Hepatotoxicity
Hepatotoxicity means chemical-induced liver damage. The liver is a remarkable organ that serves as your body’s chemical engineering and control center, playing a central role in transforming and clearing chemicals and drugs and is susceptible to the toxicity from these agents. The liver has astounding ability to adapt, to change itself, to alter activities of its enzymes and transporters, and even to regenerate rapidly if cells are killed or removed. Certain drugs when taken in overdoses and sometime even when introduced within therapeutic ranges may injure the liver. Other chemical agents such as those used in laboratories and industries, natural chemicals (e.g. microcystins) and herbal remedies can also induce hepatotoxicity. Chemicals that cause liver injury are called hepatotoxins.
Drug-induced liver injury is an uncommon but challenging clinical problem with respect to both diagnosis and management 1. Its incidence is estimated to be 14 to 19 cases per 100,000 persons, with jaundice accompanying 30% of cases 2. Drug-induced liver injury is responsible for 3 to 5% of hospital admissions for jaundice 3 and is the most frequent cause of acute liver failure in most Western countries, accounting for more than half of cases 4.
In the United States, approximately 2000 cases of acute liver failure occur annually and approved drugs account for over 50% of them (39% are due to acetaminophen, 13% are idiosyncratic reactions due to other medications) 5. Drugs account for 2-5% of cases of patients hospitalized with jaundice and approximately 10% of all cases of acute hepatitis. Internationally, data on the incidence of adverse hepatic drug reactions in the general population remain unknown.
The diagnosis of drug-induced liver injury is particularly challenging, since it is based largely on exclusion of other causes 6. The timing of the onset of injury after the implicated agent has been started (latency), resolution after the agent is stopped (“dechallenge”), recurrence on re-exposure (rechallenge), knowledge of the agent’s potential for hepatotoxicity (likelihood), and clinical features (phenotype) are the major diagnostic elements 7. With few exceptions, there are no specific diagnostic markers for drug-induced liver injury, and special tests (liver biopsy, imaging, and testing for serologic markers) are helpful mostly in ruling out other causes of liver injury. The large number of agents that can cause liver injury highlights these challenges. LiverTox, the National Institutes of Health–sponsored website on hepatotoxicity, has descriptions of more than 1200 agents (prescription and over-the-counter medications, herbal products, nutritional supplements, metals, and toxins), along with their potential to cause liver injury.12 Among the 971 prescription drugs described, 447 (46%) have been implicated in causing liver injury in at least one published case report 7.
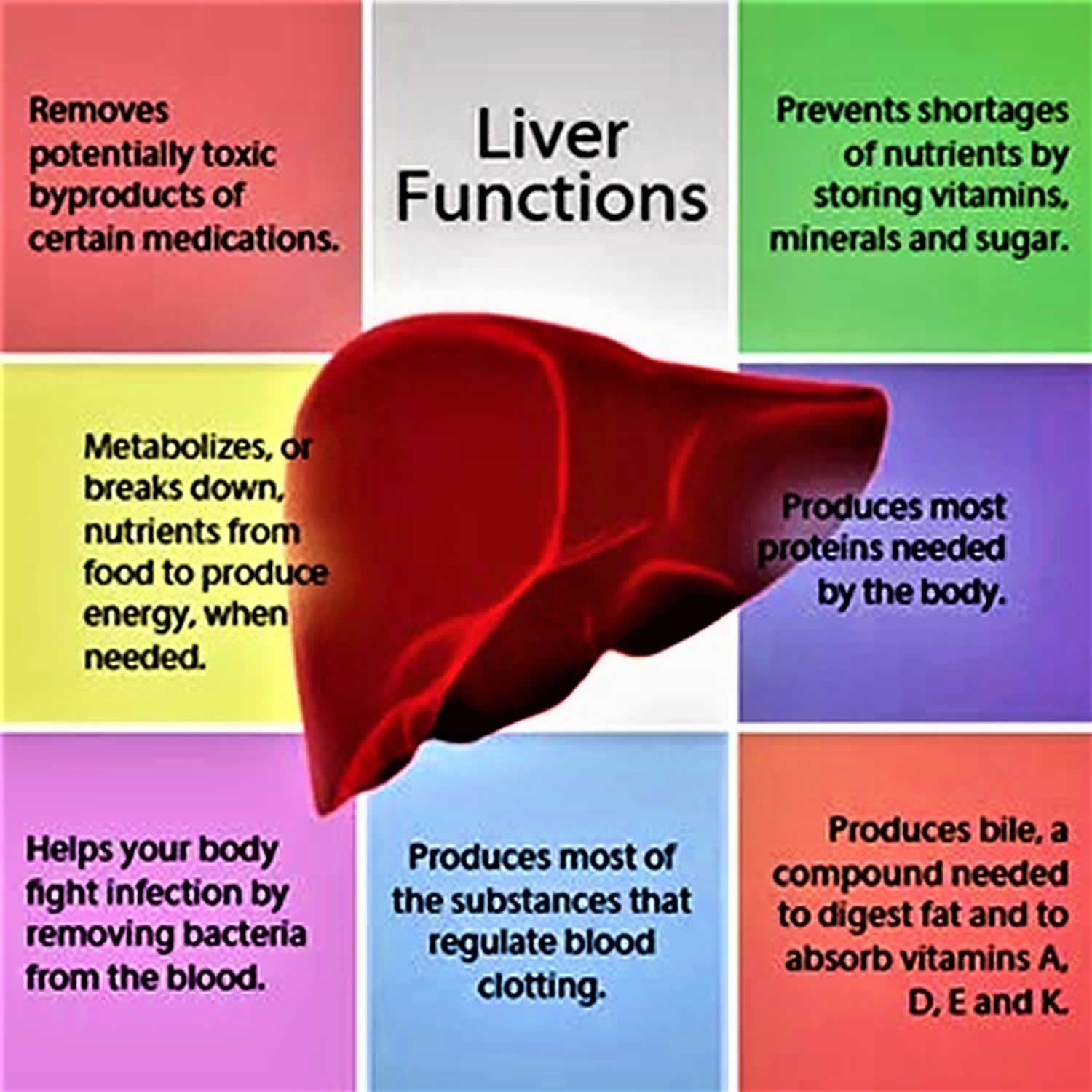
Figure 1. Liver functions
Hepatotoxicity causes
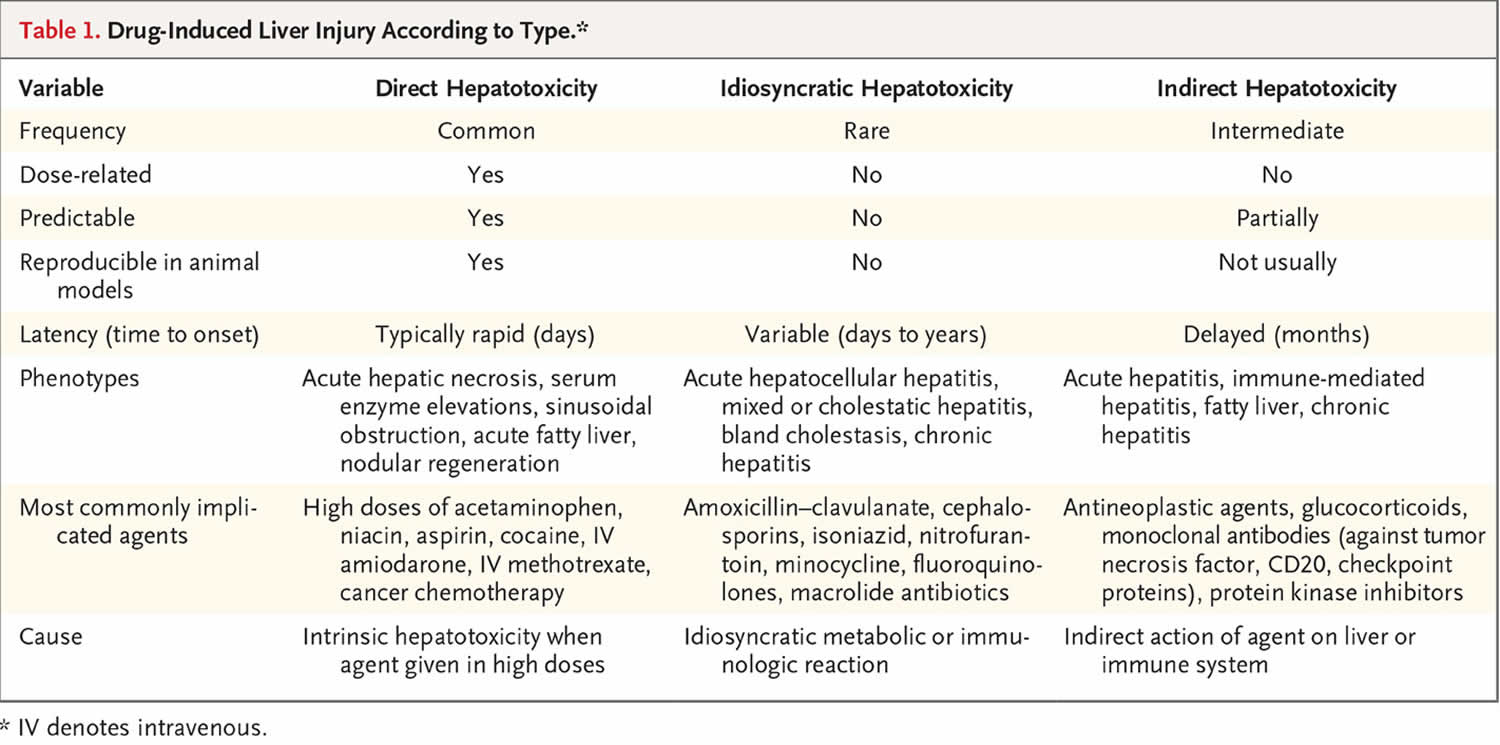
Drug-induced liver injury is typically classified as either direct or idiosyncratic 8, but indirect injury is emerging as a third type (Table 1). Direct hepatotoxicity is caused by agents that are intrinsically toxic to the liver. The injury is common, predictable, dose-dependent, and reproducible in animal models 8. The latency period is typically short, usually with an onset within 1 to 5 days after high therapeutic or supratherapeutic doses, as in the case of an intentional or accidental overdose.
Idiosyncratic hepatotoxicity is caused by agents that have little or no intrinsic toxicity and that cause liver injury only in rare cases, typically after 1 in 2000 to 1 in 100,000 patient-exposures 2. The injury is unpredictable, not dose-dependent, and not reproducible in animal models. Idiosyncratic liver injury is categorized as hepatocellular, cholestatic, or both (mixed) on the basis of the R ratio, calculated by dividing the alanine aminotransferase level by the alkaline phosphatase level from the time of initial presentation, with both values expressed as multiples of the upper limit of the normal range 9. Hepatocellular injury is defined as an R value of more than 5, cholestatic injury as a value of less than 2, and mixed injury as a value of 2 to 5.
Indirect hepatotoxicity is caused by the action of the drug (what it does) rather than by its toxic or idiosyncratic properties (what it is). Indirect injury can represent induction of a new liver condition or an exacerbation of a preexisting condition, such as induction of immune-mediated hepatitis or worsening of hepatitis B or C or fatty liver disease.
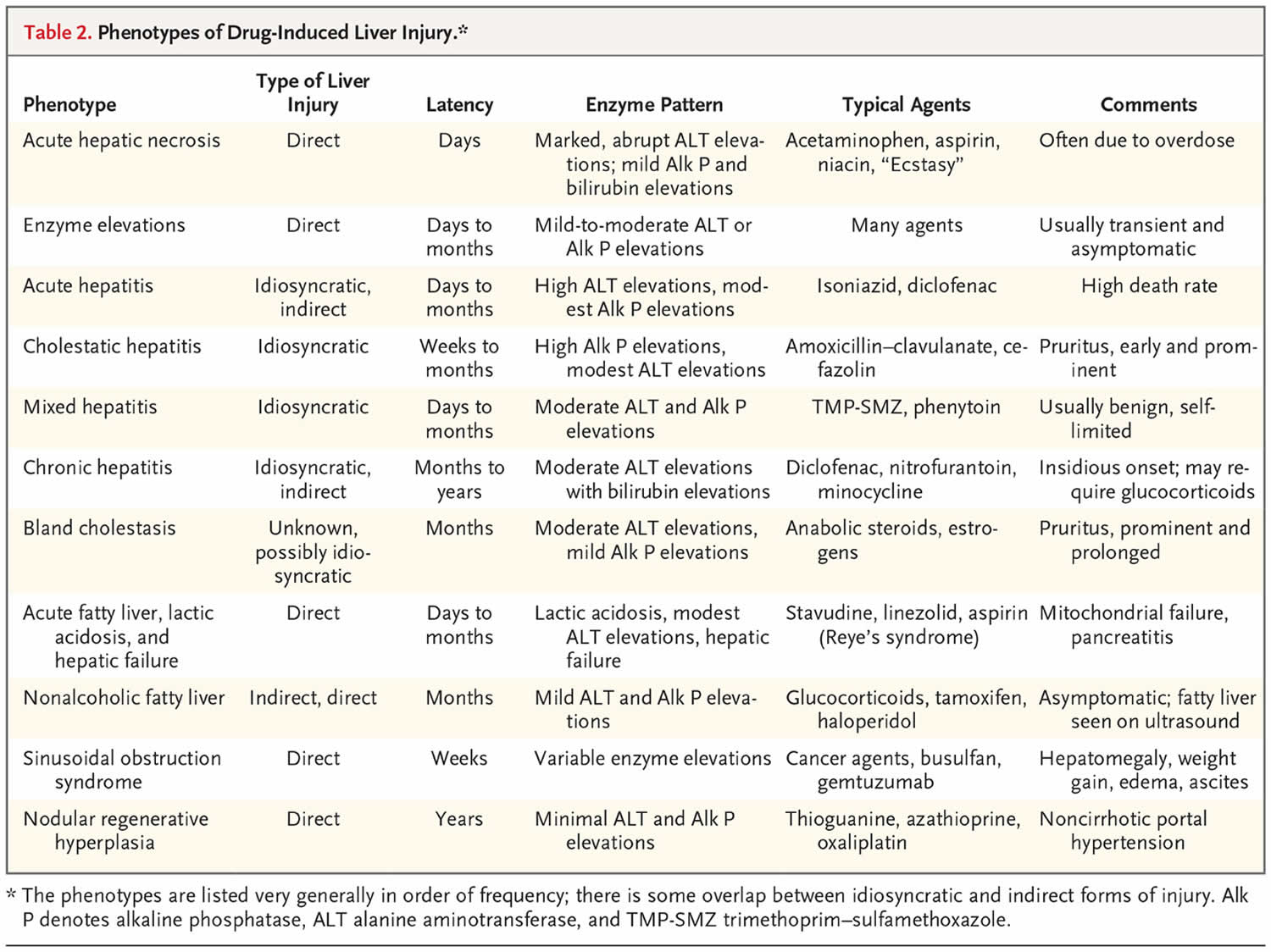
The three types of drug-induced liver injury are manifested by distinctly different patterns of clinical features (phenotypes) (Table 2) 10.
Direct hepatoxicity
Serum enzyme elevations without jaundice constitute the most common pattern of direct drug-induced liver injury, with elevations of alanine aminotransferase (ALT) or alkaline phosphatase (ALP) levels but without hyperbilirubinemia and with minimal or no symptoms 11. The elevations resolve when the drug is stopped or the dose is lowered but can also resolve spontaneously, a phenomenon referred to as adaptation 12. In some cases, adaptation does not occur, and enzyme elevations worsen and jaundice and symptoms arise. The mechanism or mechanisms underlying adaptation are unknown but may result from changes in drug-metabolizing enzyme activity, up-regulation of hepatoprotective pathways, or down-regulation of hypersensitivity reactions to the drug or its metabolites.
Acute hepatic necrosis is the most common form of clinically apparent direct hepatotoxicity. The injury occurs abruptly, soon after the medication has been started, often after exposure to a single high dose or a dose increase. Serum alanine aminotransferase levels rise to high values, whereas alkaline phosphatase (ALP) levels are minimally elevated. In severe cases, signs of hepatic failure such as coagulopathy, hyperammonemia, or coma arise within days 4. Liver histologic studies show centrilobular or panlobular necrosis with little inflammation, a pattern similar to that of ischemic hepatitis, the major disorder in the differential diagnosis. Acute hepatic necrosis can be fatal, but if it is not, recovery is rapid, and serum enzyme levels fall almost as rapidly as they rose. High doses of acetaminophen, aspirin, niacin, amiodarone, and many antineoplastic agents can cause acute hepatic necrosis 13. Typically, these drugs can be restarted at lower doses without a recurrence of injury. Poisonous mushrooms (Amanita phalloides) and other environmental toxins can cause a similar syndrome of acute hepatic necrosis.
Sinusoidal obstruction syndrome, previously known as veno-occlusive disease, is due to acute injury and loss of intrasinusoidal endothelial cells, resulting in obstruction of sinusoidal blood flow and liver injury 14. Drugs are the usual cause, the most common being myeloablative agents administered in preparation for hematopoietic cell transplantation. Symptoms of abdominal pain, increase in liver size, and weight gain, followed by jaundice, appear 1 to 3 weeks after exposure and may progress rapidly to hepatic failure. Liver histologic studies show dilatation of sinusoids and extravasation of red cells, with hepatocyte necrosis in central areas (zone 3) 15. Drugs that cause sinusoidal obstruction syndrome include alkylating agents such as busulfan or cyclophosphamide and monoclonal antibody–cytotoxic conjugates such as gemtuzumab ozogamicin 16. The syndrome can also be caused by botanicals (pyrrolizidine alkaloids) 8. Defibrotide, an antithrombotic agent, has recently been approved as therapy for severe sinusoidal obstruction syndrome with organ failure, but its use is controversial 17.
Nodular regenerative hyperplasia is usually manifested as unexplained, noncirrhotic portal hypertension with esophageal varices or ascites. Nodular regeneration can be caused by cancer chemotherapeutic agents given over a long period or in multiple courses (azathioprine, mercaptopurine, or thioguanine) 18 or by first-generation nucleoside antiretroviral agents (zidovudine, stavudine, or didanosine) 19. Nodular regenerative hyperplasia with resultant portal hypertension has also been linked to oxaliplatin infusions for metastatic colon cancer 20. The pathogenesis of nodular regeneration is unclear, but it may be the result of chronic injury to the hepatic microvasculature. Management should include withdrawal of the medication (and avoidance of similar agents) and treatment of portal hypertension.
Lactic acidosis with microvesicular steatosis and hepatic dysfunction typically occurs with nonspecific symptoms of abdominal discomfort, fatigue, and weakness, with subsequent confusion, stupor, and coma accompanied by liver injury 21. Lactic acidosis or hyperammonemia may be prominent. Jaundice arises late, and enzyme elevations are variable, sometimes markedly hepatocellular (with Reye’s syndrome triggered by aspirin) 22 and sometimes with milder, mixed patterns. The time to onset can be days (with aspirin or intravenous tetracycline) 23, weeks (with linezolid) 24, or months (with didanosine) 21. Liver biopsy shows microvesicular steatosis with minimal inflammation and necrosis. The pathogenesis of the injury is mitochondrial toxicity and failure of aerobic metabolism. Similar injury in other tissues may accompany and overshadow the liver injury (neuropathy, myopathy, and pancreatitis). Therapy should focus on withdrawal of the responsible agent, administration of glucose infusions, and correction of acidosis 21.
Idiosyncratic hepatotoxicity
Acute hepatocellular hepatitis is the most common manifestation of idiosyncratic liver injury 2. The latency period generally ranges from 5 to 90 days. The symptoms and course resemble those of acute viral hepatitis, with prominent alanine aminotransferase elevations (increased by a factor of 5 to 50), whereas alkaline phosphatase (ALP) levels are only modestly increased. Liver histologic studies show changes suggestive of acute viral hepatitis, the major disorder in the differential diagnosis, but eosinophils may be prominent. The rate of death from icteric hepatocellular injury due to medications is high, usually 10% or higher, a feature first stressed by the late Hyman J. Zimmerman, for which reason it is called Hy’s law 8. A key feature of Hy’s law is jaundice with hepatocellular rather than cholestatic injury. Drug-induced idiosyncratic acute hepatocellular injury is an important cause of acute liver failure, accounting for 11 to 15% of cases in series from the United States and Europe 4. Common causes of drug-induced idiosyncratic acute hepatocellular injury are isoniazid, nitrofurantoin, and diclofenac 25.
Chronic hepatitis is an uncommon form of drug-induced liver injury; the chronicity occurs if the agent is continued and typically resolves slowly once the agent has been stopped. Many agents that cause acute hepatocellular injury can also cause a chronic hepatocellular pattern 25. The injury arises after months or years of exposure. Autoantibodies are frequently present, and the differential diagnosis often focuses on ruling out spontaneous autoimmune hepatitis. Common causes of drug-induced, autoimmune-like chronic liver injury are nitrofurantoin, minocycline, hydralazine, methyldopa, statins, and fenofibrate 25. Glucocorticoids, which are frequently used to manage chronic hepatitis (starting dose, 20 to 60 mg of prednisone or its equivalent daily), may alleviate symptoms and speed recovery, but the injury will often resolve without intervention. If prednisone is used, the dose and duration should be kept to a minimum. Monitoring for evidence of relapse should be performed for at least 6 months after the withdrawal of glucocorticoids. Ultimately, spontaneous autoimmune hepatitis is best ruled out by evidence of resolution of the liver injury after withdrawal of the medication and, if glucocorticoids are used, by the absence of relapse when they are discontinued 26.
Cholestatic hepatitis is characterized by prominent symptoms of pruritus and jaundice accompanied by moderate-to-marked elevations in alkaline phosphatase (ALP) levels. Drug-induced cholestatic liver injury is usually self-limited, and although often protracted, it ultimately resolves 27. Liver histologic studies show bile duct injury and cholestasis in small bile canaliculi 28. Exceptions to the usual benign course occur when there is bile duct loss, which is associated with delayed resolution of jaundice and elevated enzyme levels 29. Some cases evolve into vanishing bile duct syndrome, with prolonged jaundice, liver failure, need for liver transplantation, or death. Common causes of drug-induced cholestatic hepatitis are amoxicillin–clavulanate, cephalosporins, terbinafine, azathioprine, and temozolomide 30.
Drug-induced mixed hepatitis is caused by many agents, some of which also cause hepatocellular or cholestatic hepatitis 31. The mixed forms of drug-induced liver injury tend to have the most benign outcomes, rarely leading to liver failure. Common causes of drug-induced mixed hepatitis include the fluoroquinolone and macrolide antibiotics, phenytoin, and sulfonamides 31.
All forms of idiosyncratic drug-induced hepatitis can be accompanied by immunoallergic features, such as rash, fever, and eosinophilia — signs of drug hypersensitivity 31. More extreme examples include drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, toxic epidermal necrolysis, and the Stevens–Johnson syndrome 32. Prominent causes of idiosyncratic drug-induced hepatitis with immunoallergic features include allopurinol, carbamazepine, phenytoin, sulfonamides, and macrolide antibiotics 32. Immunoallergic hepatitis is more common among black Americans than among non-Hispanic white Americans 33.
Bland cholestasis represents a distinctive phenotype of drug-induced liver injury, characterized by marked and prolonged jaundice with pruritus. In women, bland cholestasis is typically caused by estrogens or oral contraceptives 34 and in men it is typically caused by anabolic steroids, usually obtained illicitly for bodybuilding or improving athletic performance 35. Jaundice and pruritus arise within 30 to 90 days, and elevations in enzyme levels are minimal or modest, despite marked and prolonged jaundice 36. Liver biopsy shows bland cholestasis with scant inflammation and hepatocellular necrosis. The cholestasis can be prolonged, but the injury is almost always self-limited and deaths are rare. The pathogenesis remains unclear.
The pathogenesis of direct and indirect hepatotoxicity is reasonably well understood, whereas that of idiosyncratic injury is not. Genomewide association studies of large numbers of idiosyncratic cases have identified several genetic associations, most within the major histocompatibility complex (MHC) region and linked to HLA class I and II alleles. In general, the HLA associations were to uncommon alleles and were specific to selected agents, such as HLA-B*57:01 for flucloxacillin 37, HLA-A*02:01 and HLA-DRB1*15:01 for amoxicillin–clavulanate 38 and HLA-A*33:01 for fenofibrate and terbinafine 39. These associations were not reliable enough to warrant screening for HLA alleles in selecting medications, but they suggest an immunologic pathogenesis. This hypothesis is supported by the observation that implicated drugs or their metabolites bind to the active T-cell receptor groove dictated by the HLA association 40.
More recently, genomewide association studies have identified a risk allele for idiosyncratic drug-induced liver injury outside the HLA region that is linked to a missense mutation in an immunomodulatory gene encoding PTPN22 41, a protein tyrosine phosphatase that acts by down-regulating T-cell receptor signaling 42. The same missense mutation (c.C1858T, p.R620W) has also been linked to an increased risk of autoimmune diseases. This allele appears to be linked to multiple forms of idiosyncratic drug-induced liver injury.
An attractive hypothesis is that idiosyncratic drug-induced liver injury is due to a perfect storm of events, each of which is required for full expression of the injury. The production of an abnormal metabolite of the drug by the liver is followed by mild liver-cell injury and then by an immunologic response to the metabolite presented on the injured hepatocyte surface to a specific HLA-restricted T-cell receptor 1. Without adaptation, the immune recognition triggers further T-cell activation, cytokine release, and hepatocyte injury. This hypothesis may ultimately help to improve the identification of safer drugs in development.
Indirect hepatotoxicity
Indirect drug-induced liver injury results from the medication’s actions rather than from its inherent hepatotoxic effects or immunogenicity; the injury represents induction or exacerbation of a liver disease. The phenotypes are those of the underlying disease or predisposition. Fatty liver disease can be an indirect effect of drugs that cause weight gain (risperidone and haloperidol) 43 or that alter triglyceride disposition (lomitapide) 44 or insulin sensitivity (glucocorticoids). Acute hepatitis can be the indirect effect of anticancer chemotherapeutic agents that cause a reactivation of hepatitis B 45 or of antiretroviral agents that cause immune reconstitution and exacerbation of hepatitis C 46. An increasingly common form of indirect injury is immune-mediated liver injury due to various immunomodulatory agents 47, tumor necrosis factor antagonists 48 and most dramatically, antineoplastic checkpoint inhibitors 49. Many of these agents are monoclonal antibodies and are thus unlikely to cause direct or idiosyncratic liver injury. Hepatocellular or mixed hepatitis with immune features usually arises within 2 to 12 weeks after the start of therapy (or after one to three courses) and is often detected during routine monitoring at the time of each infusion. Many cases are anicteric and asymptomatic, but without intervention, the hepatitis can worsen and become life-threatening. Therapy with glucocorticoids is usually recommended 49. If the injury resolves promptly, the agent can be restarted or another agent can be substituted (infliximab can be switched to etanercept, or ipilimumab to nivolumab).
Indirect liver injury is a new and not completely accepted category of hepatotoxicity. Nevertheless, the clinical features are distinct (see Table 1). Indirect injury is much more frequent than idiosyncratic forms and is a common reaction to a whole class of medications (e.g., tumor necrosis factor antagonists and checkpoint inhibitors) rather than a rare and idiosyncratic reaction to a random, specific agent (e.g., nitrofurantoin or atorvastatin). Indirect drug-induced liver injury represents an expanded concept of hepatotoxicity and provides insights into liver conditions that are worsened (e.g., the types of immunomodulation that cause reactivation of hepatitis B) or into predispositions to liver conditions. There are plausible explanations for the pathogenesis of indirect injury, and in most instances, this type of drug-induced liver injury can be prevented or treated.
Patterns of liver injury
Chemicals produce a wide variety of clinical and pathological hepatic injury. Biochemical markers (e.g. alanine transferase, alkaline phosphatase and bilirubin) are often used to indicate liver damage. Liver injury is defined as a rise in either (a) Alanine aminotransferase (ALT) level more than three times of upper limit of normal, (b) Alkaline phosphatase (ALP) level more than twice upper limit of normal, or (c) total bilirubin level more than twice upper limit of normal when associated with increased alanine aminotransferase (ALT) or alkaline phosphatase (ALP) 50. Liver damage is further characterized into hepatocellular (predominantly initial alanine transferase elevation) and cholestatic (initial alkaline phosphatase rise) types. However they are not mutually exclusive and mixed types of injuries are often encountered.
Specific histo-pathological patterns of liver injury from drug-induced damage are discussed below.
Zonal Necrosis
This is the most common type of drug-induced liver cell necrosis where the injury is largely confined to a particular zone of the liver lobule. It may manifest as a very high level of ALT and severe disturbance of liver function leading to acute liver failure.
- Causes include: Paracetamol, carbon tetrachloride
Hepatitis
In this pattern, hepatocellular necrosis is associated with infiltration of inflammatory cells. There can be three types of drug-induced hepatitis. (A) viral hepatitis is the most common, where histological features are similar to acute viral hepatitis. (B) in focal or non-specific hepatitis, scattered foci of cell necrosis may accompany lymphocytic infiltration. (C) chronic hepatitis is very similar to autoimmune hepatitis clinically, serologically, and histologically.
Causes:
- (a) Viral hepatitis: Halothane, isoniazid, phenytoin
- (b) Focal hepatitis: Aspirin
- (c) Chronic hepatitis: Methyldopa, diclofenac
Cholestasis
Liver injury leads to impairment of bile flow and cases are predominated by itching and jaundice. Histology may show inflammation (cholestatic hepatitis) or it can be bland (without any parenchymal inflammation). On rare occasions, it can produce features similar to primary biliary cirrhosis due to progressive destruction of small bile ducts (vanishing duct syndrome).
Causes:
- (a) Bland: Oral contraceptive pills, anabolic steroid, androgens
- (b) Inflammatory: Allopurinol, co-amoxiclav, carbamazepine
- (c) Ductal: Chlorpromazine, flucloxacillin
Steatosis
Hepatotoxicity may manifest as triglyceride accumulation, which leads to either small-droplet (microvesicular) or large-droplet (macrovesicular) fatty liver. There is a separate type of steatosis by which phospholipid accumulation leads to a pattern similar to the diseases with inherited phospholipid metabolism defects (e.g., Tay–Sachs disease)
Causes:
- (a) Microvesicular: Aspirin (Reye’s syndrome), ketoprofen, tetracycline (especially if expired)
- (b) Macrovesicular: Acetaminophen, methotrexate
- (c) Phospholipidosis: Amiodarone, total parenteral nutrition
- (d) Antiviral: nucleoside analogues
- (e) Corticosteroid
- (f) Hormonal: Tamoxifen
Granuloma
Drug-induced hepatic granulomas are usually associated with granulomas in other tissues and patients typically have features of systemic vasculitis and hypersensitivity. More than 50 drugs have been implicated.
- Causes: Allopurinol, phenytoin, isoniazid, quinine, penicillin, quinidine
Vascular lesions
These result from injury to the vascular endothelium.
Causes:
- Venoocclusive disease: Chemotherapeutic agents, bush tea
- Peliosis hepatis: Anabolic steroids
- Hepatic vein thrombosis: Oral contraceptives
Neoplasm
Neoplasms have been described with prolonged exposure to some medications or toxins. Hepatocellular carcinoma, angiosarcoma, and liver adenomas are the ones usually reported.
- Causes: Vinyl chloride, combined oral contraceptive pill, anabolic steroid, arsenic, thorotrast
Hepatotoxic drugs
Drugs are an important cause of liver injury. More than 900 drugs, toxins, and herbs have been reported to cause liver injury 51 and approved drugs account for 20-40% of all instances of fulminant hepatic failure. Drug induced liver injury is responsible for 5% of all hospital admissions and 50% of all acute liver failures 52. Approximately 75% of the idiosyncratic drug reactions result in liver transplantation or death 5. Drug-induced hepatotoxicity is the most common reason cited for withdrawal from the market of an approved drug. Physicians must be vigilant in identifying drug-related liver injury because early detection can decrease the severity of hepatotoxicity if the drug is discontinued. The manifestations of drug-induced hepatotoxicity are highly variable, ranging from asymptomatic elevation of liver enzymes to fulminant hepatic failure. Knowledge of the commonly implicated agents and a high index of suspicion are essential in diagnosis.
The current major causes of clinically apparent liver injury due to prescription drugs are shown in Table 3 31. These data are based on more than 1000 cases of suspected idiosyncratic drug-induced liver injury seen at five to eight medical centers across the United States between 2004 and 2013. All cases were formally adjudicated, and the implicated agent was classified as the definite, highly likely, or probable cause. The most commonly implicated agents were amoxicillin–clavulanate, isoniazid, nitrofurantoin, trimethoprim–sulfamethoxazole, and minocycline. These medications might be the most common causes of idiosyncratic drug-induced liver injury, but liver injury in persons taking these drugs is rare. Inclusion in the top 25 implicated agents reflects not just the hepatotoxicity potential but also how commonly the drugs are used and the duration of treatment, which can range from a single intravenous infusion (cefazolin) 30, to a 3-to-14-day course (oral antibiotics), to a year or more of therapy (nitrofurantoin, minocycline, and atorvastatin) 31. The actual incidence of idiosyncratic liver injury from specific drugs is difficult to define; estimates include 1 case per 1000 exposures (isoniazid), 1 per 2500 (amoxicillin–clavulanate), 1 per 10,000 (diclofenac), 1 per 20,000 (atorvastatin), and 1 per 50,000 or more (most drugs) 2. Host and environmental factors may affect the risk, but risk factors are not well defined and are probably specific to the agent, such as male sex and older age for amoxicillin–clavulanate 27, alcoholism for isoniazid and African ancestry for phenytoin, allopurinol, and trimethoprim–sulfamethoxazole 33. Furthermore, there is little evidence that particular combinations of agents are more likely to lead to idiosyncratic hepatic injury, although combinations of hepatotoxins are fairly clear risk factors for direct injury.
A striking finding is that 9 of the top 10 causes of drug-induced liver injury are antimicrobial agents, largely antibiotics. In addition, most of the drugs have been in widespread use for decades. Among the 25 most commonly implicated agents, only 3 were introduced after 2000 (rosuvastatin [2003], duloxetine [2004], and telithromycin [2004]). The reasons that more recently approved drugs are less likely to be implicated in liver injury are not clear but may reflect improvements in drug design, preclinical screening for toxic effects, and a focus on agents with better safety profiles (those that are given in lower doses, are less likely to affect hepatic metabolism, are less lipophilic, and are less likely to interact with other drugs) 53. Another possible reason is the increased scrutiny and criteria for proof of safety required by the Food and Drug Administration 54.
Although recently approved agents may have fewer hepatotoxic effects, many are still of concern. Of note are the kinase and other targeted enzyme inhibitors, more than 50 of which have been introduced in the past two decades 10. Most are antineoplastic agents that cause transient elevations in serum enzyme levels in a sizable proportion of patients and more rarely cause icteric, clinically apparent liver injury (e.g., imatinib, nilotinib, bortezomib, pazopanib, and ribociclib) 48. Also notable are monoclonal antibodies, more than 70 of which are now available. Although these agents are frequently used for cancer chemotherapy, their use has expanded to encompass the treatment of nonmalignant conditions such as autoimmune diseases, migraines, and hypercholesterolemia, as well as management after organ transplantation. Most monoclonal antibodies do not cause liver injury, the exception being those with immunomodulatory actions.
Idiosyncratic drug reactions some inciting drugs:
- Hepatocellular injury immunoallergic: phenytoin, sulfonamides, allopurinol, halothane, diclofenac, quinolones, telithromycin,
- Hepatocellular injury metabolic: isoniazid, troglitazone, ximelagatran, bromfenac
- Cholestatic: estrogens, 17a androgens, chorpromazine, clavulinic acid, piroxicam
- Bile duct injury: carbamazepine, chorpromazine, chlorpropramide, cyproheptadine, thiabendazole, haloperidol
- Microvescicular steatosis: valproate, tetracycline, didanosine
- Phospholipidosis and pseudoalcoholic hepatitis: amiodarone, perhexiline maleate
- Chronic autoimmune-like hepatitis: dantrolene, methyldopa, nifurantoin, oxyphenisatin, propylthiouracil, tienilic acid
Common drugs that are an important cause of liver injury:
- Acetaminophen
- Atomoxetine
- Bosentan
- Erlotinib
- Felbamate
- Infliximab
- Interferon 1b/1a
- Kava
- Leflunomide
- Lipokinetix
- Natalizumab
- Nefazodone
- Nevirapine
- Pyrazinamide/rifampin
- Saquinavir
- Telithromycin
- Terbinafine
- Tolcapone
- Trovofloxacin
- Valproic acid
- Zifirlukast
Drugs withdrawn from the US:
- Bromfenac
- Pemoline
- Ximelegatran*non-US
- lumaricoxib*non-US
Herbal and dietary supplements
The role of herbal and dietary supplements in causing acute liver injury is a growing and perplexing problem. In studies from the United States, the proportions of cases of liver injury caused by herbal or dietary supplements increased from 7 to 9% in 2004–2007 to 19 to 20% in 2010–2014 55. This change probably reflects the increasing use of herbal products and dietary supplements, as well as the lack of rigorous regulatory oversight in the preparation and marketing of these products. The specific products implicated are generally not single herbs (aloe vera, saw palmetto, or black cohosh) or single nutritional substances (creatine, omega fatty acids, or vitamins) but rather are typically multiple-ingredient dietary supplements marketed for weight loss, bodybuilding, or improvements in sexual function, general well-being, or mental acuity 55. These products often have 5 to 20 ingredients, including vitamins, minerals, proteins, and herbs or botanicals of uncertain quality and concentration, often referred to as a “proprietary blend.” The specific chemical component (or components) responsible for the liver injury is rarely obvious. Most multiple-ingredient dietary supplements have commercial names, which are linked to no more than one or two cases of liver injury. Some, however, have been implicated in outbreaks (e.g., Hydroxycut and OxyELITE Pro). Once a popular proprietary supplement is implicated in liver injury, the manufacturer may alter the ingredients and continue to market the product under the same name.
Strikingly, the clinical phenotype of liver injury in most cases associated with herbal and dietary supplements is acute hepatocellular hepatitis, which is often severe, with a high rate of fulminant hepatic failure and need for liver transplantation 56. Commonly implicated components are green tea extracts (Camellia sinensis). The suspected active molecular constituents are catechins, which at high doses cause liver injury in animal models 57. The concentrations of green tea in the animal models, however, are much higher than those in commercial supplements implicated in causing injury in humans 58. In a placebo-controlled trial of green tea extract for the prevention of breast cancer, elevations in serum alanine aminotransferase levels occurred in 6.7% of recipients (36 of 538), as compared with 0.7% of controls (4 of 537) 59. The abnormalities were asymptomatic and resolved promptly with discontinuation of the supplement but recurred rapidly on readministration, suggesting that the injury was idiosyncratic and probably immune-mediated.
Table 4. Medications, herbal products and illicit drugs related to the hepatocellular-type of damage
| Compound | Other injury | Comments |
| Acarbose | Fulminant hepatic failure | |
| Allopurinol | Granuloma | Hypersensitivity |
| Amiodarone | Phospholipidosis, cirrhosis | |
| Amoxicillin, Ampicillin | ||
| Anti-HIV: (Didanosine, Zidovudine, protease inhibitors) | ||
| NSAIDs (AAS, Ibuprofen, Diclofenac, Piroxicam, Indometacin) | Nimesulide; withdrawn | |
| Asparaginase | Steatosis | |
| Bentazepam | Chronic hepatitis | |
| Chlormethizole | Cholestatic hepatitis | Fulminant hepatic failure |
| Cocaine, Ecstasy and amphetamine derivatives | FHF | |
| Diphenytoin | Hypersensitivity | |
| Disulfiram | Fulminant hepatic failure | |
| Ebrotidine | Cirrhosis | Fulminant hepatic failure |
| Fluoxetine, Paroxetine | Chronic hepatitis | |
| Flutamide | Fulminant hepatic failure | |
| Halothane | ||
| Hypolipemics; Lovastatin, Pravastatin, Simvastatin, Atorvastatin | ||
| Isoniazid | Granuloma, chronic hepatitis | Fulminant hepatic failure |
| Ketoconazole, Mebendazole, Albendazole, Pentamidine | Fulminant hepatic failure | |
| Mesalazine | Chronic hepatitis | Autoimmune features |
| Methotrexate | Steatosis, fibrosis, cirrhosis | |
| Minocycline | Chronic hepatitis, steatosis | Autoimmune features |
| Nitrofurantoin | Chronic hepatitis | |
| Nefazodone | Fulminant hepatic failure, withdrawn | |
| Omeprazole | ||
| Penicillin G | Prolonged cholestasis | |
| Pyrazinamide | ||
| Herbal remedies | Fulminant hepatic failure | |
| Germander (Teucrium chamaedrys), senna | ||
| Pennyroyal oil, kava-kava | ||
| Camellia sinnensis (green tea); Chinese herbal medicines | ||
| Risperidone | ||
| Ritodrine | ||
| Sulfasalazine | Hypersensitivity | |
| Telithromycin | ||
| Terbinafine | Cholestatic hepatitis | Fulminant hepatic failure |
| Tetracycline | Micro-steatosis | Fulminant hepatic failure |
| Tolcapone | Fulminant hepatic failure, withdrawn | |
| Topiramate | ||
| Trazodone | Chronic hepatitis | |
| Trovafloxacin | Fulminant hepatic failure, withdrawn in Europe | |
| Valproic acid | Micro-steatosis | |
| Venlafaxine | ||
| Verapamil | Granuloma | |
| Vitamin A | Fibrosis, cirrhosis | |
| Ximelagatran | Fulminant hepatic failure, discontinued |
Footnote: Features of hypersensitivity include fever, rash and eosinophilia.
[Source 60 ]Table 5. Medications associated with the cholestatic-type damage
| Compound | Other injury | Comment |
| Cholestasis without hepatitis (canalicular/bland/pure jaundice) | ||
| Estrogens, contraceptive steroids and anabolic-steroids (Budd-Chiari, adenoma, carcinoma, peliosis hepatitis, adenoma, carcinoma) | ||
| Cholestatis with hepatitis (hepatocanalicular jaundice) | ||
| Amoxicillin-clavulanic acid | Chronic cholestasis | Vanishing bile duct syndrome |
| Atorvastatin | Chronic cholestasis | |
| Azathioprine | Chronic cholestasis | |
| Benoxaprofen (withdrawn) | ||
| Bupropion | Chronic cholestasis | |
| Captopril, enalapril, fosinopril | ||
| Carbamazepine | Chronic cholestasis | Vanishing bile duct syndrome |
| Carbimazole | ||
| Cloxacillin, dicloxacillin, flucloxacillin | ||
| Clindamycin | Chronic cholestasis | |
| Ciprofloxacin, norfloxacin | ||
| Cyproheptadine | Chronic cholestasis | Vanishing bile duct syndrome |
| Diazepam, nitrazepam | ||
| Erythromycins | Chronic cholestasis | Vanishing bile duct syndrome |
| Gold compounds, penicillamine | ||
| Herbal remedies: | ||
| Chaparral leaf (Larrea tridentate); Glycyrrhizin, greater celandine (Chelidonium majus) | ||
| Irbesartan | Chronic cholestasis | |
| Lipid lowering agents (“statins”) | ||
| Macrolide antibiotics | ||
| Mianserin | ||
| Mirtazapine | Chronic cholestasis | |
| Phenotiazines (chlorpromazine) | Chronic cholestasis | |
| Robecoxib, celecoxib | ||
| Rosiglitazone, oioglitazone | ||
| Roxithromycin | Chronic cholestasis | |
| Sulfamethoxazole-trimethoprim | Chronic cholestasis | Vanishing bile duct syndrome |
| Sulfonamides | Chronic cholestasis | |
| Sulfonylureas (Glibenclamide, Chlorpropamide) | ||
| Sulindac, piroxicam, diclofenac, ibuprofen | ||
| Terbinafine | Chronic cholestasis | Vanishing bile duct syndrome |
| Tamoxifen | Hepatocellular, peliosis Chronic cholestasis | |
| Tetracycline | Chronic cholestasis | |
| Ticlopidine & Clopidogrel | Chronic cholestasis | |
| Thiabendazole | Vanishing bile duct syndrome | |
| Tricyclic antidepressants (Amitriptyline, Imipramine) | Chronic cholestasis | Vanishing bile duct syndrome |
| Sclerosing cholangitis-like | Floxuridine (intra-arterial) | |
| Cholangiodestructive (primary biliary cirrhosis) | Chlorpromazine, ajmaline | |
Footnote: Features of hypersensitivity include fever, rash and eosinophilia.
[Source 60 ]Hepatotoxicity signs and symptoms
Drug-induced hepatotoxicity may present in several ways (clinical and pathological) that simulate known forms of acute and chronic liver diseases; the severity ranging from sub-clinical elevations in liver enzyme concentrations to acute liver failure. Drugs tend to induce acute hepatitis, cholestasis or a mixed condition. A clinical picture resembling acute viral hepatitis with jaundice, malaise, anorexia, nausea and abdominal pain is the principal presentation but, because every liver cell may be the target of drug-induced toxicity, many other expressions of hepatotoxicity may be evident including chronic hepatitis, cirrhosis, sinusoidal obstruction syndrome or neoplasm 61.
Symptoms in the hepatocanalicular type of damage include abdominal pain and fever and, as such, resemble acute biliary obstruction. However, the associated hypersensitivity features that sometimes occur are an important clue toward the diagnosis of hepatotoxicity. Liver biopsy reveals variable degrees of portal inflammation and hepatocyte necrosis, in addition to marked cholestasis of centrilobular predominance 62. Older age has been found to increase the likelihood of drug-induced hepatotoxicity being expressed as cholestatic damage 63. Typical examples of drugs that cause this variety of liver damage are amoxicillin-clavulanate, macrolide antibiotics and phenothiazine neuroleptics, but many others have a similar capacity.
Hepatotoxicity diagnosis
Hepatotoxicity diagnosis remains a challenge in clinical practice due to a lack of reliable markers 60. Many other conditions lead to similar clinical as well as pathological pictures. To diagnose hepatotoxicity, a causal relationship between the use of the toxin or drug and subsequent liver damage has to be established, but might be difficult, especially when idiosyncratic reaction is suspected 64. Simultaneous use of multiple drugs may add to the complexity. As in acetaminophen toxicity, well established, dose-dependent, pharmacological hepatotoxicity is easier to spot. Several clinical scales such as CIOMS/RUCAM scale and Maria and Victorino criteria have been proposed to establish causal relationship between offending drug and liver damage. In clinical practice, physicians put more emphasis on the presence or absence of similarity between the biochemical profile of the patient and known biochemical profile of the suspected toxicity (e.g., cholestatic damage in amoxycillin-clauvonic acid) 60. Moreover, severity CANNOT be assessed by the level of serum enzyme elevation; that may indicate rate of hepatocellular injury but does not measure the ability of the liver to function and support life. The only true function tests often done are serum bilirubin concentration and plasma prothrombin time or its INR derivative.
In 1992, under the auspices of the Council for International Organizations of Medical Sciences, a working group developed and implemented a standardized method for drug causality assessment and the scales are named after the organizers of the consensus meeting: CIOMS or RUCAM (Roussel Uclaf Causality Assessment Method) 65. This method provides a standardized scoring system in which the limits and contents of most criteria were decided by consensus among experts on the basis of organ-oriented characteristics. The time-to-onset and duration are evaluated separately for hepatocellular versus cholestatic/mixed reactions since the latter can occur long after the cessation and may be resolved much more slowly (Table 3). The CIOMS/RUCAM scale provides a scoring system for 6 axes in the decision strategy. The categories of suspicion are “definite or highly probable” (score > 8), “probable” (score 6-8), “possible” (score 3-5), “unlikely” (score 1-2) and “excluded” (score ≤ 0). One of the advantages of this system that is of note is that there are very few questions that require a subjective response. The scale can assign a definitive diagnosis of drug-induced hepatotoxicity in patients even without re-challenge. Also, it performs well with newly-marketed drugs or for a previously-unreported liver injury associated with an older drug. The major drawback is its complexity. It requires training in its administration and is less efficient when a user is unfamiliar with the format. The scale may seem cumbersome and while reading across the page, care needs to be taken to not misunderstand the questions; otherwise careless errors can be made. Recently, experts have criticized the weighting attributed to certain of the risk factors (e.g., age of the patient > 55 years, alcohol consumption, pregnancy) which, at best, would be significant only for a limited number of drugs 66.
More recently, Maria and Victorino from Portugal developed a simplified scoring system to overcome the above-mentioned problems. Called the Clinical Diagnostic Scale 67 (also termed the M&V scale) it uses several features of the CIOMS/RUCAM scale while omitting and adding others (Table 6). Five components were selected for inclusion in the scale: temporal relationship between drug intake and the onset of clinical symptoms, exclusion of alternative causes, presence of extra-hepatic manifestations (e.g., rash, fever, arthralgia, eosinophilia > 6% and cytopenia), intentional or accidental re-exposure to the drug, and previous reports in the literature. The sum of the points for each parameter can vary from -6 to +20. Concordance with the five classic degrees of probability of adverse drug reactions is established on the basis of the tabulated score as follows: “definite” (score > 17), “probable” (score 14-17), “possible” (score 10-13), “unlikely” (score 6-9) and “excluded” (score < 6). The authors highlighted some limitations of the scale: The instrument performs poorly in atypical cases of drugs with unusually-long latency periods or chronic outcome. There is room for improvement in the exclusion of alternative causes of liver injury by more clearly specifying the clinical conditions to be excluded, as well as including detailed criteria for exclusion. The main advantage of the Maria and Victorino scale is its ease of application in standard clinical practice.
Table 6. Comparison of the scores for individual axes of the CIOMS and Maria and Victorino diagnostic scales
| CIOMS criteria | Score | Maria & Victorino criteria | Score |
| Chronology criterion | Chronology criterion | ||
| From drug intake until event onset | +2 to +1 | From drug intake until event onset | +1 to +3 |
| From drug withdrawal until event onset | +1 to 0 | From drug withdrawal until event onset | -3 to +3 |
| Time-course of the reaction | -2 to +3 | Time-course of the reaction | 0 to +3 |
| Risk factors | Exclusion of alternative causes | -3 to +3 | |
| Age | +1 to 0 | ||
| Alcohol | +1 to 0 | Extra-hepatic manifestations | 0 to +3 |
| Concomitant therapy | -3 to 0 | Literature data | -3 to +2 |
| Exclusion of non-drug-related causes | -3 to +2 | Re-challenge | 0 to +3 |
| Literature data | 0 to +2 | ||
| Re-challenge | -2 to +3 |
Liver histology (although not very specific and at best resulting in “compatible with”) is the ideal tool to date for defining the pattern of hepatotoxicity. However, since a liver biopsy specimen is often not available, the pattern of drug-related liver injury is, from a practical standpoint, classified according to laboratory data. This mainly includes the activity of serum alanine aminotransferase (ALT) and alkaline phosphatase (ALP) with the increase in activity being expressed with respect to the upper limit of normal and the ratio of the measured activities 68. This classification is somewhat arbitrary and insufficient in classifying all types of drug-induced liver damage (e.g. vascular lesions and chronic damage, in general), however, the system does have some prognostic value.
Acute hepatocellular (e.g., cytotoxic, cytolytic) liver injury is defined by ALT > 2-fold that of upper limit of normal (2N) or an ALT/ALP ratio ≥ 5 68. Patients with this particular type of liver damage have non-specific clinical features, and jaundice is not always evident. Sometimes there are clues of drug allergy, such as fever, rash or peripheral eosinophilia. Serum levels of aminotransferase are markedly increased. Liver histology shows variable degrees of cell necrosis and inflammation, mainly in zone 3 of the hepatic accini together with an abundance of eosinophils in the infiltrate, which is consistent with a toxic etiology 69. These expressions of hepatotoxicity are observed with many drugs. Patients with acute hepatocellular injury related to drugs are at risk of acute liver failure. The observation by Hyman Zimmerman, known as “Hy’s rule” 61, predicts a mean mortality (or its surrogate marker, liver transplantation) of 10% for jaundiced patients with acute toxic hepatocellular damage (providing total bilirubin is not elevated as a result of other causes such as biliary obstruction or Gilbert syndrome). Two recent studies 70, 71 have validated this observation using multivariate analysis, and they indicated that, apart from total bilirubin and the hepatocellular-type of injury, other variables such older age, female gender and AST levels were independently associated with a poor outcome 71.
Hepatotoxicity treatment
In most cases, liver function will return to normal if the offending drug is stopped early. Additionally, the patient may require supportive treatment. In acetaminophen toxicity, however, the initial insult can be fatal. Fulminant hepatic failure from drug-induced hepatotoxicity may require liver transplantation. In the past, glucocorticoids in allergic features and ursodeoxycholic acid in cholestatic cases had been used, but there is no good evidence to support their effectiveness.
Hepatotoxicity prognosis
An elevation in serum bilirubin level of more than 2 times upper limit of normal with associated transaminase rise is an ominous sign. This indicates severe hepatotoxicity and is likely to lead to mortality in 10% to 15% of patients, especially if the offending drug is not stopped (Hy’s Law) 72. This is because it requires significant damage to the liver to impair bilirubin excretion, hence minor impairment (in the absence of biliary obstruction or Gilbert syndrome) would not lead to jaundice. Other poor predictors of outcome are old age, female sex, high AST 73.
Acetaminophen hepatotoxicity
Acetaminophen also known as paracetamol, also known by the brand name Tylenol and Panadol, is one of the most commonly used oral analgesics and antipyretics 74. More than 60 million Americans consume acetaminophen on a weekly basis. Acetaminophen is used in many products in combination with other preparations, especially with opioids and diphenhydramine. Many people are not aware that it is contained in these combination medications 75. Acetaminophen has an excellent safety profile when administered in proper therapeutic doses, but hepatotoxicity can occur if taken in large amounts or when misused in at-risk populations. The recommended dose of acetaminophen for adults is 650 to 1000 mg every 4 to 6 hours, not to exceed 4 grams/day 74. In children, the dose is 15 mg/kg every 6 hours, up to 60 mg/kg/day 74. Acetaminophen liver toxicity develops at 7.5 to 10g/day or 140 mg/kg 76. In the United States, acetaminophen toxicity has replaced viral hepatitis as the most common cause of acute liver failure 77. Acetaminophen liver toxicity is responsible for 56,000 emergency department visits, 2600 hospitalizations, and 500 deaths per year in the United States. Fifty percent of these are unintentional overdoses 76. Acetaminophen toxicity is the second most common cause of liver transplantation worldwide and the most common in the US.
The clinical course of acetaminophen toxicity is divided into four stages:
- During the first stage (0.5 to 24 hours), the patient may be asymptomatic or may have emesis.
- In the second stage (18-72 hours), there may be emesis plus right upper quadrant pain and hypotension.
- In the third stage (72 to 96 hours), liver dysfunction is significant with renal failure, coagulopathies, metabolic acidosis, and encephalopathy. Gastrointestinal (GI) symptoms reappear, and death is most common at this stage.
- Stage 4 (4 days to 3 weeks) is marked by recovery.
Acetaminophen liver toxicity pathophysiology
Acetaminophen is rapidly absorbed from the gastrointestinal (GI) tract and reaches therapeutic levels in 30 minutes to 2 hours. Overdose levels peak at 4 hours unless there are other factors that could delay gastric emptying, such as a co-ingestion of an agent that delays gastric motility, or if the acetaminophen is in extended release form 76.
Acetaminophen metabolism primarily occurs through glucuronidation and sulfuration, both of which occur in the liver. In an overdose, these pathways are saturated, and more acetaminophen is subsequently metabolized to N-acetyl-p-benzoquinoneimine (NAPQI) by cytochrome P450. N-acetyl-p-benzoquinoneimine (NAPQI) is a toxic substance that is safely reduced by glutathione to nontoxic mercaptate and cysteine compounds, which are then renally excreted. An overdose depletes the stores of glutathione, and once they reach less than 30% of normal, NAPQI levels increase and subsequently binds to hepatic macromolecules causing hepatic necrosis. This is irreversible.
Many anti-epileptic and anti-tuberculosis medications are known to increase the activity of cytochrome P450. There is also increased the activity of this enzyme in alcoholics and smokers, although acute intoxication with alcohol or cirrhosis can decrease the activity of cytochrome P450.
Glucuronidation is dependent on carbohydrate stores, and more acetaminophen is converted to NAPQI in the malnourished patient.
There are also decreased stores of glutathione in alcoholics, and patient with AIDS.
The histological features of acetaminophen toxicity will reveal cytolysis and presence of centrilobular necrosis. The injury to the latter is chiefly due to the elevated levels of N-acetyl-p-benzoquinoneimine (NAPQI) in this zone 74.
Acetaminophen hepatotoxicity signs and symptoms
Most patients who overdose on acetaminophen will initially be asymptomatic, as clinical symptoms of end-organ toxicity do not manifest until 24-48 hours after an acute ingestion. Therefore, to identify a patient who may be at risk of hepatoxicity, the clinician should determine the time(s) of ingestion, the quantity, and the formulation of acetaminophen ingested.
Minimum toxic doses of acetaminophen for a single ingestion, posing significant risk of severe hepatotoxicity, are as follows:
- Adults: 7.5-10 g
- Children: 150 mg/kg; 200 mg/kg in healthy children aged 1-6 years
The clinical course of acetaminophen toxicity generally is divided into four phases. Physical findings may vary, depending on the degree of hepatotoxicity.
Phase 1
- 0.5-24 hours after ingestion
- Patients may be asymptomatic or report anorexia, nausea or vomiting, and malaise
- Physical examination may reveal pallor, diaphoresis, malaise, and fatigue
Phase 2
- 18-72 hours after ingestion
- Patients develop right upper quadrant abdominal pain, anorexia, nausea, and vomiting
- Right upper quadrant tenderness may be present
- Tachycardia and hypotension may indicate volume losses
- Some patients may report decreased urinary output (oliguria)
Phase 3: Hepatic phase
- 72-96 hours after ingestion
- Patients have continued nausea and vomiting, abdominal pain, and a tender hepatic edge
- Hepatic necrosis and dysfunction may manifest as jaundice, coagulopathy, hypoglycemia, and hepatic encephalopathy
- Acute renal failure develops in some critically ill patients
- Death from multiorgan failure may occur
Phase 4: Recovery phase
- 4 days to 3 weeks after ingestion
- Patients who survive critical illness in phase 3 have complete resolution of symptoms and complete resolution of organ failure
Acetaminophen hepatotoxicity diagnosis
The diagnosis of acetaminophen toxicity is based on serum levels of the drug, even if there are no symptoms, because clinical symtpoms are delayed. Other laboratory studies needed include liver function tests and PT/INR. If the ingestion is severe, liver function tests can rise within 8-12 hours of ingestion. Normally they become elevated in the second stage at 18 to 72 hours. Co-ingestions can be important, and a urine drug screen, EKG or ECG (to detect additional clues for co-ingestants), and metabolic panel may be useful. If serum levels fall into the toxic range based on the Rumack-Matthew Nomogram, then initiate treatment. The Rumack-Matthew nomogram interprets the acetaminophen concentration (in micrograms per mL), in relation to time (in hours) after ingestion, and is predictive of possible hepatotoxicity after single, acute ingestions of acetaminophen. A level greater than 140 mcg/mL at 4 hours from ingestion is considered toxic. Serum levels must be drawn between 4 to 24 hours from the time of ingestion to properly use the nomogram. It can also only be applied to a single acute ingestion 78.
For chronic acetaminophen ingestions, the Rumack-Matthew Nomogram cannot be applied. Acetaminophen levels do not correlate well with the degree of overdose. In these cases, your healthcare provider must use risk factors, lab values, and clinical suspicion to determine whether or not there was a significant ingestion. Suspect and treat an overdose if the acetaminophen level is greater than 20 mcg/mL or if liver function tests are elevated. There is usually less toxicity as the liver can regenerate its glutathione stores.
Acetaminophen hepatotoxicity treatment
The treatment of acetaminophen poisoning depends on when the drug was ingested. If the patient presents within 1 hour of ingestion, GI decontamination may be attempted. In alert patients, activated charcoal can be used. Orogastric lavage or whole bowel irrigation are not effective therapies 79.
All patients with high levels of acetaminophen need admission and treatment with N-acetyl-cysteine (N-acetyl-cysteine). This agent is fully protective against liver toxicity if given within 8 hours after ingestion. N-acetyl-cysteine works through multiple routes. It prevents binding of NAPQI to hepatic macromolecules, acts as a substitute for glutathione, is a precursor for sulfate, and reduces NAPQI back to acetaminophen. Indications for N-acetyl-cysteine include serum levels that fall in the toxic range according to the Rumack-Matthew Nomogram, an APAP level greater than 10 mcg/mL with an unknown time of ingestion, a dose of acetaminophen greater than 140 mg/kg taken more than 8 hours ago, abnormal labs with an ingestion more than 24 hours ago, and ingestion with any evidence of liver injury.
N-acetyl-cysteine can be administered both intravenously (IV) and orally. The IV form has shown to decrease the length of the hospital stay and may be better tolerated by the patient as the oral form has a foul rotten egg odor and taste. The oral form also requires 18 doses given 4 hours apart, with total treatment time being 72 hours. In comparison, the IV form requires only 20 hours of treatment. The IV form also is preferred in pregnant patients and when there is a fulminant hepatic failure.
Patients who continue to have deterioration such as renal failure, metabolic acidosis, encephalopathy, and coagulopathy should have a referral to a transplant surgeon. In patients who present 24 hours after ingestion of acetaminophen, N-acetyl-cysteine administration should still be attempted and may improve survival. At this stage, it can act as an antioxidant which diminishes hepatic necrosis, decreases neutrophil infiltration, improves microcirculatory blood flow, and increases tissue oxygen delivery. Hemodialysis can also be an effective treatment, especially with concurrent renal failure.
There is no need to adjust the dose for patients with alcoholism or the chronically ill, and it is safe in pregnancy. Repeat acetaminophen levels are also not needed after treatment has begun.
N-acetyl-cysteine should be continued past 72 hours if there is a fulminant hepatic failure until the patient receives a liver transplant, recovers, or dies.
Acetaminophen hepatotoxicity prognosis
If the patient is diagnosed and treated promptly, the mortality for acetaminophen toxicity is less than 2%. However, if patients present late and have developed severe liver failure, the mortality is high. About 1% to 3% of patients with severe liver failure need to undergo a liver transplant as a life-saving measure 80.
In general, children less than 6 years of age have a better prognosis than adults, chiefly because of their greater capacity to detoxify acetaminophen. The overall prognosis of patients depends on the following criteria:
- Creatinine levels more than 3.4 mg/dL
- Arterial pH remaining less than 7.3 despite adequate fluid hydration
- Prothrombin time more than 1.8 times control or an INR of more than 6.5
- Development of grade 3 or 4 encephalopathy.
- Kullak-Ublick GA, Andrade RJ, Merz M, et al. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut 2017;66:1154-1164.[↩][↩]
- Björnsson ES, Bergmann OM, Björnsson HK, Kvaran RB, Olafsson S. Incidence, presentation, and outcomes in patients with drug-induced liver injury in the general population of Iceland. Gastroenterology 2013;144:1419-1425.[↩][↩][↩][↩]
- Vuppalanchi R, Liangpunsakul S, Chalasani N. Etiology of new-onset jaundice: how often is it caused by idiosyncratic drug-induced liver injury in the United States? Am J Gastroenterol 2007;102:558-562.[↩]
- Reuben A, Koch DG, Lee WM. Drug-induced acute liver failure: results of a U.S. multicenter, prospective study. Hepatology 2010;52:2065-2076.[↩][↩][↩]
- Drug-Induced Hepatotoxicity. https://emedicine.medscape.com/article/169814-overview[↩][↩]
- Drug-Induced Liver Injury — Types and Phenotypes. N Engl J Med 2019; 381:264-273 DOI: 10.1056/NEJMra1816149 https://www.nejm.org/doi/full/10.1056/NEJMra1816149[↩][↩][↩]
- Björnsson ES, Hoofnagle JH. Categorization of drugs implicated in causing liver injury: critical assessment based on published case reports. Hepatology 2016;63:590-603.[↩][↩]
- Zimmerman HJ. Hepatotoxicity: the adverse effects of drugs and other chemicals on the liver. 2nd ed. Philadelphia: Lippincott Williams & Wilkins, 1999.[↩][↩][↩][↩]
- Danan G, Benichou C. Causality assessment of adverse reactions to drugs — I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol 1993;46:1323-1330.[↩]
- LiverTox: clinical and research information on drug-induced liver injury. Bethesda, MD: National Institutes of Health https://www.LiverTox.nih.gov[↩][↩]
- Navarro VJ, Senior JR. Drug-related hepatotoxicity. N Engl J Med 2006;354:731-739.[↩]
- Watkins PB. Idiosyncratic liver injury: challenges and approaches. Toxicol Pathol 2005;33:1-5.[↩]
- Larson AM, Polson J, Fontana RJ, et al. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology 2005;42:1364-1372.[↩]
- McDonald GB. Hepatobiliary complications of hematopoietic cell transplantation, 40 years on. Hepatology 2010;51:1450-1460.[↩]
- DeLeve LD, Shulman HM, McDonald GB. Toxic injury to hepatic sinusoids: sinusoidal obstruction syndrome (veno-occlusive disease). Semin Liver Dis 2002;22:27-42.[↩]
- Battipaglia G, Labopin M, Candoni A, et al. Risk of sinusoidal obstruction syndrome in allogeneic stem cell transplantation after prior gemtuzumab ozogamicin treatment: a retrospective study from the Acute Leukemia Working Party of the EBMT. Bone Marrow Transplant 2017;52:592-599.[↩]
- Richardson PG, Riches ML, Kernan NA, et al. Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure. Blood 2016;127:1656-1665.[↩]
- Suárez Ferrer C, Llop Herrera E, Calvo Moya M, et al. Idiopathic portal hypertension regarding thiopurine treatment in patients with inflammatory bowel disease. Rev Esp Enferm Dig 2016;108:79-83.[↩]
- Cotte L, Bénet T, Billioud C, et al. The role of nucleoside and nucleotide analogues in nodular regenerative hyperplasia in HIV-infected patients: a case control study. J Hepatol 2011;54:489-496.[↩]
- Morris-Stiff G, White AD, Gomez D, et al. Nodular regenerative hyperplasia (NRH) complicating oxaliplatin chemotherapy in patients undergoing resection of colorectal liver metastases. Eur J Surg Oncol 2014;40:1016-1020.[↩]
- McKenzie R, Fried MW, Sallie R, et al. Hepatic failure and lactic acidosis due to fialuridine (FIAU), an investigational nucleoside analogue for chronic hepatitis B. N Engl J Med 1995;333:1099-1105.[↩][↩][↩]
- Wei C-M, Chen H-L, Lee P-I, Chen C-M, Ma C-Y, Hwu W-L. Reye’s syndrome developing in an infant on treatment of Kawasaki syndrome. J Paediatr Child Health 2005;41:303-304.[↩]
- Peters RL, Edmondson HA, Mikkelsen WP, Tatter D. Tetracycline-induced fatty liver in nonpregnant patients: a report of six cases. Am J Surg 1967;113:622-632.[↩]
- Su E, Crowley K, Carcillo JA, Michaels MG. Linezolid and lactic acidosis: a role for lactate monitoring with long-term linezolid use in children. Pediatr Infect Dis J 2011;30:804-806.[↩]
- de Boer YS, Kosinski AS, Urban TJ, et al. Features of autoimmune hepatitis in patients with drug-induced liver injury. Clin Gastroenterol Hepatol 2017;15(1):103-112.e2.[↩][↩][↩]
- Björnsson E, Talwalkar J, Treeprasertsuk S, et al. Drug-induced autoimmune hepatitis: clinical characteristics and prognosis. Hepatology 2010;51:2040-2048.[↩]
- deLemos AS, Ghabril M, Rockey DC, et al. Amoxicillin-clavulanate-induced liver injury. Dig Dis Sci 2016;61:2406-2416.[↩][↩]
- Kleiner DE, Chalasani NP, Lee WM, et al. Hepatic histological findings in suspected drug-induced liver injury: systematic evaluation and clinical associations. Hepatology 2014;59:661-670.[↩]
- Bonkovsky HL, Kleiner DE, Gu J, et al. Clinical presentations and outcomes of bile duct loss caused by drugs and herbal and dietary supplements. Hepatology 2017;65:1267-1277.[↩]
- Alqahtani SA, Kleiner DE, Ghabril M, Gu J, Hoofnagle JH, Rockey DC. Identification and characterization of cefazolin-induced liver injury. Clin Gastroenterol Hepatol 2015;13(7):1328-1336.e2.[↩][↩]
- Chalasani N, Bonkovsky HL, Fontana R, et al. Features and outcomes of 889 patients with drug-induced liver injury: the DILIN Prospective Study. Gastroenterology 2015;148(7):1340-52.e7.[↩][↩][↩][↩][↩]
- Devarbhavi H, Raj S, Aradya VH, et al. Drug-induced liver injury associated with Stevens-Johnson syndrome/toxic epidermal necrolysis: patient characteristics, causes, and outcome in 36 cases. Hepatology 2016;63:993-999.[↩][↩]
- Chalasani N, Reddy KRK, Fontana RJ, et al. Idiosyncratic drug induced liver injury in African-Americans is associated with greater morbidity and mortality compared to Caucasians. Am J Gastroenterol 2017;112:1382-1388.[↩][↩]
- Pauli-Magnus C, Meier PJ, Stieger B. Genetic determinants of drug-induced cholestasis and intrahepatic cholestasis of pregnancy. Semin Liver Dis 2010;30:147-159.[↩]
- Robles-Diaz M, Gonzalez-Jimenez A, Medina-Caliz I, et al. Distinct phenotype of hepatotoxicity associated with illicit use of anabolic androgenic steroids. Aliment Pharmacol Ther 2015;41:116-125.[↩]
- Stolz A, Navarro V, Hayashi PH, et al. Severe and protracted cholestasis in 44 young men taking bodybuilding supplements: assessment of genetic, clinical and chemical risk factors. Aliment Pharmacol Ther 2019;49:1195-1204.[↩]
- Daly AK, Donaldson PT, Bhatnagar P, et al. HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat Genet 2009;41:816-819.[↩]
- Lucena MI, Molokhia M, Shen Y, et al. Susceptibility to amoxicillin-clavulanate-induced liver injury is influenced by multiple HLA class I and II alleles. Gastroenterology 2011;141:338-347.[↩]
- Ahmad J, Odin JA, Hayashi PH, et al. Identification and characterization of fenofibrate-induced liver injury. Dig Dis Sci 2017;62:3596-3604.[↩]
- Kim SH, Saide K, Farrell J, et al. Characterization of amoxicillin- and clavulanic acid-specific T cells in patients with amoxicillin-clavulanate-induced liver injury. Hepatology 2015;62:887-899.[↩]
- Cirulli ET, Nicoletti P, Abramson K, et al. A missense variant in PTPN22 is a risk factor for drug-induced liver injury. Gastroenterology 2019;156(6):1707-1716.e2.[↩]
- Stanford SM, Bottini N. PTPN22: the archetypal non-HLA autoimmunity gene. Nat Rev Rheumatol 2014;10:602-611.[↩]
- Kumra S, Herion D, Jacobsen LK, Briguglia C, Grothe D. Case study: risperidone-induced hepatotoxicity in pediatric patients. J Am Acad Child Adolesc Psychiatry 1997;36:701-705.[↩]
- Sacks FM, Stanesa M, Hegele RA. Severe hypertriglyceridemia with pancreatitis: thirteen years’ treatment with lomitapide. JAMA Intern Med 2014;174:443-447.[↩]
- Di Bisceglie AM, Lok AS, Martin P, Terrault N, Perrillo RP, Hoofnagle JH. Recent US Food and Drug Administration warnings on hepatitis B reactivation with immune-suppressing and anticancer drugs: just the tip of the iceberg? Hepatology 2015;61:703-711.[↩]
- Kim HN, Harrington RD, Shuhart MC, et al. Hepatitis C virus activation in HIV-infected patients initiating highly active antiretroviral therapy. AIDS Patient Care STDS 2007;21:718-723.[↩]
- Nociti V, Biolato M, De Fino C, et al. Liver injury after pulsed methylprednisolone therapy in multiple sclerosis patients. Brain Behav 2018;8(6):e00968-e00968.[↩]
- Ghabril M, Bonkovsky HL, Kum C, et al. Liver injury from tumor necrosis factor-α antagonists: analysis of thirty-four cases. Clin Gastroenterol Hepatol 2013;11(5):558-564.e3.[↩][↩]
- Huffman BM, Kottschade LA, Kamath PS, Markovic SN. Hepatotoxicity after immune checkpoint inhibitor therapy in melanoma: natural progression and management. Am J Clin Oncol 2018;41:760-765.[↩][↩]
- Mumoli N, Cei M, Cosimi A (2006). “Drug-related hepatotoxicity”. N. Engl. J. Med. 354 (20): 2191–3, author reply 2191–3. doi:10.1056/NEJMc060733[↩]
- Friedman, Scott E.; Grendell, James H.; McQuaid, Kenneth R. (2003). Current diagnosis & treatment in gastroenterology. New York: Lang Medical Books/McGraw-Hill. pp. p664–679. ISBN 0-8385-1551-7.[↩]
- Ostapowicz G, Fontana RJ, Schiødt FV; et al. (2002). “Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States”. Ann. Intern. Med. 137 (12): 947–54. https://www.ncbi.nlm.nih.gov/pubmed/12484709[↩]
- Regev A. Drug-induced liver injury and drug development: industry perspective. Semin Liver Dis 2014;34:227-239.[↩]
- Avigan MI. DILI and drug development: a regulatory perspective. Semin Liver Dis 2014;34:215-226.[↩]
- Navarro VJ, Khan I, Björnsson E, Seeff LB, Serrano J, Hoofnagle JH. Liver injury from herbal and dietary supplements. Hepatology 2017;65:363-373.[↩][↩]
- Navarro VJ, Barnhart H, Bonkovsky HL, et al. Liver injury from herbals and dietary supplements in the U.S. Drug-Induced Liver Injury Network. Hepatology 2014;60:1399-1408.[↩]
- Bonkovsky HL. Hepatotoxicity associated with supplements containing Chinese green tea (Camellia sinensis). Ann Intern Med 2006;144:68-71.[↩]
- Zheng EX, Rossi S, Fontana RJ, et al. Risk of liver injury associated with green tea extract in SLIMQUICK(®) weight loss products: results from the DILIN prospective study. Drug Saf 2016;39:749-754.[↩]
- Dostal AM, Samavat H, Bedell S, et al. The safety of green tea extract supplementation in postmenopausal women at risk for breast cancer: results of the Minnesota Green Tea Trial. Food Chem Toxicol 2015;83:26-35.[↩]
- Andrade RJ, Robles M, Fernández-Castañer A, López-Ortega S, López-Vega MC, Lucena MI. Assessment of drug-induced hepatotoxicity in clinical practice: a challenge for gastroenterologists. World J Gastroenterol. 2007;13(3):329–340. doi:10.3748/wjg.v13.i3.329 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4065885[↩][↩][↩][↩][↩]
- Zimmerman HJ. Hepatotoxicity. The adverse effects of Drugs and Other Chemicals on the Liver. 2nd ed. Philadelphia: Lippincott Williams & Wilkins; 1999.[↩][↩]
- Goodman ZD. Drug hepatotoxicity. Clin Liver Dis. 2002;6:381–397.[↩]
- Lucena MI, Andrade RJ, Fernández MC, Pachkoria K, Pelaez G, Durán JA, Villar M, Rodrigo L, Romero-Gomez M, Planas R, et al. Determinants of the clinical expression of amoxicillin-clavulanate hepatotoxicity: a prospective series from Spain. Hepatology. 2006;44:850–856.[↩]
- Arundel C, Lewis JH (2007). “Drug-induced liver disease in 2006”. Curr. Opin. Gastroenterol. 23 (3): 244–54. doi:10.1097/MOG.0b013e3280b17dfb[↩]
- Danan G, Benichou C. Causality assessment of adverse reactions to drugs–I. A novel method based on the conclusions of international consensus meetings: application to drug-induced liver injuries. J Clin Epidemiol. 1993;46:1323–1330.[↩]
- Lee WM, Senior JR. Recognizing drug-induced liver injury: current problems, possible solutions. Toxicol Pathol. 2005;33:155–164.[↩]
- Maria VA, Victorino RM. Development and validation of a clinical scale for the diagnosis of drug-induced hepatitis. Hepatology. 1997;26:664–669.[↩]
- Bénichou C. Criteria of drug-induced liver disorders. Report of an international consensus meeting. J Hepatol. 1990;11:272–276.[↩][↩]
- Goodman ZD. Drug hepatotoxicity. Clin Liver Dis. 2002;6:381–397[↩]
- Andrade RJ, Lucena MI, Fernández MC, Pelaez G, Pachkoria K, García-Ruiz E, García-Muñoz B, González-Grande R, Pizarro A, Durán JA, et al. Drug-induced liver injury: an analysis of 461 incidences submitted to the Spanish registry over a 10-year period. Gastroenterology. 2005;129:512–521.[↩]
- Björnsson E, Olsson R. Outcome and prognostic markers in severe drug-induced liver disease. Hepatology. 2005;42:481–489.[↩][↩]
- Hy’s law. Hepatology. 2004 Feb;39(2):574-8. https://doi.org/10.1002/hep.20081[↩]
- Andrade RJ, Lucena MI, Kaplowitz N, et al. (2006). “Outcome of acute idiosyncratic drug-induced liver injury: Long-term follow-up in a hepatotoxicity registry”. Hepatology. 44 (6): 1581–8. doi:10.1002/hep.21424[↩]
- Agrawal S, Khazaeni B. Acetaminophen Toxicity. [Updated 2018 Nov 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441917[↩][↩][↩][↩]
- Athersuch TJ, Antoine DJ, Boobis AR, Coen M, Daly AK, Possamai L, Nicholson JK, Wilson ID. Paracetamol metabolism, hepatotoxicity, biomarkers and therapeutic interventions: a perspective. Toxicol Res (Camb). 2018 May 08;7(3):347-357.[↩]
- Ye H, Nelson LJ, Gómez Del Moral M, Martínez-Naves E, Cubero FJ. Dissecting the molecular pathophysiology of drug-induced liver injury. World J. Gastroenterol. 2018 Apr 07;24(13):1373-1385.[↩][↩][↩]
- Lee WM. Acute liver failure. Semin Respir Crit Care Med. 2012 Feb. 33 (1):36-45.[↩]
- McGill MR, Jaeschke H. Biomarkers of drug-induced liver injury: progress and utility in research, medicine, and regulation. Expert Rev. Mol. Diagn. 2018 Sep;18(9):797-807.[↩]
- Alempijevic T, Zec S, Milosavljevic T. Drug-induced liver injury: Do we know everything? World J Hepatol. 2017 Apr 08;9(10):491-502.[↩]
- Chiew AL, Gluud C, Brok J, Buckley NA. Interventions for paracetamol (acetaminophen) overdose. Cochrane Database Syst Rev. 2018 Feb 23;2:CD003328[↩]




{kind=link}