoma is presented only in case reports and reviews of wide series are very rare.
The causes of breast hamartomas is not clear but they are thought to result from dysgenesis 6 rather than a true tumorous process. However, female sex steroid hormones 7 have been implicated in the development of breast hamartomas. In one study, Herbert et al. 8 reported estrogen receptor (ER) and progesterone receptor (PR) positivity in epithelial cells and stromal cells in all 24 cases with breast hamartomas. Additionally, there are no clear data on the source of smooth muscle for myoid hamartomas, but this muscle could derive from vessels, the nipples, undifferentiated breast stromal tissue or myoepithelial cells 9. Another hypothesized smooth muscle source is the metaplasia of breast stromal cells 9 into smooth muscle cells. The existence of CD34 on smooth muscles 10 is an important sign of the metaplasia of stromal cells into smooth muscle cells, but there are no clear data on this subject. Myoid hamartomas stain strongly positive for SMA, desmin and vimentin 11 by immunohistochemical staining. In our study, only 1 patient of 27 was diagnosed with myoid hamartoma and immunohistochemical staining for desmin, SMA and CD34 was positive, consistent with the literature.
Breast hamartomas have a typical mammographic appearance. The radiolucent lesions include fat; various amounts of fibrous and adenomatous tissues, with smooth rims; and occasionally, a thin capsule 12. US evaluates breast hamartomas as having sonolucent fat and a heterogeneous internal echo pattern with echogenic fibrous components 13. Alongside US, MRI characteristically shows a smooth and well-defined hypointense rim, internal heterogeneous enhancement and the presence of fat density 7. In the present study, US was used for all patients as the first imaging procedure and in addition to US, mammography was utilized for 6 patients. MRI was used only for 1 patient with invasive ductal carcinoma.
The pathologic features of hamartomas are not well known. The original definition consisted of a fibrous fatty stroma including various amounts of epithelial elements, as well as nodular lesions. The fibrous and fatty tissues were used for the early classification of hamartomas: McGuire et al. 14 classified breast hamartomas as fibrous, fatty or fibrous-fatty. Jones et al. 15 suggested 4 classification groups for breast hamartomas: encapsulated fibrocystic changes, fibroadenoma with a fibrous stroma, fibroadenoma-like and surrounded adenolipoma. These classifications were not widely accepted. No criteria with detailed explanations of breast hamartomas are used by pathologists. Fechner et al. 16 described the lobular distribution and the presence of fat in breast hamartomas as distinguishing features compared with fibroadenomas. A fibrotic stroma surrounding the lobules and extending into the interlobular areas, causing obliteration, is the most frequently observed feature; most authors 17 refer to this feature as interlobular fibrosis. However, interlobular fibrosis is not specific to breast hamartomas. In sclerosing hyperplasia, an increasing number of interlobular glands are observed as round masses with interlobular fibrosis, which can mimic breast hamartomas. The absence of fatty tissue in the stromal structure is often associated with fibroadenomas, so this condition can be distinguished from sclerosing lobular hyperplasia and breast hamartomas 7. Fischer et al. 17 defined the presence of a pseudoangiomatous stroma particularly in breast hamartomas. This condition was revealed to be a consistent observation in subsequent reports and the incidence rate 7 varied between 16 and 71% 7.
Malignancies associated with breast hamartomas 18 are rarely observed, and only a few studies have reported invasive breast cancer associated with breast hamartoma. Invasive ductal carcinoma has been identified in only 1 case and lobular carcinoma in situ in 1 other case. Although epithelial hyperplasia is not a characteristic feature of breast hamartomas, ductal epithelial hyperplasia was identified in 6 cases (22.2%). Wahner-Rodler et al. 19 reported this incidence rate as 26% in their large case series. These high coincidence rates are very important. Patients with a certain or suspected diagnosis of hamartoma must be operated and these patients should never be followed, even if their tumors are small.
PTEN hamartoma tumor syndrome
PTEN hamartoma tumor syndrome refers to a spectrum of conditions that are characterized by multiple hamartomas. These conditions include 20:
- Cowden syndrome –
- Cowden syndrome is a multiple hamartoma syndrome with a high risk for benign and malignant (cancerous) tumors of the thyroid, breast, and uterus. Affected individuals usually have macrocephaly, trichilemmomas, and papillomatous papules, and present by the late 20s. The lifetime risk of developing breast cancer is 85%, with an average age of diagnosis between 38 and 46 years. The lifetime risk for thyroid cancer (usually follicular, rarely papillary, but never medullary thyroid cancer) is approximately 35%. The risk for endometrial cancer may approach 28%.
- Bannayan-Riley-Ruvalcaba syndrome – characterized by macrocephaly (large head size), hamartomas of the intestines (called hamartomatous intestinal polyps), and dark freckles on the penis.
- Proteus syndrome – characterized by overgrowth of the bones, skin, and other tissues.
- Proteus-like syndrome – people with many of the signs and symptoms associated with Proteus syndrome, but who do not meet the diagnostic criteria.
Cowden syndrome was estimated to affect 1 in 200,000 individuals; this study was conducted just as PTEN was discovered. However, because the disorder is difficult to recognize, researchers believe it is under-diagnosed, making it difficult to determine its true frequency in the general population. Men and women are affected equally with PTEN hamartoma tumor syndrome. PTEN hamartoma tumor syndrome is not more commonly found in persons of a particular racial or ethnic group.
Individuals with a variety of clinical diagnoses who ultimately have been found to carry a germline PTEN mutation as the underlying cause are said to have PTEN hamartoma tumor syndrome. When the strictest diagnostic criteria are used, patients with a personal and family history of Cowden syndrome features have up to an 85% chance to have a PTEN mutation. Patients with features of Bannayan-Riley-Ruvalcaba syndrome and with features reminiscent of but not meeting diagnostic criteria for Proteus syndrome (called Proteus-like syndrome) have also been found to have an underlying PTEN hamartoma tumor syndrome diagnosis. Once thought to be completely separate conditions, patients with features of Cowden syndrome or Bannayan-Riley-Ruvalcaba syndrome and an underlying PTEN mutation are unified as all having PTEN hamartoma tumor syndrome, with CS being a diagnosis traditionally given to adults and Bannayan-Riley-Ruvalcaba syndrome being first described in the pediatrics literature. This makes sense given that many of the characteristics first associated with Cowden syndrome tend to not appear until adulthood. The symptoms vary greatly from patient to patient, even among individuals in the same family.
PTEN hamartoma tumor syndrome is caused by changes (mutations) in the PTEN gene and is inherited in an autosomal dominant manner, which means it can be passed down in a 50-50 fashion. Treatment is based on the signs and symptoms present in each person 21.
PTEN hamartoma tumor syndrome causes
PTEN hamartoma tumor syndrome is caused by a germline mutation of PTEN, a tumor suppressor gene. PTEN stands for phosphatase tensin homologue. A tumor suppressor is a gene that slows down cell division, repairs damage to the DNA of cells, and tells cells when to die, a normal process called apoptosis. Mutations in a tumor suppressor gene often lead to cancer. The PTEN gene regulates the production of an enzyme (protein tyrosine phosphatase) which is believed to be important in stopping cell growth and starting apoptosis. Researchers believe that the PTEN gene plays a broad role in the development of human malignancies.
PTEN hamartoma tumor syndrome is inherited in an autosomal dominant pattern. Dominant genetic disorders occur when only a single copy of an abnormal gene is necessary to cause a particular disease. The abnormal gene can be inherited from either parent or can be the result of a mutated (changed) gene in the affected individual. The risk of passing the abnormal gene from an affected parent to an offspring is 50% for each pregnancy. The risk is the same for males and females.
PTEN hamartoma tumor syndrome signs and symptoms
The primary findings in PTEN hamartoma tumor syndrome include increased risk for certain types of cancer, benign tumors and tumor-like malformations (hamartomas), and neurodevelopmental disorders. The symptoms of PTEN hamartoma tumor syndrome vary greatly from person to person and can develop at any age.
Cancer in PTEN hamartoma tumor syndrome
Previous data, which focused only on patients with a clinical diagnosis of Cowden syndrome without understanding whether an underlying PTEN mutation was present, estimated lifetime breast cancer risk to be 25-50% and risk for non-medullary thyroid cancer to be 10%. Risks for endometrial (uterine) and renal cell (kidney) cancer were thought to be increased, but an exact risk level was undetermined.
Current data focusing on patients known to have PTEN hamartoma tumor syndrome provide the following lifetime risk estimates, with the majority of diagnoses occurring after age 30:
| Cancer | Lifetime Risk with PTEN hamartoma tumor syndrome (%) | Average Age at presentation |
| Breast | 85 | 40s |
| Thyroid | 35 | 30s-40s* |
| Renal Cell | 34 | 50s |
| Endometrial | 28 | 40s-50s |
| Colon | 9 | 40s |
| Melanoma | 6 | 40s |
Footnote: *Earliest age for thyroid cancer in PTEN hamartoma tumor syndrome is as early as 7 years old
Benign tumors in PTEN hamartoma tumor syndrome
Benign skin or oral lesions are very common and tend to appear in early adulthood. The most common types of benign skin lesions seen in PTEN hamartoma tumor syndrome include:
- Lipomas – benign fatty tumors which can appear just under the skin or elsewhere (breast area, GI tract)
- Acral keratosis – rough patches of skin most often seen on the extremities (arms, hands, legs, feet)
- Papillomatous skin papules – wart-like lesions which can appear anywhere, with feet and hands commonly being affected
- Mucosal papillomas – Benign overgrowth of tissue affecting the tongue, gums, or inside the nose
- Trichilemmomas – Benign tumor of the hair follicle
- Fibromas – another kind of overgrowth involving the skin and other connective tissue; may also affect tissue covering organs, such as the ovaries.
Gastrointestinal polyps are very common in adults with PTEN hamartoma tumor syndrome. Among patients who had undergone colonoscopy as part of their clinical care, >90% were found to have polyps with a mix of histological subtypes. The most common types of polyps found are hyperplastic or hamartomatous, which rarely develop into malignancy; however, adenomas, which may develop into a cancer, were also identified. Many polyps were very small and did not cause noticeable symptoms such as pain or rectal bleeding. Supported by this evidence, colorectal surveillance should be offered to any PTEN mutation carrier.
Benign breast, thyroid, and uterine lesions are also common in persons with PTEN hamartoma tumor syndrome. Some women have severe fibrocystic disease or changes which lead to multiple breast biopsies and complications with imaging. Multinodular goiter and Hashimoto’s thyroiditis may develop in children and adults. Uterine fibroids may appear and cause bleeding or discomfort to the extent that a hysterectomy is indicated without an underlying cancer diagnosis.
Vascular tumors, including hemangiomas, arteriovenous malformations, and developmental venous anomalies, have also been observed in patients with PTEN hamartoma tumor syndrome. Treatment of some lesions has been complicated by tendency for regrowth and scarring.
A small percentage of adults develop a rare tumor known as a cerebellar dysplastic gangliocytoma (Lhermitte-Duclos syndrome). Symptoms of Lhermitte-Duclos syndrome include increased intracranial pressure, impaired ability to coordinate voluntary movements (ataxia), and seizures. It is rare when a person with adult-onset Lhermitte-Duclos does not have an underlying PTEN mutation, and observing this tumor type is an automatic indicator for PTEN testing.
Neurodevelopmental problems in PTEN hamartoma tumor syndrome
Macrocephaly (large head size) is found in 94% of measured patients with PTEN hamartoma tumor syndrome and can be a helpful screening tool to identify patients for PTEN testing. In most patients, large head size is caused by overgrowth of brain tissue. The head shape also tends to be longer than wide (dolicocephaly).
Autism and other developmental disorders, such as intellectual disability and developmental delays, have been observed in patients with PTEN hamartoma tumor syndrome. In previous case series, up to 17% of children presenting with macrocephaly and an autism spectrum disorder alone were found to have an underlying PTEN mutation.
PTEN hamartoma tumor syndrome diagnosis
A diagnosis of PTEN hamartoma tumor syndrome may be suspected based upon a thorough clinical evaluation, a detailed patient history and the presence of characteristic findings. Recently, a mutation risk calculator has been developed which can estimate the risk for adults to have a PTEN mutation based on their personal history characteristics; this tool is available online at (http://www.lerner.ccf.org/gmi/ccscore/). The diagnosis can only be confirmed when a mutation of the PTEN gene is identified.
PTEN hamartoma tumor syndrome treatment
Individuals with PTEN mutations should undergo cancer surveillance and screening at the time of diagnosis as follows to enable healthcare providers to detect any tumors at the earliest, most treatable stages. Current suggested screening by age includes:
Pediatric (below age 18)
- Yearly thyroid ultrasound starting at the time of diagnosis
- Yearly skin check with physical examination
- Consider neurodevelopmental evaluation
Adults
- Monthly breast self-examination
- Yearly thyroid ultrasound and dermatologic evaluation
- Women: breast screening (at minimum mammogram) yearly beginning at age 30; MRI may also be incorporated
- Women: annual transvaginal ultrasound and/or endometrial biopsy beginning at age 30
- Colonoscopy beginning at age 35-40; frequency dependent on degree of polyposis identified
- Biannual (every other year) renal imaging (CT or MRI preferred) beginning at age 40
For patients with a family history of a particular cancer type, screening may be considered 5-10 years prior to the youngest diagnosis in the family. For example, a patient whose mother developed breast cancer at 30 may begin breast surveillance at age 25-30.
Additional treatment for PTEN hamartoma tumor syndrome is symptomatic and supportive. Various techniques may be used to treat the mucocutaneous symptoms of Cowden syndrome including topical agents, the use of extreme cold to destroy affected tissue (cryosurgery), the removal of tissue or growths by through a process called curettage, in which a surgical tool shaped like a spoon (curette) is used to scrape away affect tissue, or destroying affected tissue by exposing it to laser beams (laser ablation). Genetic counseling may be of benefit for affected individuals and their families.
Pulmonary hamartoma
Pulmonary hamartoma is benign malformation of common lung tissue, to include cartilage, epithelium, fat, or muscle. Pulmonary hamartoma is the most common benign pulmonary neoplasm in adults 1. However, pulmonary hamartoma is much more rare in children 22. Pulmonary hamartoma lesions are usually located in the peripheral parenchyma but have in rare cases been present in the central chest wall. Lesions tend to grow at the same rate as the surrounding tissue. Therefore neoplastic pressure erosion of adjacent structures is typically not noted.
Pulmonary hamartomas occur with an incidence of 0.025 to 0.032% within the adult population. They usually present in the fifth and sixth decade of life, with males being four times more likely to be affected than females 23. Although still an uncommon occurrence, these lesions are the most common benign pulmonary neoplasm, accounting for an estimated 77% of benign lung nodules and 8% of solitary lung lesions 24. Most hamartomas occur in the peripheral parenchyma, with exceptions observed in the central chest wall. Additionally, approximately 10 % of lesions present endobronchially 24. Within the pediatric population, pulmonary hamartomas are significantly rarer 23.
Historically, no specific risk factors have been identified for pulmonary hamartoma. Occasionally, a relationship between lesions that have undergone sarcomatous transformation and specific genomic alterations has been described 25, but there are currently no screening guidelines specifically designed for the early diagnosis of pulmonary hamartoma.
Pulmonary hamartoma histopathology
Also known as chondroid hamartomas, the histological makeup of these tumors is a mixture of mature mesenchymal tissue, like adipose tissue, cartilage, bone, or smooth muscle bundles, and fibromyxoid tissue, with varying proportions of each component. They are non-invasive, slow-growing, nodular lesions, sometimes displaying cleft-like spaces within, lined by respiratory epithelium 23.
Pulmonary hamartoma signs and symptoms
In adults the majority of parenchymal hamartomas produce no symptoms, often being incidental findings. Depending on the location and size of the lesion, however, patients can still develop an array of complaints, including persistent coughing or wheezing, dyspnea, hemoptysis, rhonchi, higher likelihood of pneumonia, atelectasis or even pneumothorax. Endobronchial masses additionally pose the danger of airway obstruction. Hamartomas that have become symptomatic require a thorough diagnostic approach, and surgical resection can become necessary 23.
Pulmonary hamartoma diagnosis
Pulmonary hamartomas are often incidental findings on imaging and can mimic pulmonary malignancies. Once found, or in the case of asymptomatic hamartoma, there are multiple diagnostic strategies available to determine the nature of the lesion. On imaging, e.g., chest X-ray or CT scans, masses present as coin- shaped and solitary, with well-defined edges, typically measuring less than 4 cm in diameter. Calcification is present in 25% to 30% of patients. The “popcorn” or “comma-shaped” appearance of calcification is pathognomonic for hamartomas 26. While CT imaging remains the gold-standard, further diagnostic measures can become necessary. FDG-PET scan can be useful to determine the rate of FDG uptake, and therefore metabolic rate, of lesions with an indeterminate risk of malignancy 26. Especially in the case of lesions with an absent adipose component, or those lacking the characteristic calcification pattern, biopsy becomes mandatory to rule out underlying malignancy. Bronchoscopy with fine needle aspiration (FNA) is commonly the strategy of choice, though aspirations can be scant due to the density of the lesions. Masses with the typical coin appearance that fulfill CT criteria for hamartoma (less than 4cm in size, well-defined edges, detectable calcification or fatty component) should remain subject to conservative follow up with periodical observation. Resection is reserved for fast-growing or symptomatic masses or those in which the possibility of malignancy cannot be excluded 27.
Pulmonary hamartoma treatment
Surgery remains the only definitive curative option available. In the event of surgery, preservation of functional lung tissue is the primary goal. Therefore, enucleation and wedge resections are the most common surgical choice, with more radical lobectomy or total pneumonectomy reserved for particularly deep lesions, multiple or large lesions that are making wedge resection impossible, or lesions adhering severely to the hilum of the lung. To avoid overlooking underlying malignant potential, obtaining intraoperative frozen sections is generally recommended 28.
Hypothalamic hamartoma
Hypothalamic hamartomas are very rare, tumor-like malformations that occur during fetal development and are present at birth and can occur in people of any gender or ethnicity. Hypothalamic hamartomas are very rare, with approximately one tumor occurring in every 200,000 people. Hypothalamic hamartoma lesions usually do not change in size or spread to other locations. Both the type and severity of symptoms vary greatly among patients with hypothalamic hamartomas 29. Some individuals can go years with very few symptoms, or ones that are so mild, they are often missed by both parents and medical professionals. However, for most people with hypothalamic hamartoma, the most common symptoms are frequent daily gelastic seizures (spontaneous laughing, giggling and/or smirking) or dacrystic seizures (crying or grunting); developmental delays; and/or precocious puberty 30. Additional symptoms may include cognitive impairment; emotional and behavioral difficulties; and endocrine disturbances. These symptoms often start early in life but are frequently misdiagnosed. For some patients, endocrine (hormonal) disturbances such as central precocious puberty may be the only symptom. These symptoms often start early in life – for some, they become apparent shortly after birth, but are frequently misdiagnosed. A misdiagnosis or delay in proper diagnosis may result in unnecessary procedures, a decline in the overall quality of life for the individual, and cognitive decline that cannot be regained. Treatments are now available for some patients that can eliminate or reduce the hypothalamic hamartoma in size; eliminate or significantly reduce the frequency of seizures; and halt cognitive declines.
Hypothalamic hamartomaspatients can often be treated successfully with medications. For some, however, hypothalamic hamartoma can be disabling. For those with hypothalamic hamartoma and epilepsy, it is common for the disorder to progress and for different types of seizures to develop. The seizures associated with hypothalamic hamartoma often cannot be well-controlled with the standard seizure medications. For some, additional treatment such as surgical removal, radiosurgery, or thermoablation may be indicated. Though hypothalamic hamartomas can occur in patients with certain genetic disorders (such as Pallister-Hall syndrome), the majority of cases are sporadic.
Hypothalamic hamartoma causes
The underlying cause for hypothalamic hamartomas remains unknown. Over 95% of cases are sporadic (that is, there is no prior family history and the identified patient remains the only affected individual in the family). A defect in factors that regulate fetal development of the hypothalamus is most likely.
However, hypothalamic hamartoma can also occur in patients with identified genetic disorders. Of these, Pallister-Hall syndrome accounts for the majority. This is a rare dysmorphology syndrome that can include hypothalamic hamartoma, deformities of the hands and feet (postaxial polydactyly and syndactyly), abnormalities of the larynx (bifid epiglottis), imperforate anus, and others. Pallister-Hall syndrome is known to be associated with a genetic mutation in the GLI3 gene, which acts as a transcription factor (a regulator protein for turning on and off gene expression) in the sonic hedgehog intracellular signaling pathway. This is a genomic (or genome-wide) mutation, which is to say, a mutation present in every cell in the body.
Knowing that Pallister-Hall syndrome is due to a mutation in GLI3, researchers have examined the possibility that a somatic (tumor-only) mutation of GLI3 is responsible for sporadic hypothalamic hamartoma cases. Using tissue removed at surgery, it has been discovered that up to 25% of patients have a somatic mutation of GLI3 in hypothalamic hamartoma tissue. More recently, advanced genotyping has shown that several other genes within the sonic hedgehog pathway can also have somatic mutations that result in hypothalamic hamartoma. With current (2017) genotyping technology, somatic mutations can be identified in approximately 40% of hypothalamic hamartoma lesions. It is also likely that other, as yet unidentified mutations in other genes can be responsible. At this time, mutation analysis (genotyping) of hypothalamic hamartoma lesions is not recommended for routine clinical care of patients with hypothalamic hamartoma.
Hypothalamic hamartoma signs and symptoms
The symptoms of a hypothalamic hamartoma can include:
- Seizures
- Precocious (early) puberty
- Hormone imbalances
- Cognitive impairment
- Behavioral problems
- Emotional difficulties
Seizures caused by a hypothalamic hamartoma usually begin in infancy, most often as brief and frequent gelastic (or laughing) seizures. You may not be able to tell the difference between a gelastic seizure and normal laughter.
The difficulty in identifying gelastic seizures usually delays the diagnosis of epilepsy caused by a hypothalamic hamartoma. Doctors sometimes can have difficulty recognizing gelastic seizures because they are rare, and children affected by them generally develop normally.
Between the ages of four and 10 years old, you may notice the seizures becoming more disabling, with the emergence of different seizure types, such as:
- Complex partial seizures (diminished or altered consciousness with involuntary but coordinated movements)
- Generalized convulsions (involuntary shaking, twitching, and disorganized movements)
- Drop or atonic seizures (brief loss of muscle tone leading to falls)
In this stage, your child may display signs of progressive cognitive impairment, worsening school performance, and the following behavioral problems:
- Tantrums
- Rage attacks
- Social isolation
Some children with a hypothalamic hamartoma may have endocrine (hormonal) disturbances. Central precocious puberty (early puberty) is the most common sign of these disturbances, and may be the only symptom of hypothalamic hamartoma that your child has.
Not all people with a hypothalamic hamartoma have the same symptoms or the same age of onset. While you might notice gelastic seizures in your newborn, it is also possible that seizures will not develop until later in life. Seizures caused by a hypothalamic hamartoma generally do not respond to medication.
Hypothalamic hamartomas diagnosis
Hypothalamic hamartomas are diagnosed by assessment of neurological symptoms, magnetic resonance imaging (MRI), and neurological tests. Early detection can improve the likelihood of successful treatment, especially for children who have worsening seizures or whose learning skills or behavior have begun to deteriorate.
MRI is used to locate a hypothalamic hamartoma and confirm the diagnosis. Hypothalamic hamartomas can be small and difficult to detect, so interpretation by a doctor that specializes in neuroradiology is desirable.
Electroencephalography (EEG) may also be used to identify seizure patterns that suggest a hypothalamic hamartoma
Hypothalamic hamartoma treatment
Central precocious puberty can usually be treated successfully with medication. Surgery is not required for most patients with central precocious puberty. Effective treatment consists of administration of gonadotropin-releasing hormone (GnRH) agonists, such as leuprolide acetate [Lupron], which have the effect of feedback inhibition of the pulsatile (pulse-like) release of GnRH that is required to trigger puberty. Leuprolide acetate is customarily administered as a once-monthly intramuscular injection for the duration of time that puberty needs to be suppressed. Once discontinued, puberty occurs normally. Long-term follow-up studies for hypothalamic hamartoma patients with a history of successfully treated central precocious puberty show that normal adult height and normal reproductive function are expected.
Treatment options that avoid once-monthly injections have also been developed. Consultation with a pediatric endocrinologist experienced in the treatment of central precocious puberty is important to review the treatment options and for discussion of possible side effects.
Gelastic seizures usually do not respond to anti-epilepsy drugs (AEDs). Exceptions to this appear to be rare. Additionally, while the other seizure types that occur later in childhood may be helped with anti-epilepsy drug therapy, it is unlikely that these seizures will be completely controlled with anti-epilepsy drugs (“treatment-resistant”). Consequently, surgical treatment is usually required, as discussed below.
The timing of surgical intervention (including gamma knife radiosurgery) is a major decision point facing the patient, family and doctor. Surgical intervention carries the risk of complications and should not be performed until the degree of clinical severity calls for it. For example, relatively brief and infrequent gelastic seizures are usually not significantly disabling. If the child is making good developmental progress, a decision to withhold surgical intervention may be appropriate. However, under these circumstances, the clinical course needs to be observed carefully for any adverse changes in symptoms, such as worsening of seizures, slowing of developmental progress, or emergence of psychiatric symptoms.
However, our current understanding of epilepsy associated with hypothalamic hamartoma also argues against excessive delay in surgical treatment, since it appears that some patients undergo a process known as secondary epileptogenesis, in which uncontrolled seizures from the original location can provoke a process by which seizures begin to arise from a second, distant location elsewhere in the brain. When this occurs (a process that is likely to occur over a period of years rather than months) then surgery removing the hypothalamic hamartoma lesion may be less successful for completely controlling seizures. Outcome studies of surgery for hypothalamic hamartoma associated with epilepsy have shown that success in controlling seizures is inversely related to the patent’s age. That is, older patients are less likely to gain complete seizure control. A recent research study on hypothalamic hamartoma patients undergoing surgery has also shown that a higher likelihood of cognitive improvement after surgery correlates with younger age at the time of surgery.
Gelastic seizures begin in the hypothalamic hamartoma lesion and removing (or otherwise damaging or ablating) the hypothalamic hamartoma can cure seizures. Until recently, hypothalamic hamartoma with epilepsy was considered not treatable, since surgery on the hypothalamus was too dangerous. However, since 2000, several different treatment approaches have been developed that are recognized as effective and safe. These treatment decisions (selecting one of several surgical options including radiosurgery) are highly individualized to the unique circumstances of each patient. This includes an assessment of their clinical course and symptoms, but also the exact anatomy of their hypothalamic hamartoma lesion. Consequently, consultation at a referral center that specializes in the treatment of hypothalamic hamartoma is highly recommended.
A brief discussion of each of these treatment modalities follows. The relative merits of any of these therapies for an individual patient require consultation with a specialist in the field of hypothalamic hamartoma treatment.
Surgical removal
Neurosurgeons use the transcallosal approach to remove hypothalamic hamartomas. This approach uses the natural gap between the two hemispheres of your brain to create an avenue to access and remove the tumor.
Neurosurgeons also use more traditional subtemporal or orbitozygomatic approaches if they offer the best chance of success. This surgery begins on the side of your head and passes under your brain and above the base of your skull to reach the lesion.
Less-invasive endoscopic surgery can be considered when the size and anatomical positioning of the lesion allows.
Laser generated stereotactic thermoablation
Laser generated stereotactic thermoablation is a new minimally invasive technology that uses energy in the form of light to destroy tumors or damaged tissue. The energy from the laser causes the temperature of the target tissue—in this case a hypothalamic hamartoma—to rise, destroying it.
Computerized safety mechanisms automatically turn the laser off if it approaches brain tissue surrounding the tumor. Magnetic resonance images (MRI) are also used further improve the accuracy of the procedure and guide the placement of the laser.
Gamma knife radiosurgery
Gamma Knife radiosurgery uses many beams of radiation that converge on a single point to shrink tumors. Each individual beam is not powerful enough to destroy healthy tissue, but at the point where the beams meet sufficient energy is concentrated to destroy tumor cells.
References- Lundeen KS, Raj MS, Ludhwani D. Pulmonary Hamartoma. [Updated 2019 Mar 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK539806
- Leiter Herrán F, Restrepo CS, Alvarez Gómez DI, Suby-Long T, Ocazionez D, Vargas D. Hamartomas from head to toe: an imaging overview. Br J Radiol. 2017 Mar;90(1071):20160607
- Hamartoma. https://emedicine.medscape.com/article/1254012-overview
- Sevim Y, Kocaay AF, Eker T, et al. Breast hamartoma: a clinicopathologic analysis of 27 cases and a literature review. Clinics (Sao Paulo). 2014;69(8):515–523. doi:10.6061/clinics/2014(08)03 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4129555
- Mammary hamartoma–a review of 35 cases. Fisher CJ, Hanby AM, Robinson L, Millis RR. Histopathology. 1992 Feb; 20(2):99-106.
- Guray M, Sahin AA. Benign breast diseases: classification, diagnosis, and management. Oncologist. 2006;11(5):435–49
- Tse GM, Law BK, Ma TK, Chan AB, Pang LM, Chu WC, et al. Hamartoma of the breast: a clinicopathological review. J Clin Pathol. 2002;55(12):951–4
- Herbert M, Sandbank J, Liokumovich P, Yanai O, Pappo I, Karni T, et al. Breast hamartomas: clinicopathological and immunohistochemical studies of 24 cases. Histopathology. 2002;41(1):30–4
- Kajo K, Zubor P, Danko J. Myoid (Muscular) Hamartoma of the Breast: Case Report and Review of the Literature. Breast Care (Basel) 2010;5(5):331–4
- Murugesan JR, Joglekar S, Valerio D, Bradley S, Clark D, Jibril JA. Myoid hamartoma of the breast: case report and review of the literature. Clin Breast Cancer. 2006;7(4):345–6
- Mizuta N, Sakaguchi K, Mizuta M, Imai A, Nakatsukasa K, Morita M, et al. Myoid hamartoma of the breast that proved difficult to diagnose: a case report. World J Surg Oncol. 2012;10:12
- Altermatt HJ, Gebbers JO, Laissue JA. Multiple hamartomas of the breast. Appl Pathol. 1989;7(2):145–8
- Kopans DB. Pathologic, mammographic and sonographic correlation. In: Kopans D B, editor. Breast Imaging. 2nd ed. Boston, Massachusetts: Lippincott-Raven; 1998. pp. 558–560
- McGuire LI, Cohn D. Hamartoma of the breast. Aust N Z J Surg. 1991;61(9):713–6.
- Jones MW, Norris HJ, Wargotz ES. Hamartomas of the breast. Surg Gynecol Obstet. 1991;173(1):54–6
- Fechner RE. Fibroadenoma and related lesions. In: Page D L, Anderson T J, editors. Diagnostic Histopathology of the Breast. Edinburgh: Churchill Livingstone; 1987. pp. 72–85.
- Fisher CJ, Hanby AM, Robinson L, Millis RR. Mammary hamartoma—a review of 35 cases. Histopathology. 1992;20(2):99–106
- Kai M, Tada K, Tamura M, Gomi N, Horii R, Akiyama F, et al. Breast cancer associated with mammary hamartoma. Breast Cancer. 2012;19(2):183–6
- Wahner-Roedler DL, Sebo TJ, Gisvold JJ. Hamartomas of the breast: clinical, radiologic, and pathologic manifestations. Breast J. 2001;7(2):101–5
- PTEN Hamartoma Tumor Syndrome. https://rarediseases.org/rare-diseases/pten-hamartoma-tumor-syndrome
- Eng C. PTEN Hamartoma Tumor Syndrome. 2001 Nov 29 [Updated 2016 Jun 2]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1488
- Ahmed S, Arshad A, Mador MJ. Endobronchial hamartoma; a rare structural cause of chronic cough. Respir Med Case Rep. 2017;22:224-22
- Saadi MM, Barakeh DH, Husain S, Hajjar WM. Large multicystic pulmonary chondroid hamartoma in a child presenting as pneumothorax. Saudi Med J. 2015 Apr;36(4):487-9
- Ahmed S, Arshad A, Mador MJ. Endobronchial hamartoma; a rare structural cause of chronic cough. Respir Med Case Rep. 2017;22:224-227
- Itoga M, Kobayashi Y, Takeda M, Moritoki Y, Tamaki M, Nakazawa K, Sasaki T, Konno H, Matsuzaki I, Ueki S. A case of pulmonary hamartoma showing rapid growth. Case Rep Med. 2013;2013:231652
- Lu Z, Qian F, Chen S, Yu G. Pulmonary hamartoma resembling multiple metastases: A case report. Oncol Lett. 2014 Jun;7(6):1885-1888
- Singh H, Khanna SK, Chandran V, Jetley RK. PULMONARY HAMARTOMA. Med J Armed Forces India. 1999 Jan;55(1):79-80
- Guo W, Zhao YP, Jiang YG, Wang RW, Ma Z. Surgical treatment and outcome of pulmonary hamartoma: a retrospective study of 20-year experience. J. Exp. Clin. Cancer Res. 2008 May 31;27:8
- Hypothalamic Hamartoma. https://rarediseases.org/rare-diseases/hypothalamic-hamartoma
- Understanding hypothalamic hamartoma. http://www.hopeforhh.org/what-is-hh/




{kind=link}