Mikulicz syndrome
Mikulicz syndrome also known as IgG4-related dacryoadenitis and sialadenitis, dacryosialoadenopathy, Mikulicz’s disease, Mikulicz-Radecki syndrome or Mikulicz-Sjogren syndrome, is an immunoglobulin G4 (IgG4)-related disease characterized by inflammation of the lacrimal glands (which produce tears), parotid glands, and submandibular glands (two of the major salivary glands) 1. In some cases, it also affects other glands or organs 2. Mikulicz syndrome is usually painless, mainly causing mouth and eye dryness, and swelling over the affected glands 2, but can also affect other organs such as the lungs, liver, and kidneys. When other organs are affected, it can be accompanied by complications such as autoimmune pancreatitis, retroperitoneal fibrosis, and tubulointerstitial nephritis 1.
The first descriptions of Mikulicz’ syndrome or IgG4-related dacryoadenitis and sialadenitis were published in the late 19th century. Johann von Mikulicz first presented Mikulicz disease in 1888 at a meeting of the Society for Scientific Medicine in Königsberg 3. He later published a report of this sentinel case detailing a 42 year-old man with an inflammatory disease of the salivary glands with initial symptoms of lacrimal gland swelling followed by submandibular and parotid swelling 3. Mikulicz syndrome is now considered part of the IgG4-related autoimmune disease spectrum, which can include inflammatory disorders of the pancreas, thyroid gland, pachymeninges, pituitary gland and infundibulum 4. Cranial nerve involvement has also been described and there is a postulated association with inflammatory pseudotumor 5.
Mikulicz disease is the second most common autoimmune disease after rheumatoid arthritis (RA), more common in the Northern than the Southern Hemisphere 6. The incidence of Mikulicz disease and Mikulicz syndrome is unknown but thought to be quite rare 4. Patients are primarily women, nine times more likely to be affected than men and Mikulicz disease usually presents in the 4th and 5th decades of life, with symptoms appearing to increase with age 7.
The underlying cause of Mikulicz syndrome or IgG4-related disease is still not known 8.
Mikulicz syndrome treatment involves corticosteroids, which are usually effective 9. Medicines that suppress the immune system (immunosuppressants) may also be used in cases that do not respond to corticosteroids 9.
Figure 1. Mikulicz’s disease
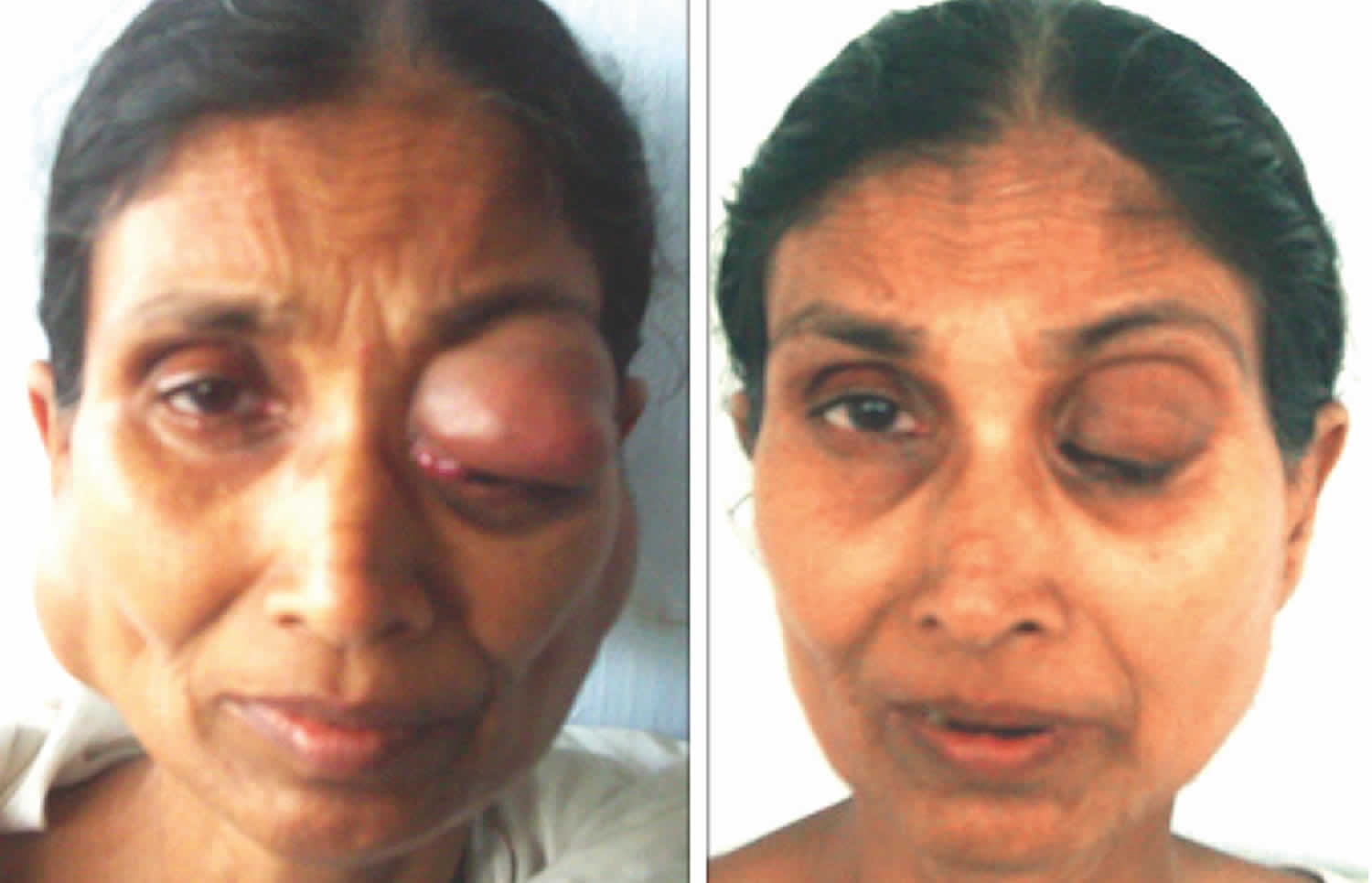
Footnote: A 61-year-old female patient presented with a swell-ing on the right parotid and submandibular regions along with another swelling in the left upper eyelid of 1.5 years’ duration. Both the swelling gradually increased in size with mild pain, which was referred to the right ear. This was followed by another swelling involving the left parotid and submandibular regions and a painless swelling in the left groin for 1 year. Local examination revealed multiple, firm, mobile, nontender, well-defined swelling over both parotid and submandibular regions, right side of the neck, over both the upper eye-lids, and both sides of the groin. The patient had gradu-ally diminishing vision with dryness and irritation of both eyes and associated dryness of the mouth. An oral examination revealed no other abnormality except dryness in the oral mucosa. The patient also had difficultly in talking and hoarseness of voice. Histopathological examination from left parotid and labial biopsy showed lymphoid infiltration and fibrosis consistent with Mikulicz disease.
[Source 6 ]Figure 2. Mikulicz syndrome MRI image
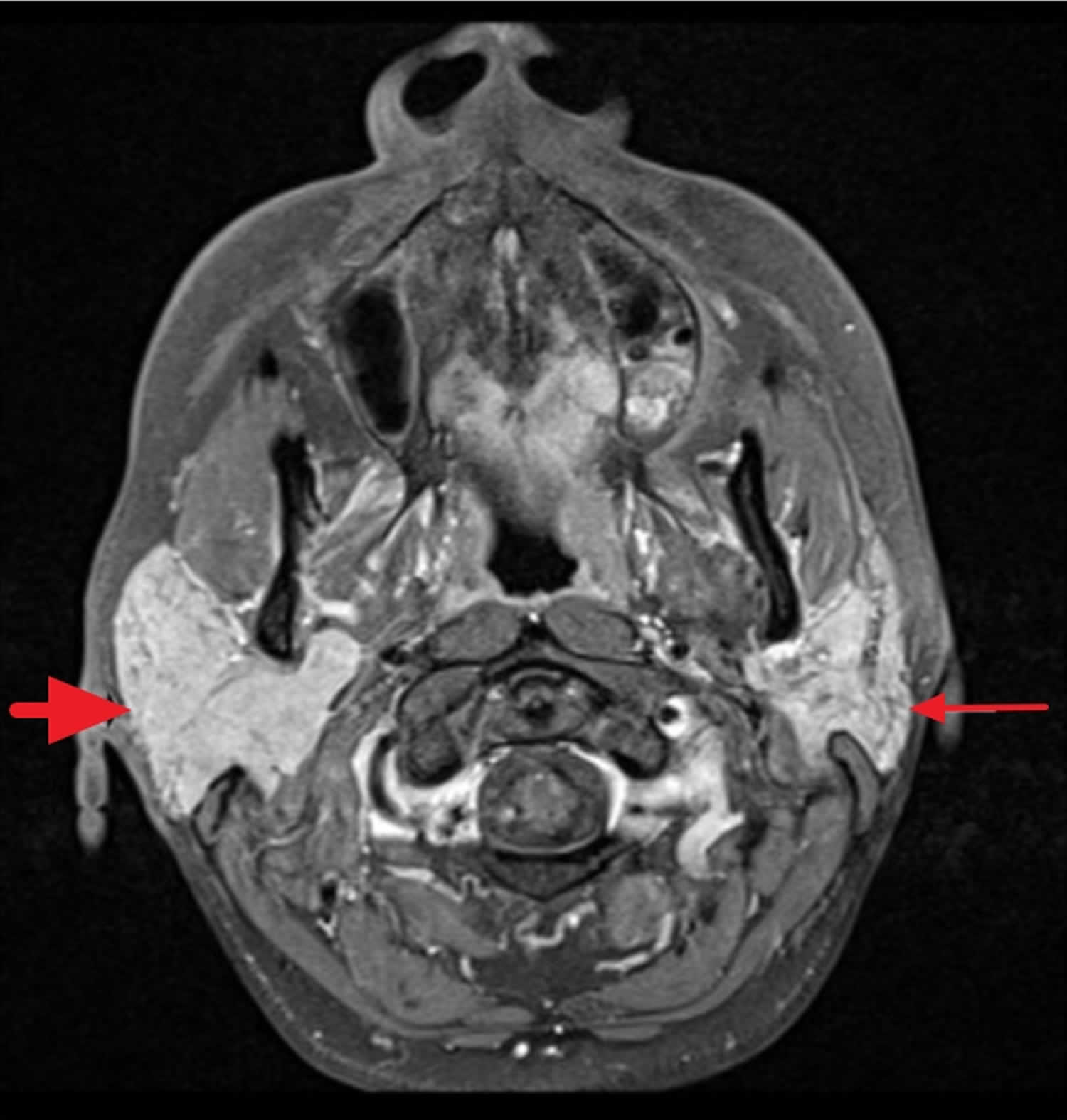
Footnote: A 43-year-old female with Mikulicz syndrome. Axial T1 fat saturated sequence with intravenous contrast demonstrating diffuse enlargement of the right parotid gland (thick short arrow) with homogeneous and normal signal intensity in the right parotid gland without a focal parotid lesion. The left parotid gland is normal in size and signal intensity (thin long arrow). Cranial nerves 5 and 7 were unremarkable (not shown).
[Source 4 ]What is IgG4?
IgG4 is a subclass of immunoglobulin G (IgG), which is the most common form of immunoglobulin. IgG accounts for 75% of antibodies circulating in the blood, which are an essential part of the secondary immune response to infection and toxins. IgG is made by plasma cells, a specific type of B lymphocyte.
There are four subclasses of IgG. Subclass IgG4 is the least common of these, accounting for about 4% of IgG in serum. IgG4 has a unique structure. Its specific biological role is uncertain. However, IgG4 is known to play a role in protection against type 1 hypersensitivity reactions, for example to bee venom, and in the pathogenesis of autoimmune blistering diseases such as pemphigus vulgaris, helminth infections, and malignancy including melanoma.
IgG4-related disease is a newly-described rare syndrome consisting of many disease entities that were previously thought to be unrelated. These conditions have the pathological features of:
- Inflammatory pseudotumours, that is, organomegaly or nodules within an organ
- Lymphoplasmacytic infiltration of tissues with many IgG4-positive plasma cells
- Storiform fibrosis (scarring involving cells arranged like a cartwheel on histology).
Elevated serum IgG4 is present in 60–70% patients.
Mikulicz syndrome causes
The cause of Mikulicz syndrome is unknown. Multiple causes account for Mikulicz syndrome that includes Sjogren syndrome, sarcoidosis, lymphoma, and tuberculosis. According to the criteria of Schaffer and Jacobsen 10, Mikulicz disease is of unknown etiology and follows a benign course, whereas Sjogren syndrome is associated with some other disorders, such as leukemia, lymphosarcoma, tuberculosis, and sarcoidosis.
Mikulicz syndrome pathology
- Mikulicz’s disease may be an autoimmune disorder: antinuclear antibodies and autoantibodies to pancreatic antigens have been found in autoimmune pancreatitis.
- Mikulicz syndrome has features of an allergic disorder: abnormally high Th2 cytokines in tissues, raised IgE and increased T-reg lymphocytes in the blood, and raised eosinophil count in 40% of patients. Cytokines IL-10 (interleukin 10) and TGF-β (transforming growth factor Beta) are known to support IgG4 production and are at elevated levels in IgG4-related disease.
Little is known about the initiation process of immunoglobulin G4-related disease or how and why these specific organ infiltrations occur. There is some evidence from Asian populations, that certain HLA alleles – particularly DRB1*0405 and DQB*0401 – increase the susceptibility to type 1 autoimmune pancreatitis 11. Further, few single-nucleotide polymorphisms (SNP) in genes related to immune system such as CTLA-4 and Fc- receptor-like 3 have been linked to immunoglobulin G4-related disease 12. In autoimmune pancreatitis, autoantibodies against the plasminogen- binding protein of H. pylori were found in the majority of patients in one study 13. Plasminogen- binding protein of H. pylori shares homology to a protein expressed in pancreatic acinar cells. These authors 9 hypothesized, that through molecular mimicry this immune response could lead to immunoglobulin G4-related disease.
It is suspected but unclear, whether immunoglobulin G4-related disease truly belongs to the group of autoimmune disorders. There is evidence of autoantibodies such as ANA, rheumatoid factor, and others in some patients 14. However, these autoantibodies are far beyond from being specific for immunoglobulin G4-related disease. The same is true for organ- specific autoantibodies to proteins expressed for instance in the pancreas or bile ducts 15.
In contrast to many autoimmune disorders, immunoglobulin G4-related disease seems to have a skewed T- cell response towards a TH2 phenotype. Increased tissue levels of TH2-cytokines such as Interleukin-4 (IL-4), −5 (IL-5) and −13 (IL-13) were found in immunoglobulin G4-related disease 16. Further, 40% of immunoglobulin G4-related disease patients have increased serum IgE levels and allergic diseases are common 17. Moreover, tissue eosinophilia is typical for immunoglobulin G4-related disease 18. Some studies also suggested an increased activation of regulatory T cells (Treg), which might be due to the over-expression of transforming growth factor β (TGF-β), an important regulator of Treg development 19. Previously it could be demonstrated that the expression of IgG4 class switch-related molecules is different in labial salivary glands and peripheral blood mononuclear cells. In glands of immunoglobulin G4-related disease patients upregulated Treg cytokines such as IL-10 and TGF-β appear to play pathogenic roles in IgG4-specific class-switch recombination and fibrosis. Activation-induced cytidine deaminase, as a marker of nonspecific immunoglobulin class-switch recombination, was also found to be increased in labial salivary glands of immunoglobulin G4-related disease patients and could contribute to upregulation of IgG4-specific class-switch recombination. IgG4 class-switch recombination seemed to be mainly upregulated in affected organs 20.
Well-fitting to a TH2 immune response, immunoglobulin G4-related disease is characterized by both systemic and localized IgG4 production. It is currently unclear, whether these antibodies are acting as pathogenic antibodies or are just a bystander phenomenon. In healthy individuals, IgG4 is the least abundant IgG subclass molecule accounting for less than 5% of total IgG 21. In addition, IgG4 itself seems to be unable to activate the classic complement pathway. Another characteristic feature of IgG4 antibodies is the possible occurrence of Fab arm exchange of these antibodies 22. This is due to weak bonds between the heavy chain of the IgG4 antibodies thus allowing the Fab arm exchange and formation asymmetric bispecific antibodies. This further hampers antigen cross-linking and immune complex formation of IgG4 antibodies. Nonetheless, there is evidence of importance and pathogenicity of IgG4 antibodies in human diseases. Certain autoantibody immune mediated diseases such as membranous glomerulonephropathy and thrombotic thrombocytopenic purpura seem to be mediated by antibodies of the IgG4 type 23.
Interestingly, low C3 and C4 complement levels can be observed in one third of immunoglobulin G4-related disease patients with renal involvement indicating immune complex formation 24. In type 1 autoimmune pancreatitis, tissue deposition of C3c, IgG4 and IgG can be observed at the basement membrane of pancreatic and bile ducts and of acini 25. C3 and IgG deposition can also be observed at the tubular basement membrane in IgG4- related tubulointerstitial nephritis 24. Thus, immune complex mediated tissue damage may occur at least in some organ manifestations of immunoglobulin G4-related disease.
Recent insights into the pathophysiology of immunoglobulin G4-related disease suggest that one of the major drivers of the disease appear to be plasmablasts. Oligoclonal expanded plasmablasts with extensive somatic hypermutation were found to be increased in immunoglobulin G4-related disease patients with active disease. Of interest, depletion of B cells with rituximab leads to a reduction of these plasmablasts and correlates with disease remission 26. Compared to other inflammatory diseases before treatment and healthy controls, circulating plasmablasts are elevated in patients with active immunoglobulin G4-related disease, even in those patients with normal serum IgG4 concentrations. This observation poses plasmablasts as potential biomarkers for diagnosis and assessment of treatment response 27.
Mikulicz syndrome symptoms
Classic symptoms of Mikulicz’s syndrome are dry mouth (xerostomia) and parotid enlargement 4. Dry eyes and lacrimal gland enlargement are also commonly reported symptoms. Parotid and lacrimal gland enlargement has been reported with both bilateral and unilateral distribution, in concurrence, or in isolation 28. Mikulicz syndrome is usually painless, mainly causing mouth and eye dryness, and swelling over the affected glands 2, but can also affect other organs such as the lungs, liver, and kidneys. When other organs are affected, it can be accompanied by complications such as autoimmune pancreatitis, retroperitoneal fibrosis, and tubulointerstitial nephritis 1. Dry mouth (xerostomia) leads to sore mouth, inability to speak, hoarseness, and dif-ficulty in swallowing and chewing. Decreased mucin production predisposes to loss of taste, bacterial and yeast infection with increased risk of caries 29 and oral candidiasis in upto 80% 30.
Mikulicz syndrome diagnosis
Diagnosis of Mikulicz disease can be done upon the following criteria: (1) Visual confirmation of symmetrical and persistent swelling in more than two lacrimal and major salivary glands; (2) prominent mononuclear infiltration of lacrimal and major salivary glands; and (3) exclusion of sarcoidosis and other lymphoproliferative disease 31.
The diagnosis of immunoglobulin G4-related disease can be difficult, as multiple organs may be involved simultaneously. Diagnostic criteria have not been fully developed. Investigation in suspected IgG4-related disease requires a combination of clinical, endoscopic, radiological and serological tests looking for organ involvement and end-organ damage (eg, hormonal abnormalities). Tissue diagnosis requires a biopsy of affected organ tissues.
Laboratory findings in immunoglobulin G4-related disease are often inconspicuous. Inflammatory markers such as ESR and CRP may be highly elevated, but can be normal despite active disease in a substantial proportion of patients. Anti-nuclear antibodies, anti-SS-A as well as anti-SS-B antibodies are negative in the majority of patients, while low complement levels (C3 and C4) are not uncommon 32. Polyclonal hypergammaglobulinemia is often found in immunoglobulin G4-related disease. Increased serum IgE levels and allergic diseases are present in about one third of patients 17. IgG subclass analyses reveal highly elevated serum IgG4 levels in many but not all patients. It should be underlined that IgG4 levels can be substantially misleading when they are used as a sole criterion for diagnosis (or exclusion) of immunoglobulin G4-related disease. On the one hand, a number of other diseases, such as cancer, infections and autoimmune diseases, including vasculitis, are associated with increased IgG4 levels 33. On the other hand, a number of immunoglobulin G4- related disease patients may have normal IgG4 levels. Thus, the sensitivity of IgG4 in immunoglobulin G4- related disease was found to be 90% and the specificity 60% in one study 34. In another study the positive predictive value of an elevated serum IgG4 for immunoglobulin G4- related disease was found to be as poor as 10% 35.
In some patients with active immunoglobulin G4-related disease IgG4 levels were found to be inappropriately low due to the prozone effect, a situation where too many antibodies prevent agglutination of antigenic particles in the test system. If disease activity is high, but IgG4 levels are low, sample dilution may lead to correction of the prozone effect and reveal – appropriately – much higher IgG4 levels in particular patients 36.
While the location of Mikulicz’s disease manifestation may vary, the histological pattern is – with some exceptions – more or less uniform and shares particular features:
- (i) storiform fibrosis (resembling the spokes of a cartwheel),
- (ii) a dense lymphoplasmatic infiltrate with an increased number of IgG4+ plasma cells (at least > 10/high power field – depending on the particular organ) and/or an increased IgG4/IgG ratio (usually >40%) and
- (iii) an obliterative phlebitis.
Tissue eosinophilia is also very characteristic. The presence of neutrophils, granulomas, neutrophilic microabscesses, and necrotizing vasculitis strongly argues against immunoglobulin G4- related disease 37 In some organs, immunoglobulin G4- related disease can manifest with distinct histopathological features, e.g. in kidney and lymph nodes (see section on clinical manifestations). Recently, a Japanese study group proposed diagnostic criteria for immunoglobulin G4 related disease in general (Figure 3) 38. In addition, there have been proposals for diagnostic criteria of organ- specific immunoglobulin G4- related disease manifestations such as in kidney or pancreatic disease 39.
Figure 3. Comprehensive diagnostic criteria for IgG4- related disease
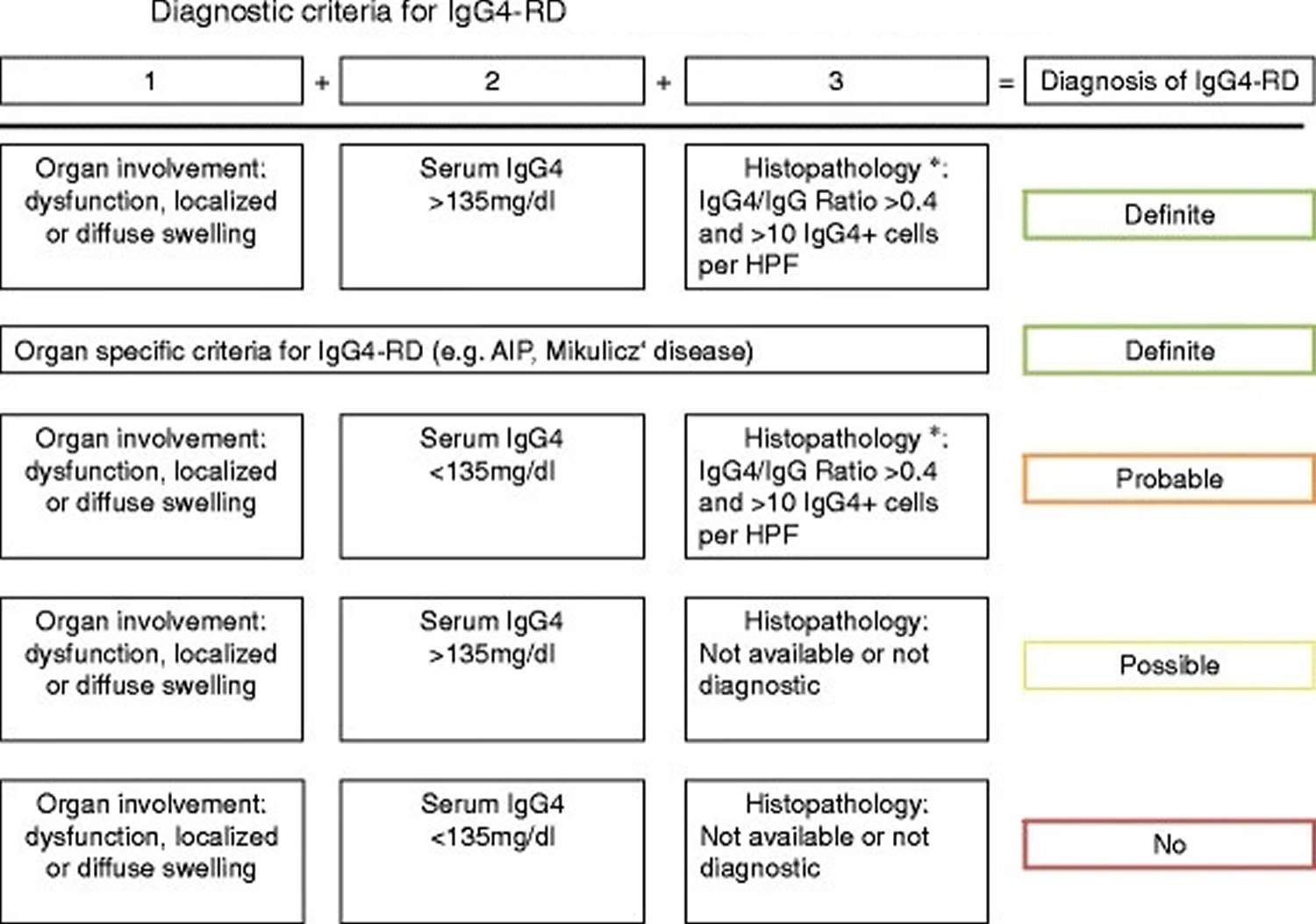
Footnote: *Histopathology suggestive of IgG4-related disease, e.g. lymphoplasmacytic infiltration and fibrosis.
[Source 38 ]Mikulicz syndrome treatment
Mikulicz syndrome is treated with corticosteroid therapy in the acute phase, often prednisone 40 mg per day for 2–4 weeks followed by a gradual tapering of the dose. In patients that cannot be taken off prednisone, a steroid-sparing agent like azathioprine or mycophenolate may be used. Rituximab, a B cell-depleting monoclonal antibody, has been used with some success.
Surgical excision of the affected gland has also been performed.
Organ-specific replacement therapy may be required:
- Thyroxine for thyroiditis (thyroid disease) causing hypothyroidism
- Pancreatic enzyme replacement for pancreatic insufficiency
- Insulin for diabetes mellitus
- Hormone replacement for hypopituitarism: hydrocortisone, thyroxine, growth hormone, desmopressin and sex hormones (testosterone for men; oestrogen and progesterone for women).
Sarcoidosis treatment depends on patient symptoms. Most patients (>75%) require only symptomatic therapy with nonsteroidal anti-inflammatory drugs (NSAIDs). Approximately 10% need treatment for extrapulmonary disease and 15% are treated for persistent pulmonary disease. Oral prednisone given daily is the mainstay of treatment for people with chronic disease 40.
Mikulicz syndrome prognosis
The long-term prognosis of Mikulicz syndrome is unknown. It is not known if IgG4-related disease leads to an increased risk of other forms of cancer.
Major causes for morbidity and mortality are significant organ involvement such as:
- Liver cirrhosis
- Portal hypertension
- Biliary obstruction
- Retroperitoneal fibrosis
- Aortic dissection from an aneurysm
- Diabetes mellitus
- Pancreatic insufficiency
Among Sjogren syndrome patients, the incidence of non-Hodgkin’s lymphoma is 43.8 fold higher than in the general population, although these statistics may be in question given the recently elucidated distinction between Mikulicz syndrome and Sjogren syndrome 41.
- IgG4-related dacryoadenitis and sialadenitis. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=79078[↩][↩][↩]
- Takano K, Yamamoto M, Takahashi H, Himi T. Recent advances in knowledge regarding the head and neck manifestations of IgG4-related disease. Auris Nasus Larynx. February, 2017; 44(1):7-17. http://www.sciencedirect.com/science/article/pii/S0385814616303285[↩][↩][↩]
- Mikulicz J: Über eine eigenartige symmetrische Erkrankung der Tränen und Mundspeicheldrüsen. In Beitr Chir Fortsch Gewidmet Theodor Billroth. Stuttgart: ; 1892:610–630.[↩][↩]
- Rao D, Natter P, Fernandes R, Wang ZB, Sandhu SJS. A Case Report of Mikulicz Syndrome. J Radiol Case Rep. 2017;11(7):1-7. Published 2017 Jul 31. doi:10.3941/jrcr.v11i7.3147 J Radiol Case Rep. 2017 Jul; 11(7): 1–7. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5743147[↩][↩][↩][↩]
- Toyoda K, Oba H, Kutomi K, et al. MR imaging of IgG4-related disease in the head and neck and brain. AJNR Am J Neuroradiol. 2012;33(11):2136–2139[↩]
- Bhattacharjee, A., Uddin, S., Prakash, R., Rathor, A., & Kalita, S. (2015). Diagnostic and Management Challenges in Mikulicz Syndrome. International Journal of Head and Neck Surgery, 6, 139-145. https://pdfs.semanticscholar.org/3e37/c20d2adffab90985f8edd4a36f6cd4ce360b.pdf[↩][↩]
- Pillemar SR, Matteson EL, Jacobsson LT, Martens PB, Melton LJ 3rd, O’Fallon WM, Fox PC. Incidence of physician-diagnosed primary Sjögren syndrome in residents of Olmsted County, Minnesta. Mayo Clin Proc 2001 Jun;76(6):593-599.[↩]
- Kamekura R, Takano K, Yamamoto M, et al. Cutting Edge: A Critical Role of Lesional T Follicular Helper Cells in the Pathogenesis of IgG4-Related Disease. J Immunol. 2017;199(8):2624-2629. doi:10.4049/jimmunol.1601507 https://doi.org/10.4049/jimmunol.1601507[↩]
- Pieringer, H., Parzer, I., Wöhrer, A. et al. IgG4- related disease: an orphan disease with many faces. Orphanet J Rare Dis 9, 110 (2014). https://doi.org/10.1186/s13023-014-0110-z[↩][↩][↩]
- Schaffer A, Jacobsen A. Mickulicz’s syndrome – a report of ten cases. Am J Dis Child 1927 Sep;34(3):327-346.[↩]
- Kawa S, Ota M, Yoshizawa K, Horiuchi A, Hamano H, Ochi Y, Nakayama K, Tokutake Y, Katsuyama Y, Saito S, Hasebe O, Kiyosawa K: HLA DRB10405-DQB10401 haplotype is associated with autoimmune pancreatitis in the Japanese population. Gastroenterology. 2002, 122: 1264-1269.[↩]
- Chang MC, Chang YT, Tien YW, Liang PC, Jan IS, Wei SC, Wong JM: T-cell regulatory gene CTLA-4 polymorphism/haplotype association with autoimmune pancreatitis. Clin Chem. 2007, 53: 1700-1705.[↩]
- Frulloni L, Lunardi C, Simone R, Dolcino M, Scattolini C, Falconi M, Benini L, Vantini I, Corrocher R, Puccetti A: Identification of a novel antibody associated with autoimmune pancreatitis. N Engl J Med. 2009, 361: 2135-2142.[↩]
- Smyk DS, Rigopoulou EI, Koutsoumpas AL, Kriese S, Burroughs AK, Bogdanos DP. Autoantibodies in autoimmune pancreatitis. Int J Rheumatol. 2012;2012:940831. doi:10.1155/2012/940831[↩]
- Nishimori I, Miyaji E, Morimoto K, Nagao K, Kamada M, Onishi S: Serum antibodies to carbonic anhydrase IV in patients with autoimmune pancreatitis. Gut. 2005, 54: 274-281.[↩]
- Müller T, Beutler C, Picó AH, Otten M, Dürr A, Al-Abadi H, Guckelberger O, Meyer Zum Büschenfelde D, Jöhrens K, Volkmann M, Lankisch T, Voigtländer T, Anders M, Shibolet O, Jefferson DM, Podolsky DK, Fischer A, Veltzke-Schlieker W, Adler A, Baumgart DC, Sturm A, Wiedenmann B, Schott E, Berg T: Increased T-helper 2 cytokines in bile from patients with IgG4-related cholangitis disrupt the tight junction-associated biliary epithelial cell barrier. Gastroenterology. 2013, 144: 1116-28.[↩]
- Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH: Prevalence of atopy, eosinophilia, and IgE elevation in IgG4-related disease. Allergy. 2013, [Epub ahead of print][↩][↩]
- Khosroshahi A, Deshpande V, Stone JH: The Clinical and Pathological Features of IgG(4)-Related Disease. Curr Rheumatol Rep. 2011, 13: 473-481.[↩]
- Tanaka A, Moriyama M, Nakashima H, Miyake K, Hayashida JN, Maehara T, Shinozaki S, Kubo Y, Nakamura S: Th2 and regulatory immune reactions contribute to IgG4 production and the initiation of Mikulicz disease. Arthritis Rheum. 2012, 64: 254-263.[↩]
- Tsuboi H, Matsuo N, Iizuka M, et al. Analysis of IgG4 class switch-related molecules in IgG4-related disease. Arthritis Res Ther. 2012;14(4):R171. Published 2012 Jul 23. doi:10.1186/ar3924[↩]
- Yount WJ, Dorner MM, Kunkel HG, Kabat EA: Studies on human antibodies. VI. Selective variations in subgroup composition and genetic markers. J Exp Med. 1968, 127: 633-646.[↩]
- van der Neut KM, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, den Bleker TH, Wiegman L, Vink T, Aarden LA, De Baets MH, van de Winkel JG, Aalberse RC, Parren PW: Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007, 317: 1554-1557.[↩]
- Beck LH, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, Klein JB, Salant DJ: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009, 361: 11-21.[↩]
- Kawano M, Mizushima I, Yamaguchi Y, et al. Immunohistochemical Characteristics of IgG4-Related Tubulointerstitial Nephritis: Detailed Analysis of 20 Japanese Cases. Int J Rheumatol. 2012;2012:609795. doi:10.1155/2012/609795[↩][↩]
- Detlefsen S, Bräsen JH, Zamboni G, Capelli P, Klöppel G: Deposition of complement C3c, immunoglobulin (Ig)G4 and IgG at the basement membrane of pancreatic ducts and acini in autoimmune pancreatitis. Histopathology. 2010, 57: 825-835.[↩]
- Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, Deshpande V, Stone JH, Pillai S: De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014, [Epub ahead of print][↩]
- Wallace ZS, Mattoo H, Carruthers M, Mahajan VS, Della Torre E, Lee H, Kulikova M, Deshpande V, Pillai S, Stone JH: Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2014, [Epub ahead of print][↩]
- Morgan WS, Castleman B. A clinicopathologic study of Mikulicz’s disease. Am J Pathol. 1953;29(3):471–503.[↩]
- Van der Reijden WA, Vissink A, Veerman ECI, Amerongen AVN. Treatment of oral dryness related complaints in Sjögren’s syndrome. Ann Rheum Dis 1999;58(8):465-473.[↩]
- Rehman HU. Sjögren’s syndrome. Yonsei Med J 2003 Dec 30;44(6):947-954.[↩]
- Carbone A, Gloghini A, Ferlito A. Pathological features of lym-phoid proliferations of the salivary glands: lymphoepithelial sialadenitis versus low-grade B-cell lymphoma of the malt type. Ann Otol Rhinol Laryngol 2000 Dec;109(12 Pt 1):1170-1175.[↩]
- Takahashi H, Yamamoto M, Tabeya T, Suzuki C, Naishiro Y, Shinomura Y, Imai K: The immunobiology and clinical characteristics of IgG4 related diseases. J Autoimmun. 2012, 39: 93-96.[↩]
- Ebbo M, Grados A, Bernit E, et al. Pathologies Associated with Serum IgG4 Elevation. Int J Rheumatol. 2012;2012:602809. doi:10.1155/2012/602809[↩]
- Carruthers MN, Khosroshahi A, Augustin T, Deshpande V, Stone JH: The diagnostic utility of serum IgG4 concentrations in IgG4-related disease. Ann Rheum Dis. 2014, [Epub ahead of print][↩]
- Ngwa TN, Law R, Murray D, Chari ST: Serum Immunoglobulin G4 Level Is a Poor Predictor of Immunoglobulin G4-Related Disease. Pancreas. 2014, 43: 704-707.[↩]
- Khosroshahi A, Cheryk LA, Carruthers MN, Edwards JA, Bloch DB, Stone JH: Brief Report: spuriously low serum IgG4 concentrations caused by the prozone phenomenon in patients with IgG4-related disease. Arthritis Rheumatol. 2014, 66: 213-217.[↩]
- Deshpande V: The pathology of IgG4-related disease: critical issues and challenges. Semin Diagn Pathol. 2012, 29: 191-196.[↩]
- Umehara H, Okazaki K, Masaki Y, Kawano M, Yamamoto M, Saeki T, Matsui S, Yoshino T, Nakamura S, Kawa S, Hamano H, Kamisawa T, Shimosegawa T, Shimatsu A, Nakamura S, Ito T, Notohara K, Sumida T, Tanaka Y, Mimori T, Chiba T, Mishima M, Hibi T, Tsubouchi H, Inui K, Ohara H: Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012, 22: 21-30.[↩][↩]
- Kawano M, Saeki T, Nakashima H, Nishi S, Yamaguchi Y, Hisano S, Yamanaka N, Inoue D, Yamamoto M, Takahashi H, Nomura H, Taguchi T, Umehara H, Makino H, Saito T: Proposal for diagnostic criteria for IgG4-related kidney disease. Clin Exp Nephrol. 2011, 15: 615-626.[↩]
- Yee AMF. Sarcoidosis: Rheumatology perspective. Best Pract Res Clin Rheumatol. 2016;30(2):334–356.[↩]
- Kassan SS, Thomas TL, Moutsopoulos HM, et al. Increased risk of lymphoma in sicca syndrome. Ann Intern Med. 1978;89(6):888–892.[↩]





{kind=link}