Hyperpituitarism
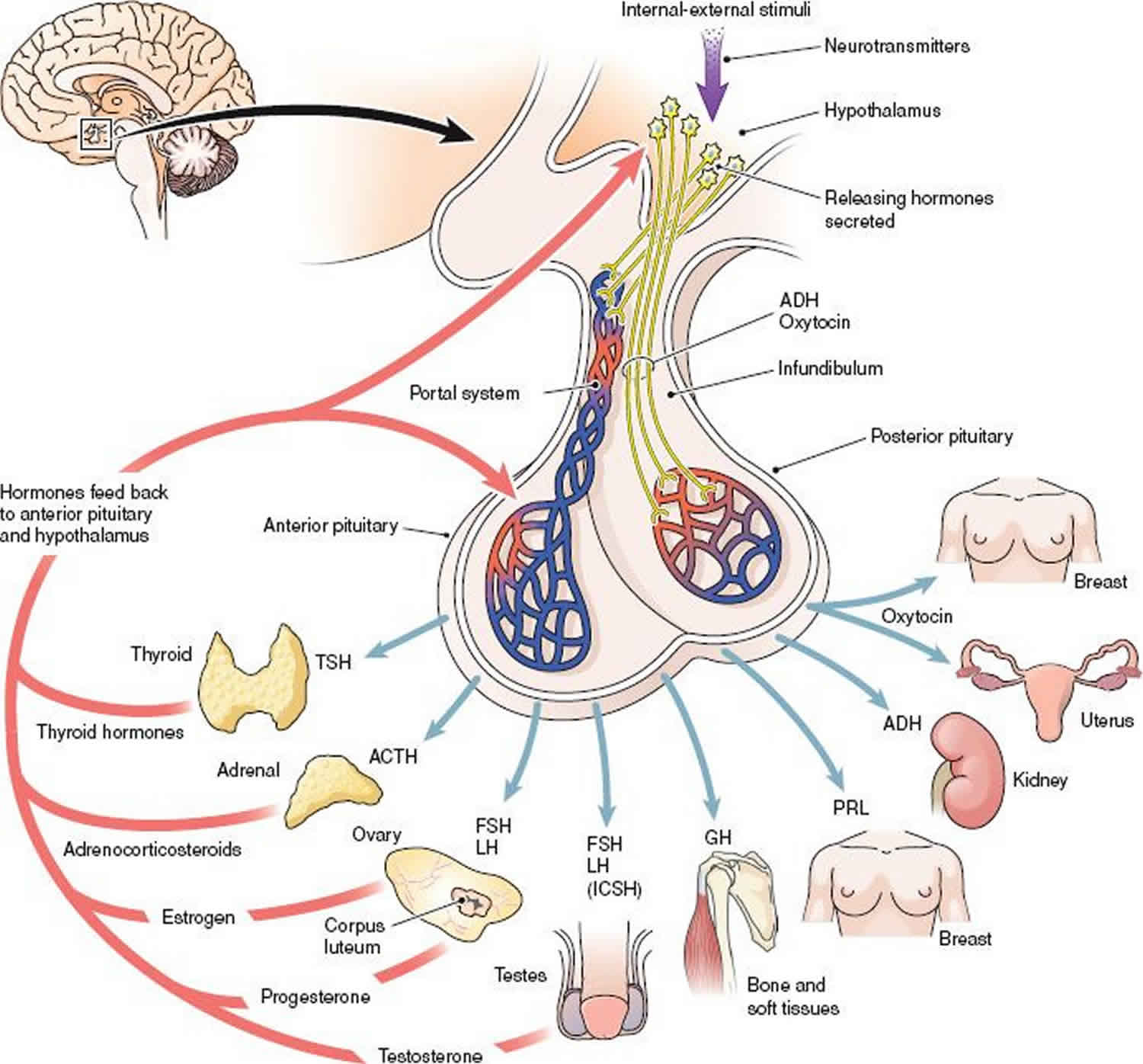
Hyperpituitarism is defined as an excessive secretion or production of 1 or more of the hormones produced by the pituitary gland 1. The pituitary gland produces and secretes various hormones that play a vital role in regulating endocrine function within the body. The pituitary gland consists of 2 lobes: an anterior and a posterior lobe. Hormones produced by the anterior lobe of the pituitary gland include growth hormone (GH), thyroid-stimulating hormone (TSH), luteinizing hormone (LH), follicular stimulating hormone (FSH), adrenocorticotropic hormone (ACTH) and prolactin. Hormones stored and released from the posterior pituitary are antidiuretic hormone (ADH) or vasopressin and oxytocin. ADH (antidiuretic hormone) and oxytocin are produced by neurosecretory cells in the hypothalamus. Trophic hormones produced by the hypothalamus stimulate the production of different anterior pituitary hormones which in turn stimulate the production of hormones at the level of the target organ. Negative feedback by hormones produced by the target organ and tissue inhibit further production of the related pituitary hormones.
Figure 1. Pituitary hormones
Hyperpituitarism causes
Prolactin-secreting pituitary tumors are known as prolactinomas. They are the most frequent secretory tumor of the pituitary gland accounting for at least 40% of all pituitary tumors. These are reported more commonly in women. Other causes of increased prolactin, apart from pituitary adenomas, include craniopharyngioma and other sellar or parasellar masses, infiltration of the hypothalamus, and head trauma. Mild elevations in prolactin may also be seen in patients with hypothyroidism, polycystic ovarian disease, and secondary to use of certain medications such as antipsychotic agents, opioid analgesics, antidepressants, among others. Physiological causes of increased prolactin levels include stimulation of the nipple and sexual intercourse 2.
Excessive production of growth hormone (GH) leads to the clinical conditions acromegaly and gigantism. The incidence of acromegaly is 5 cases per million per year, and the prevalence is 60 cases per million. Over 95% of patients with acromegaly have a growth hormone secreting pituitary adenoma derived from the somatotroph cell line. In less than 5% of cases, acromegaly is a result of excessive growth hormone releasing hormone (GHRH) secretion from a hypothalamic or a neuroendocrine tumor. Ectopic production of growth hormone is extremely rare 3.
Excessive adrenocorticotropic hormone (ACTH) production leads to the presence of hypercortisolism (Cushing syndrome). The incidence of Cushing syndrome is 2 to 3 cases per million. Pituitary adenomas (Cushing disease) accounts for greater than 80% of adrenocorticotropic hormone (ACTH) dependent hypercortisolism, and 10% to 20% are secondary to the ectopic production of ACTH. Cushing disease is more common in females than in males with a ratio of 3:1.4.
Pituitary adenomas that produce thyroid-stimulating hormone (TSH) represent less than 1% of functioning pituitary adenomas, with the Swedish National Registry reporting an incidence rate of 0.15 cases per million inhabitants per year. Co-secretion of growth hormone and prolactin is common.
The presence of a pituitary adenoma with pancreatic endocrine tumors and the parathyroid tumor is associated with multiple endocrine neoplasia (MEN) type 1 syndromes.
Hyperpituitarism symptoms
Presentation of patients with hyperpituitarism will depend on the hormone or hormones that are produced in excess, whether there is a mass pituitary lesion present, and whether there are associated features of hypopituitarism. Local effects of a pituitary tumor such as visual field disturbances and headache may be present in the presence of a mass lesion. Pituitary tumors can be microadenomas less than 10 mm or macroadenomas greater than 10 mm. Macroadenomas usually cause hypopituitarism or panhypopituitarism.
Excessive production of prolactin may occur in isolation or can occur with increased production of other hormones such as GH. Clinical features include infertility and galactorrhoea and secondary amenorrhea in females, loss of libido, and impotence in males. In males, the tumors are usually macroadenomas presenting with features of extension beyond the pituitary.
Excessive production of growth hormone
Adults
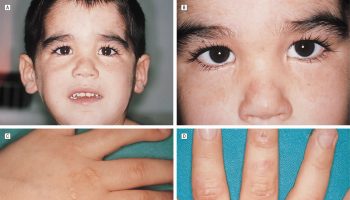
Excessive production of growth hormone (GH) in adults results in the condition known as acromegaly. Features include:
- Skeletal overgrowth of flat bones like the mandible in a condition known as prognathism as well as the growth of bones in the feet with a resultant increase in shoe size
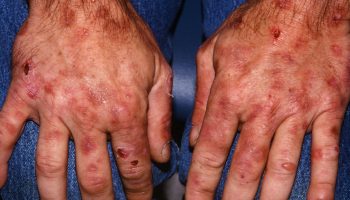
- Overgrowth of skin and subcutaneous tissue
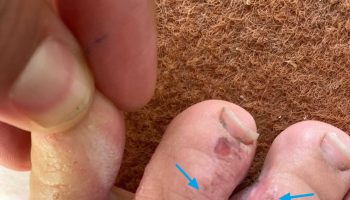
- Increased presence of skin tags; macroglossia; cardiomyopathy; peripheral neuropathy
- Carpal tunnel syndrome caused by compression of median nerve by increased soft tissue growth
- Metabolic disturbances that include glucose intolerance or frank diabetes mellitus as well as hypertension
- Osteoarthritis
- Excessive sweating
Children
In children, prior to fusion of epiphyseal plates in long bones (for example femur and tibia), excessive growth hormone (GH) production results in gigantism.
Excessive production of thyroid-stimulating hormone (TSH) causes secondary hyperthyroidism. Clinical features are those of thyroid hormone excess and include weight loss, heat intolerance, anxiety, menstrual disturbances, and palpitations. A diffuse goiter may also be present. Hyperthyroid symptoms with TSHomas are often mild to moderate.
Excessive production of adrenocorticotropic hormone (ACTH) by the pituitary results in Cushing disease. Clinical features include central obesity, skeletal muscle wasting, the presence of buffalo hump, striae, excessive bruising, menstrual abnormalities, increased blood pressure, glucose intolerance, as well as depression and psychosis.
Hyperpituitarism diagnosis
Laboratory investigations
The initial tests should be directed at the suspected excess hormone. Additionally, the possible deficiencies of other pituitary hormones should also be considered and relevant testing performed 4.
Prolactin
Basal levels of prolactin are useful with values of greater than 200 ng/mL associated with the presence of a prolactinoma. Prolactin levels generally are in parallel to the tumor size. Prolactin (PRL) may be increased due to other causes not related to pituitary disease as described above. MRI and basal prolactin levels are sufficient for the diagnosis. Provocative tests are not indicated. If prolactin levels are normal with clinical features then it is possible that there is assay interference, the “hook effect” and samples should be diluted to provide a reliable result since the excess antigen, prolactin is causing a prozone effect. Rarely patients have an increase in prolactin due to macroprolactinemia which is a harmless condition and can be diagnosed by contacting the laboratory for precipitating the big prolactin.
Growth hormone (Acromegaly/Gigantism)
Random growth hormone levels are usually not recommended due to the diurnal and pulsatile nature of secretion that occurs during a day. Dynamic function (suppression testing) forms the cornerstone of laboratory investigations of growth hormone excess. The oral glucose suppression test involves intake of 75 gm of glucose with measurement of growth hormone levels at zero, 60, and 120 minutes. A growth hormone level of less than 1 ug/L usually excludes the diagnosis of acromegaly. The presence of increased IGF-1 levels accompanies excessive growth hormone production. IGF-1 measurement does not require the use of fasting sample or suppression testing as a random sample adequately reflects the influence of growth hormone excess/deficiency. Thus it is recommended as the initial screening tests for acromegaly. It is vital that the relevant age and sex-matched reference intervals be used when interpreting IGF1 results. As the liver under the action of growth hormone produces IGF1, significant liver disease may result in decreased IGF1 production. Other conditions affecting IGF1 results include renal failure, hypothyroidism, poorly controlled diabetes mellitus and severe malnutrition. Around 30% of patients with Acromegaly can have increased prolactin levels.
Adrenocorticotropic hormone (Cushing Disease)
The laboratory investigation of Cushing syndrome includes confirmation of excessive corticosteroid production followed by identification of ACTH dependent/independent production and determination of the site of adrenocorticotropic hormone (ACTH) production (whether ectopic production is present).
Tests for confirmation of hypercortisolism
- Midnight serum cortisol greater than 5 ug/dl or salivary cortisol greater than 0.15 ug/dl supports a diagnosis of Cushing syndrome. Cushing syndrome is associated with a loss of circadian rhythm of cortisol production, and there is a loss of the normal nadir of cortisol at midnight.
- Twenty-four-hour urine free cortisol which measures unbound cortisol excreted in the urine. The unbound fraction is the active fraction in serum and comprises 5% to 10% of the total circulating cortisol. It cannot be used in patients with significant renal impairment.
- Overnight dexamethasone suppression test in which 1 mg of dexamethasone is administered between 11 pm and midnight, and the serum cortisol is measured the next morning between 8 am, and 9 am. In normal individuals, the cortisol is suppressed less than 1.8 ug/dl. This test has a high sensitivity, but specificity is comparatively low. Drugs affecting the absorption and metabolism of dexamethasone in the liver may affect results. False positives for the overnight dexamethasone suppression test may occur in individuals with obesity, depression, alcoholism, and high estrogen states (pseudo-Cushing syndrome). Also, phenytoin and phenobarbital therapy (both enhance the clearance of dexamethasone) can result in false positives.
- Low-dose dexamethasone suppression test in which 0.5 mg of dexamethasone is taken orally every 6 hours for 48 hours from 9 am on day zero. The serum cortisol is measured between 8 am, and 9 am at 24 and 48 hours respectively.
Determination of ACTH Dependence/Independence
- ACTH levels: Increased ACTH levels in the presence of elevated cortisol production indicate the presence of an ACTH dependent cause of Cushing syndrome. If ACTH is greater than 15 ng/L to 20 ng/L, it is an ACTH dependent cause of Cushing syndrome. It has also been noted that patients with ectopic ACTH production usually have higher levels of ACTH than patients with Cushing disease (pituitary cause).
- High dose dexamethasone suppression test: 2 mg of dexamethasone is administered every 6 hours for 48 hours or with a single dose of 8 mg. Eighty percent of patients with Cushing Disease will suppress to less than 50% of baseline cortisol levels.
- Corticotrophin-releasing hormone (CRH) stimulation test: Corticotrophin-releasing hormone (CRH) is produced by the hypothalamus and stimulates the production of ACTH by the pituitary. In this test, corticotrophin-releasing hormone (CRH) is administered intravenously, and ACTH and cortisol are measured at baseline and short intervals after that. A rise in ACTH of greater than 40% and cortisol levels of greater than 20% indicate an ACTH dependent cause most likely Cushing disease as ectopic sources of ACTH are not usually responsive to corticotrophin-releasing hormone (CRH) stimulation.
The corticotrophin-releasing hormone (CRH) stimulation test may be accompanied by bilateral inferior petrosal sinus sampling to confirm the presence of pituitary lesion causing Cushing syndrome. During this invasive procedure, the inferior petrosal sinuses into which the pituitary venous blood drains is catheterized by a radiologist. ACTH is measured at baseline and following stimulation with 100 micrograms of corticotrophin-releasing hormone (CRH) from both inferior petrosal sinus and peripheral veins. Measurement of prolactin is also performed to confirm catheter is in the correct position. A ratio of inferior petrosal sinus to peripheral ACTH of 2:1 before CRH stimulation and 3:1 after stimulation indicates the pituitary cause of ACTH production consistent with Cushing’s Disease has also been used to assist in lateralization of the pituitary lesion.
Thyroid-stimulating hormone (TSH)
Secondary hyperthyroidism presents with increased or unsuppressed thyroid-stimulating hormone (TSH) levels and elevated thyroid hormone (free and total T4 and T3) levels. This picture is also seen in the presence of laboratory interference and thyroid hormone resistance both of which are more commonly occurring than TSH producing pituitary tumors. Since glycoprotein hormones have alpha and beta subunits, tumors can produce an excess of alpha subunit in TSHoma.
Thyroid releasing hormone (TRH) if available, stimulation test can be used to verify if the thyroid-stimulating hormone (TSH) is of pituitary origin. Following administration of thyroid releasing hormone (TRH), TSH is measured. Patients with a TSH producing tumor have an impaired response.
FSH, LH, estradiol, and testosterone
FSH, LH, estradiol, and testosterone levels may be useful in ascertaining deficiency of these hormones secondary to a pituitary tumor. Rarely elevated FSH and LH may be associated with a gonadotropin cell adenoma.
Imaging studies
Pituitary imaging studies using unenhanced or gadolinium-enhanced MRI are preferred to CT scans for visualization of pituitary adenomas. Often pituitary adenomas are discovered incidentally when imaging studies have been done for other reasons.
Hyperpituitarism treatment
Management of hyperpituitarism will depend on the cause and the hormone or hormones affected.
Pharmacological treatment includes the use of somatostatin analogs and competitive receptor antagonists. For prolactinomas, medical therapy is the usual choice and consists of dopamine agonists such as bromocriptine and cabergoline.
Surgical management
Resection of pituitary and tumors is technically challenging because of the anatomical location. Minimally invasive procedures are increasingly utilized. These include trans-sphenoidal surgery for acromegaly, prolactinoma-macroadenoma and Cushing syndrome with an adenoma. However, as many patients with pituitary tumors often present late with large tumors complete resection is often not possible.
Radiotherapy
Conventional radiotherapy may be used to reduce tumor size however pituitary damage and resultant hypopituitarism may result.
Hyperpituitarism prognosis
The prognosis for hyperfunctioning pituitary tumors in children is very good. Medical therapy or transsphenoidal surgery are preferred methods of treatment.
- Gounden V, Basit H, Jialal I. Hyperpituitarism. [Updated 2019 May 14]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2019 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK482233[↩]
- Ioachimescu AG, Fleseriu M, Hoffman AR, Vaughan Iii TB, Katznelson L. Psychological effects of dopamine agonist treatment in patients with hyperprolactinemia and prolactin-secreting adenomas. Eur. J. Endocrinol. 2019 Jan 01;180(1):31-40.[↩]
- Leonart LP, Borba HHL, Ferreira VL, Riveros BS, Pontarolo R. Cost-effectiveness of acromegaly treatments: a systematic review. Pituitary. 2018 Dec;21(6):642-652.[↩]
- Leonart LP, Ferreira VL, Tonin FS, Fernandez-Llimos F, Pontarolo R. Medical Treatments for Acromegaly: A Systematic Review and Network Meta-Analysis. Value Health. 2018 Jul;21(7):874-880.[↩]


{kind=link}