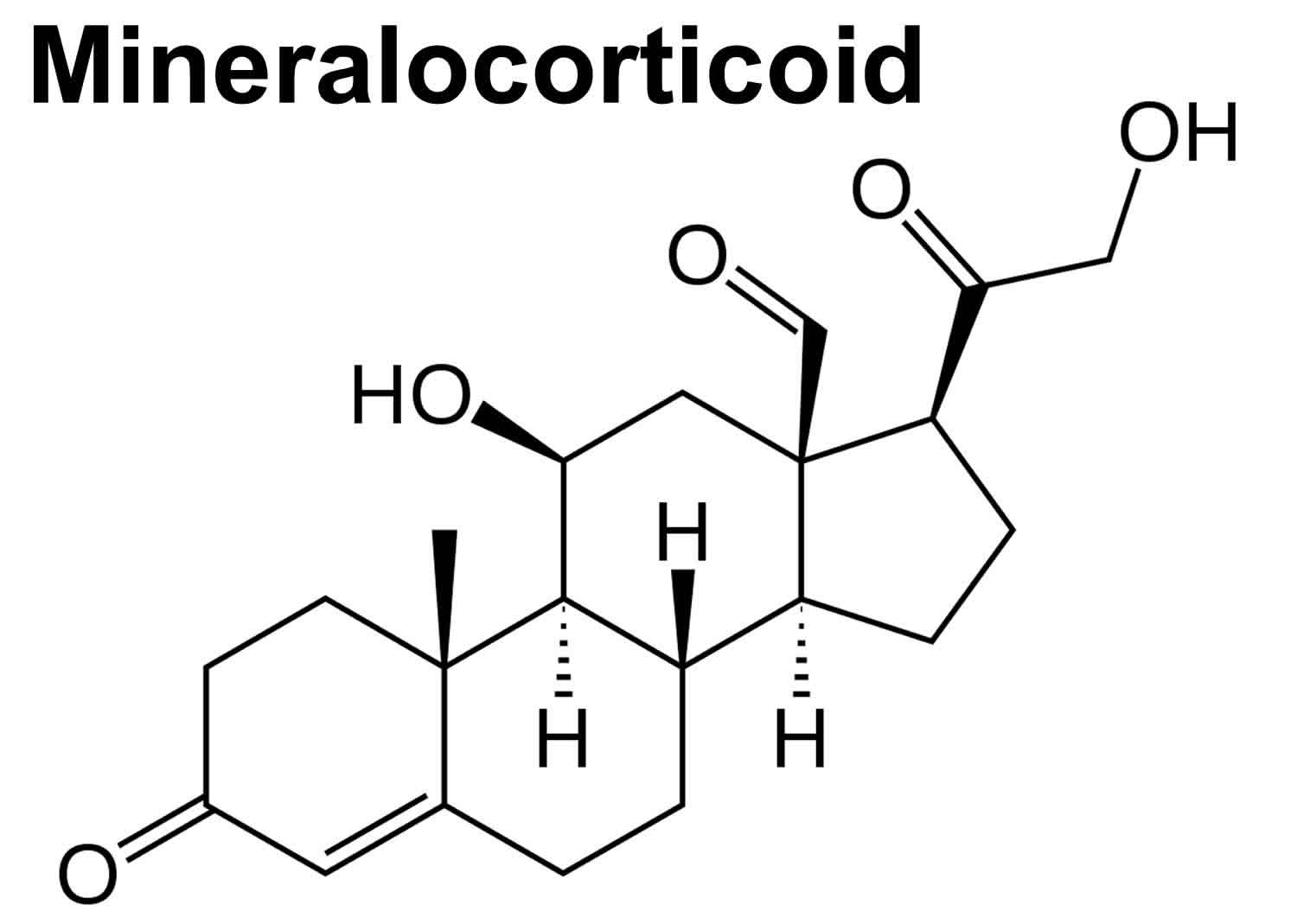
What is mineralocorticoid
Mineralocorticoids are hormones produced in the adrenal cortex zona glomerulosa that include corticosterone, 11-deoxycorticosterone, and more importantly, aldosterone, that act on the kidney to increase sodium reabsorption and potassium excretion 1. Water reabsorption follows increased sodium reabsorption, resulting in an increase in effective circulating volume and therefore increased blood pressure. Mineralocorticoids achieve this via increased synthesis of epithelial sodium channels and sodium-potassium ATPases on the principal cells of the distal nephron 2.
Mineralocorticoids also promote potassium ion secretion at the principal cells because of the gradients produced by the above channels. In high potassium states, aldosterone synthesis is increased to promote potassium excretion. Lastly, mineralocorticoids promote hydrogen ion secretion at the intercalated cells 3.
Interestingly, 11-deoxycorticosterone and corticosterone also have mineralocorticoid effects 1. These are weaker than aldosterone but can produce a strong mineralocorticoid effect when present in excess levels, as in some forms of congenital adrenal hyperplasia (CAH), for example, 11-beta-hydroxylase deficiency resulting in hypertension 2.
Physiologic effects of mineralocorticoids
Mineralocorticoids play a critical role in regulating concentrations of minerals – particularly sodium and potassium – in extracellular fluids to influence salt and water balances (electrolyte balance and fluid balance). As described above, loss of mineralocorticoid hormones leads rapidly to life-threatening abnormalities in electrolyte and fluid balance.
The major target of aldosterone is the distal tubule of the kidney, where it stimulates exchange of sodium and potassium. Three primary physiologic effects of aldosterone result:
- Increased resorption of sodium: sodium loss in urine is decreased under aldosterone stimulation.
- Increased resorption of water, with consequent expansion of extracellular fluid volume. This is an osmotic effect directly related to increased resorption of sodium.
- Increased renal excretion of potassium.
Knowing these effects should quickly suggest the cellular mechanism of action this hormone. Aldosterone stimulates transcription of the gene encoding the sodium-potassium ATPase, leading to increased numbers of “sodium pumps” in the basolateral membranes of tubular epithelial cells. Aldosterone also stimulates expression of a sodium channel which facilitates uptake of sodium from the tubular lumen.
Aldosterone has effects on sweat glands, salivary glands and the colon which are essentially identical to those seen in the distal tubule of the kidney. The major net effect is again to conserve body sodium by stimulating its resorption or, in the case of the colon, absorption from the intestinal lumen. Conservation of water follows conservation of sodium.
Control of aldosterone secretion
Control over aldosterone secretion is truly multifactorial and tied into a spider web of other factors which regulate fluid and electrolyte composition and blood pressure. If the major effects of aldosterone are considered, it is rather easy to predict factors which stimulate or suppress aldosterone secretion.
The two most significant regulators of aldosterone secretion are:
- Concentration of potassium ions in extracellular fluid: Small increases in blood levels of potassium strongly stimulate aldosterone secretion.
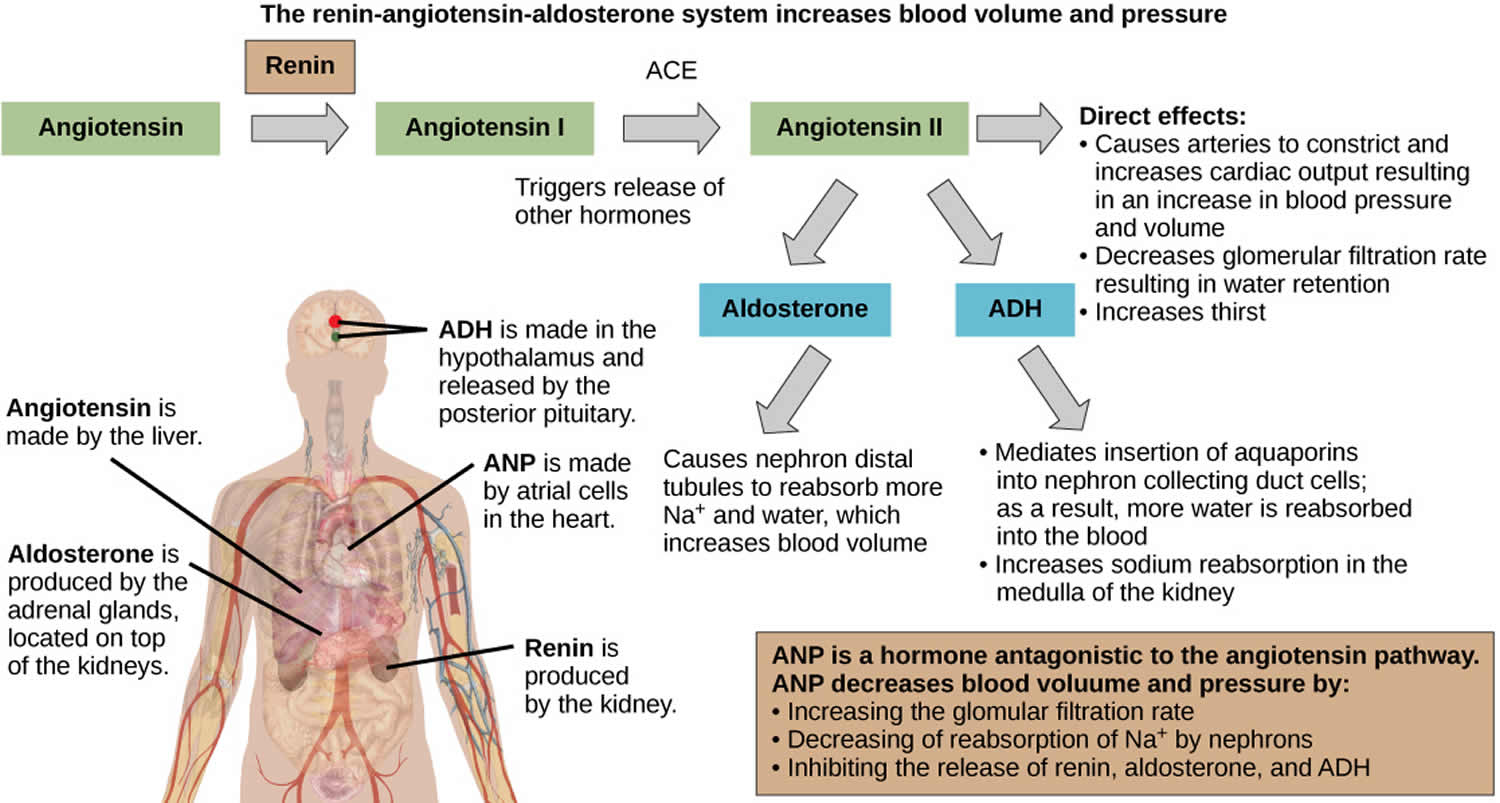
- Angiotensin 2: Activation of the renin-angiotensin system as a result of decreased renal blood flow (usually due to decreased vascular volume) results in release of angiotensin 2, which stimulates aldosterone secretion.
Other factors which stimulate aldosterone secretion include adrenocorticotropic hormone (ACTH) (short-term stimulation only) and sodium deficiency. Factors which suppress aldosterone secretion include atrial naturetic hormone, high sodium concentration and potassium deficiency.
Figure 1. Renin angiotensin aldosterone system

Mineralocorticoid excess
Hyperaldosteronism is a disorder in which the adrenal gland releases too much of the hormone aldosterone into the blood.
Hyperaldosteronism can be primary or secondary.
Aldosterone excess is most commonly observed in two conditions: elevated plasma potassium (hyperkalemia) and low vascular volume. This should make sense considering that plasma potassium and angiotensin 2 are the major factors that regulate aldosterone secretion, as described above. Importantly, it is now recognized that roughly 1 in 10 cases of primary hypertension in humans is associated with hyperaldosteronism, due most commonly to aldosterone-secreting adrenal tumors or mutations in potassium channels.
Mineralocorticoid excess causes
Primary hyperaldosteronism is due to a problem of the adrenal glands themselves, which causes them to produce too much aldosterone, causing you to lose potassium and retain sodium. The excess sodium in turn holds on to water, increasing your blood volume and blood pressure. Primary aldosteronism leads to high blood pressure.
In contrast, with secondary hyperaldosteronism, a problem elsewhere in the body causes the adrenal glands to release too much aldosterone. These problems can be with genes, diet, or a medical disorder such as with the heart, liver, kidneys, or high blood pressure.
Common conditions causing the overproduction of aldosterone include:
- A benign growth in an adrenal gland (aldosterone-producing adenoma) — a condition also known as Conn’s syndrome
- Overactivity of both adrenal glands (idiopathic hyperaldosteronism)
Most cases of primary hyperaldosteronism are caused by a noncancerous (benign) tumor of the adrenal gland. The condition is most common in people 30 to 50 years old.
In rare cases, primary aldosteronism may be caused by:
- A cancerous (malignant) growth of the outer layer (cortex) of the adrenal gland (adrenal cortical carcinoma)
- A rare type of primary aldosteronism called glucocorticoid-remediable aldosteronism that runs in families and causes high blood pressure in children and young adults
Diagnosis and treatment of primary aldosteronism are important because people with this form of high blood pressure have a higher risk of heart disease and stroke. Also, the high blood pressure associated with primary aldosteronism may be curable.
Options for people with primary aldosteronism include medications, lifestyle modifications and surgery.
Mineralocorticoid excess symptoms
Primary and secondary hyperaldosteronism have common symptoms, including:
- Moderate to severe high blood pressure
- High blood pressure that takes several medications to control (resistant hypertension)
- High blood pressure along with a low potassium level in the blood (hypokalemia)
- Feeling tired all the time
- Headache
- Muscle weakness
- Numbness
Have your blood pressure checked regularly, especially if you have risk factors for high blood pressure. Ask your doctor about the possibility of having primary aldosteronism if:
- You’re age 45 or older
- You have a family history of high blood pressure
- You have high blood pressure that began at age 44 or younger
- You’re overweight
- You have a sedentary lifestyle
- You use tobacco
- You drink a lot of alcohol
- You have dietary imbalances (too much salt, not enough potassium)
Mineralocorticoid excess complications
Primary aldosteronism can lead to high blood pressure and low potassium levels. These complications in turn can lead to other problems.
Problems related to high blood pressure
Persistently elevated blood pressure can lead to problems with your heart and kidneys, including:
- Heart attack
- Heart failure
- Left ventricular hypertrophy — enlargement of the muscle that makes up the wall of the left ventricle, one of your heart’s pumping chambers
- Stroke
- Kidney disease or kidney failure
- Premature death
High blood pressure caused by primary aldosteronism carries a higher risk of cardiovascular complications than do other types of high blood pressure. This excess risk is due to the high aldosterone levels, which can cause heart and blood vessel damage independent of complications related to high blood pressure.
Problems related to low potassium levels (hypokalemia)
Some, but not all, people with primary aldosteronism have low potassium levels (hypokalemia). Mild hypokalemia may not cause any symptoms, but very low levels of potassium can lead to:
- Weakness
- Cardiac arrhythmias
- Muscle cramps
- Excess thirst or urination
Mineralocorticoid excess diagnosis
A variety of tests are available to help diagnose primary aldosteronism.
Screening test
Initially, your doctor is likely to measure the levels of aldosterone and renin in your blood. Renin is an enzyme released by your kidneys that helps regulate blood pressure. The combination of a very low renin level with a high aldosterone level suggests that primary aldosteronism may be the cause of your high blood pressure.
Confirmation tests
If the aldosterone-renin test suggests that you might have primary aldosteronism, you’ll need another test to confirm the diagnosis, such as one of the following:
- Oral salt loading. You’ll follow a high-sodium diet for three days before your doctor measures aldosterone and sodium levels in your urine.
- Saline infusion test. Your aldosterone levels are tested after sodium mixed with water (saline) is infused into your bloodstream for several hours.
- Fludrocortisone suppression test. After you’ve followed a high-sodium diet and taken fludrocortisone — which mimics the action of aldosterone — for several days, aldosterone levels in your blood are measured.
Additional tests
If you receive a diagnosis of primary aldosteronism, your doctor will run additional tests to determine whether the underlying cause is an aldosterone-producing adenoma or overactivity of both adrenal glands. Tests may include:
- Abdominal computerized tomography (CT) scan. A CT scan can help identify a tumor on your adrenal gland or an enlargement that suggests overactivity. You may still need additional testing after a CT scan because this imaging test may miss small but important abnormalities or find tumors that don’t produce aldosterone.
- Adrenal vein sampling. A radiologist draws blood from both your right and left adrenal veins and compares the two samples. Aldosterone levels that are significantly higher on one side indicate the presence of an aldosteronoma on that side. Similar aldosterone levels on both sides point to overactivity in both glands. This test involves placing a tube in a vein in your groin and threading it up to the adrenal veins. Though essential for determining the appropriate treatment, this test carries the risk of bleeding or a blood clot in the vein.
Mineralocorticoid excess treatment
Treatment for primary aldosteronism depends on the underlying cause, but its basic goal is to normalize or block the effect of high aldosterone levels and prevent the potential complications of high blood pressure and low potassium levels.
Treatment for an adrenal gland tumor
An adrenal gland tumor may be treated with surgery or medications and lifestyle changes.
- Surgical removal of the adrenal gland. Surgical removal of the adrenal gland containing the tumor (adrenalectomy) is usually recommended because it may permanently resolve high blood pressure and potassium deficiency, and it can bring aldosterone levels back to normal. Blood pressure usually drops gradually after a unilateral adrenalectomy. Your doctor will follow you closely after surgery and progressively adjust or eliminate your high blood pressure medications. An adrenalectomy carries the usual risks of abdominal surgery, including bleeding and infection. However, adrenal hormone replacement is not necessary after a unilateral adrenalectomy because the other adrenal gland is able to produce adequate amounts of all the hormones on its own.
- Aldosterone-blocking drugs. If you’re unable to have surgery or prefer not to, primary aldosteronism caused by a benign tumor can also be treated with aldosterone-blocking drugs (mineralocorticoid receptor antagonists) and lifestyle changes. But high blood pressure and low potassium will return if you stop taking your medications.
Treatment for overactivity of both adrenal glands
A combination of medications and lifestyle modifications can effectively treat primary aldosteronism caused by overactivity of both adrenal glands (bilateral adrenal hyperplasia).
- Medications. Mineralocorticoid receptor antagonists block the action of aldosterone in your body. Your doctor may first prescribe spironolactone. This medication helps correct high blood pressure and low potassium, but it may cause problems. In addition to blocking aldosterone receptors, spironolactone blocks androgen and progesterone receptors and may inhibit the action of these hormones. Side effects can include male breast enlargement (gynecomastia), decreased sexual desire, impotence, menstrual irregularities and gastrointestinal distress. A newer, more expensive mineralocorticoid receptor antagonist called eplerenone acts just on aldosterone receptors, but eliminates the sex hormone side effects associated with spironolactone. Your doctor may recommend eplerenone if you have serious side effects with spironolactone. You may also need other medications for high blood pressure.
- Lifestyle changes. High blood pressure medications are more effective when combined with a healthy diet and lifestyle. Work with your doctor to create a plan to reduce the sodium in your diet and maintain a healthy body weight. Getting regular exercise, limiting the amount of alcohol you drink and stopping smoking also may improve your response to medications.
Home remedies
A healthy lifestyle is essential for keeping blood pressure low and maintaining long-term heart health. Here are some healthy lifestyle suggestions:
- Follow a healthy diet. Limit the sodium in your diet by focusing on fresh foods and reduced-sodium products, avoiding condiments, and removing salt from recipes. Diets that also emphasize a healthy variety of foods — including grains, fruits, vegetables and low-fat dairy products — can promote weight loss and help lower blood pressure. Try the Dietary Approaches to Stop Hypertension (DASH) diet — it has proven benefits for your heart.
- Achieve a healthy weight. If your body mass index (BMI) is 25 or more, losing as few as 10 pounds (4.5 kilograms) may reduce your blood pressure.
- Exercise. Regular aerobic exercise can help lower blood pressure. You don’t have to hit the gym — taking vigorous walks most days of the week can significantly improve your health. Try walking with a friend at lunch instead of dining out.
- Don’t smoke. Quitting smoking will improve your overall cardiovascular health. Nicotine in tobacco makes your heart work harder by constricting your blood vessels and increasing your heart rate and blood pressure. Talk to your doctor about medications that can help you stop smoking.
- Limit alcohol and caffeine. Both substances can raise your blood pressure, and alcohol can interfere with the effectiveness of some blood pressure medications. Ask your doctor whether moderate alcohol consumption is safe for you.
Mineralocorticoid deficiency
A deficiency in aldosterone can occur by itself or, more commonly, in conjunction with a glucocorticoid deficiency, and is known as hypoadrenocorticism or Addison’s disease. Without treatment by mineralocorticoid replacement therapy, a lack of aldosterone is lethal, due to electrolyte imbalances and resulting hypotension and cardiac failure.
Mineralocorticoid deficiency causes
Various syndromes are characterized by or associated with hypoaldosteronism. Hypoaldosteronism is classified in three large categories, defective stimulation of aldosterone secretion, primary defects in adrenal synthesis or secretion of aldosterone and aldosterone resistance, according to their pathophysiology and summarized in Table 1.
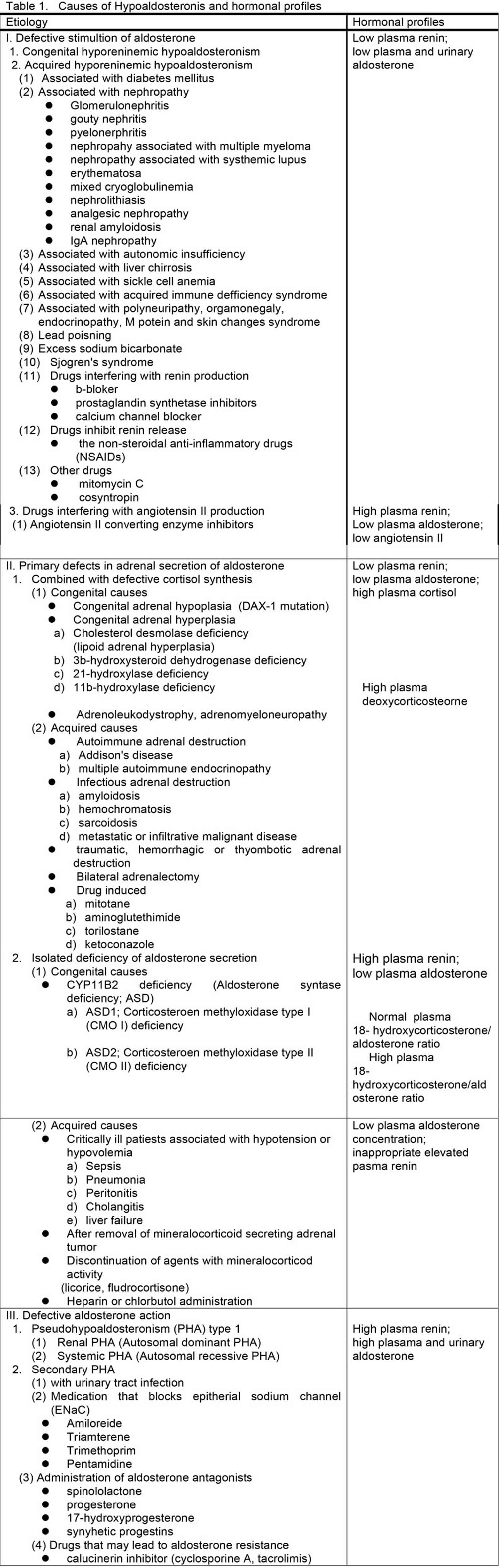
Defective stimulation of aldosterone secretion
The first category of conditions, which is characterized by defective stimulation of aldosterone secretion, includes the syndromes of congenital and acquired hyporeninemic hypoaldosteronism. One of these conditions is due to a defect of renin secretion such as hyporeninemia resulting from β-blockers, prostaglandin synthetase inhibitors, and calcium channel blockers. Another condition is due to decrease in the conversion of angiotensin I to angiotensin II mediated by converting enzyme inhibitor medications and is associated with hyperreninemia.
Primary defects in adrenal biosynthesis or secretion of aldosterone
The second category of conditions, which are characterized by primary defects in adrenal synthesis or secretion of aldosterone, includes all causes of primary adrenal insufficiency and primary hypoaldosteronism caused by aldosterone synthase (CYP11B2) deficiency or as an acquired state. Primary adrenal insufficiency causes include congenital adrenal hypoplasia, congenital adrenal hyperplasia, adrenoleukodystrophy/adrenomyeloneuropathy, acquired adrenal insufficiency due to autoimmune, infectious and infiltrative disease, bilateral adrenalectomy and use of adrenolytic agents and enzyme inhibitors that block cortisol and aldosterone biosynthesis. These conditions are usually combined with defective cortisol synthesis. Aldosterone synthase (CYP11B2) deficiency (ASD) leads to reduced aldosterone production associated with low or high levels of 18-hydroxycorticosterone, referred to as corticosterone methyloxidase I (CMO I) or corticosterone methyloxidase II (CMO II) deficiency, respectively. Several conditions may be associated with aldosterone biosynthetic activity. Heparin suppresses aldosterone synthesis. Critically ill patients with persistent hypovolemia and hypotension also have inappropriately low plasma aldosterone concentrations in relation to the activity of the renin-angiotensin system. Isolated primary hypoaldosteronism in occasionally associated with metastatic cancer of the adrenal gland.
Defective aldosterone actions
The third category which is characterized by defective aldosterone action includes syndromes of aldosterone resistance such as pseudohypoaldosteronism type 1 and sodium-wasting states resulting from excessive amounts of circulating mineralocorticoid antagonists, such as spinololactone and its analogues, and synthetic progestin or natural agonists, such as progesterone or 17-hydroxyprogesterone. These mineralocorticoid antagonists may antagonize aldosterone at the levels of mineralocorticoid receptor 5 and frequently, these states are compensated for by elevated concentrations of plasma aldosterone.
Mineralocorticoid deficiency symptoms
Hyporeninemic hypoaldosteronism
The most common form of isolated hypoaldosteronism is caused by impaired renin release from the kidney. Hudson et al. 6 first described this syndrome in 1957, however, hyporeninemia was first recognized in 1972 7. The typical patient is 50 to 70 years old and usually presents with chronic and asymptomatic hyperkalemia and mild to moderate renal insufficiency with a 40-70% decrease in the glomerular filtration rate when compared to that of age matched healthy subjects. Hyperchloremic metabolic acidosis is seen in approximately half of the patients. This acidosis is classified as a renal tubular acidosis type 4 8. The acidosis is a consequence of decreased renal ammonia neogenesis, reduced hydrogen ion-secretory capacity in the distal nephron and mild reduction in the proximal tubular threshold for bicarbonate reabsorption. Occasionally, muscle weakness or cardiac arrhythmias are present in some patients. More than a half of the patients have diabetes mellitus 9. Other frequently associated states include autonomic neuropathy, hypotension and various nephropathies such as glomerulonephritis, gouty nephropathy and pyelonephritis 10. Also, this syndrome is associated with nephropathies of multiple myeloma and systemic lupus erythematosus, mixed cryoglobulinemia, nephrolithiasis, analgesic nephropathy, renal amyloidosis, IgA nephropathy, cirrhosis, sickle cell anemia, acquired immune deficiency syndrome (AIDS), polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes (POEMS) syndrome, lead poisoning, excess sodium bicarbonate and Sjögren’s symdrome 11. Moreover, this syndrome occurs transiently in association with use of non-steroidal anti-inflammatory drugs, cyclosporin A, mitomycin C, cosyntropin and other agents in susceptible individuals 12.
Pathophysiology
Urinary aldosterone excretion is low under basal conditions and fails to increase after sodium restriction. Plasma renin activity is also low and does not increase appropriately during sodium restriction, periods of prolonged upright posture, or diuretic administration 13. Interstitial renal disease and damage to the juxtaglomerular apparatus seems the most likely cause for the primary defect in renin generation or release and secondary deficiency of aldosterone. However, in some patients with this syndrome there is an absent or blunted aldosterone response to angiotensin 2 14, suggesting a coexisting primary defect in aldosterone secretion or it reflects atrophy of the zona glomerulosa caused by chronic renin deficiency.
There are various mechanisms to be explained the hyporeninemia. First possible mechanism is the hypervolemia. The expanded extracellular fluid volume due to hypertension may suppress renin. In fact, long-term sodium restriction and diuretic administration increase plasma renin activity in these patients, however, the increments of plasma renin activity are less than those of normal subjects 15. A second possible mechanism is insufficiency of the autonomic nervous system, particularly in patients with diabetic neuropathy. Impaired adrenergic response to postural change may contribute to insufficient renin release. Besides, these patients exhibit decreased sensitivity to β-adrenergic agonists, suggesting defects in both production and action of catecholamines 16. A third proposed mechanism is secretion of abnormal forms of renin, such as a defect in the conversion of prorenin to renin. Insufficiency of autonomic nervous system may be associated with impaired conversion of prorenin to renin. Indeed, diabetic patients with autonomic neuropathy have elevated plasma levels of prorenin 17. A fourth possibility is prostaglandin deficiency. Production of prostaglandin I2 (prostacyclin), which mediates renin release 18, is apparently diminished in patients with hyporeninemic hypoaldosteronism as assessed by measurement of the stable urinary metabolite 6-keto-prostaglandin F1α 19. Furthermore, the prostaglandin I2 in these patients was unresponsive to the potent stimulators norepinephrine and calcium. Prostaglandin I2 deficiency may cause hyporeninemic hypoaldosteronism by causing defects in the conversion of prorenin to renin 20 and renin release.
Diagnosis
The diagnosis of hyporeninemic hypoaldosteronism must be considered in any patient with unexplained hyperkalemia. Excess potassium intake from food or drugs does not cause sustained hyperkalemia, if renal function is normal. Renal function should be evaluated and drugs that impair renal potassium excretion should be excluded as a cause. The clinical diagnosis is confirmed by low plasma renin activity and low plasma concentrations or urinary aldosterone excretion under conditions that activate the renin-angiotensin-aldosterone axis suc h as maintenance of upright posture and/or furosemide administration. A low random plasma renin concentration associated with a normal ratio of aldosterone to plasma renin activity is also useful for the diagnosis 21.
Therapy
The therapeutic approach should be chosen after taking into consideration the age of the patients and other concurrent disorders. Only monitoring potassium concentrations is enough for patients with moderate hyperkalemia and without electro-cardiographic changes. Drugs that promote hyperkalemia, such as β-adrenergic antagonists, cyclooxygenase inhibitors, angiotensin-converting enzyme inhibitors, heparin and potassium-sparing diuretics, should be avoided. Dietary potassium intake should be reduced, if possible. Diuretics are the initial treatment for patients who have disorders associated with sodium retention, such as hypertension and congestive heart failure. Mineralocorticoid replacement with fludrocortisone is reserved for patients with severe hyperkalemia without hypertension and congestive heart failure.
Primary hypoaldosteronism: Aldosterone Synthase Deficiency
Congenital hypoaldosteronism is a rare inherited disorder transmitted as either an autosomal recessive or autosomal dominant trait with mixed penetrance. This disorder was previously termed “corticosterone methyloxidase (CMO)” deficiency and subdivided into two types according to the relative levels of aldosterone and its precursors in an affected person. Patients with “corticosterone methyloxidase I (CMO I)” deficiency have elevated serum levels of corticosterone and low levels of 18-hydroxycorticosterone and aldosterone. In contrast, patients with “corticosterone methyloxidase II (CMO II)” deficiency have high levels of 18-hydroxycorticosterone, the immediate precursor of aldosterone 22. With greater understanding of structure-activity relationships in the CYP11B2 enzyme, this disorder may be better considered a spectrum of hormonal deficiencies, depending on the nature of the CYP11B2 gene defect (88.6). Because two steps of aldosterone biosynthesis from corticosterone previously proposed to be catalyzed by separate enzymes, CMO I and II, previously, are known to involve only one enzyme substarate interaction 23. Isolated aldosterone deficiency results from loss of activity of aldosterone synthase encoded by CYP11B2 gene 24. Therefore, the term aldosterone synthase deficiency type 1 (ASD1) and type 2 (ASD2) reflects more appropriately the molecular basis of this disease. In both Aldosterone Synthase Deficiency-1 and Aldosterone Synthase Deficiency-2, glomerulosa zone corticosterone is increased and aldosterone decreased, but 18-hydroxycorticosterone is increased in Aldosterone Synthase Deficiency-2 25. ASD1 is associated with loss of both 18-hydroxilation and 18-oxidation enzyme activities. In ASD2, the ability to convert corticosterone (B) to 18-hydorxytetrahydro11-dehydrocorticosterone (18-OH-B) is preserved with failure of further oxidation of 18-hhdroxicorticosrerone to aldosterone 26. The deficiency of aldosterone is much more severe in ASD1. In contrast, aldosterone may reach normal levels under intense stimulation of rennin-angiotensin system in ASD2 25. The clinical presentations of these deficiencies are otherwise similar. The clinical presentation is typical of aldosterone deficiency, including electrolyte abnormalities such as a variable degree of hyponatremia, hyperkalemia and metabolic acidosis, with poor growth in childhood, but they usually no symptoms in adults 22. In infants, it is characterized by recurrent dehydration, salt wasting and failure to thrive. These symptoms are present generally within the first 3 months of life, and most often after the first 5 days of life. A modest uremia with a normal creatinine level reflects dehydration in the presence of intrinsically normal renal function. Plasma renin activity is invariably elevated.
Diagnosis and Therapy
The diagnosis can be established by measuring the appropriate corticosteroids or their major metabolic products, such as 11-deoxycorticosterone, corticosterone, 18-hydroxycorticosterone, 18-hydroxy-deoxycorticosterone and aldosterone levels in plasma. The ratio of plasma 18-hydroxycorticosterone to plasma aldosterone differentiates the two disorders; it is less than 10 in ASD1 (CMO I deficiency) and more than 100 in ASD2 (CMO II deficiency) 27. Patients with ASD2 (CMO II deficiency) tend to have increased plasma cortisol levels that may result from increased adrenal sensitivity to ACTH induced by the increased plasma angiotensin II levels in response to sodium depletion 28.
Both forms of the syndrome are treated by replacement of mineralocorticoid with the usual dosage of fludrocortisone (0.1-0.3 mg/ day). Almost infants and children require oral sodium supplementation (2 g/day as NaCl alone or in combination with NaHCO3), although some infants with severe symptom need intravenous fluids. Oral sodium supplementation may be discontinued once plasma rennin activity has decreased to normal, but mineralocorticoid replacement is usually maintained through childhood 26.
Addison’s disease
Addison’s disease is also called adrenal insufficiency, is an uncommon disorder that affects your body’s adrenal glands. Addison’s disease damages your adrenal glands. It causes your body to shut down production of the hormones cortisol and, often, aldosterone. Addison’s disease commonly affects people 30 to 50 years of age. However, it can occur at any age and affects both men and women and can be life-threatening.
Addison’s disease treatment involves taking hormones to replace those that are missing.
What causes Addison’s disease?
Addison’s disease is caused by injury to your adrenal glands or when your glands are affected by another medical condition. This is called adrenal insufficiency.
There are 2 types of adrenal insufficiency:
Primary adrenal insufficiency
This occurs through damage to your adrenal glands by an autoimmune disease (when your body attacks its own immune system). Other causes of primary adrenal insufficiency include:
- Tuberculosis (or other infections) of the adrenal glands
- Cancer of the adrenal glands
- Spread of cancer to the adrenal glands
- Bleeding of the adrenal glands. In this case, you may have an addisonian crisis without any previous symptoms.
Secondary adrenal insufficiency
The pituitary gland makes a hormone called adrenocorticotropic hormone (ACTH). ACTH in turn stimulates the adrenal cortex to produce its hormones. Benign pituitary tumors, inflammation and prior pituitary surgery are common causes of not producing enough pituitary hormone.
Too little ACTH can lead to too little of the glucocorticoids and androgens normally produced by your adrenal glands, even though your adrenal glands themselves aren’t damaged. This is called secondary adrenal insufficiency. Mineralocorticoid production is not affected by too little ACTH.
Most symptoms of secondary adrenal insufficiency are similar to those of primary adrenal insufficiency. However, people with secondary adrenal insufficiency don’t have hyperpigmentation and are less likely to have severe dehydration or low blood pressure. They’re more likely to have low blood sugar.
A temporary cause of secondary adrenal insufficiency occurs when people who take corticosteroids (for example, prednisone) to treat chronic conditions, such as asthma or arthritis, stop taking the corticosteroids all at once rather than tapering off.
Symptoms of Addison’s disease
Symptoms appear over a period of several months. They are difficult to diagnose because they are vague. Symptoms include:
- fatigue
- muscle weakness
- joint or muscle pain
- fever
- weight loss
- nausea, vomiting, and or diarrhea (leading to dehydration)
- headache
- sweating
- changes in mood or personality, such as irritability, anxiety, or depression
- loss of appetite
- darkening of the skin (called hyperpigmentation)
- lightheadedness or fainting when standing up, due to low blood pressure
- cravings for salty food
- sudden, severe pain in the abdomen (lower stomach), lower back, or legs
- confusion or slurred speech
- sluggish movements
- seizures
- high fever.
When symptoms appear suddenly, or quickly get worse, it is called acute adrenal failure. Sometimes it’s called Addisonian crisis. This can lead to death if not treated. If you have any of the following symptoms, contact your doctor or go to an emergency room immediately.
- Sudden, intense pain in your lower back, abdomen, or legs
- Severe weakness
- Severe abdominal pain, vomiting and diarrhea, leading to dehydration
- Lower than normal blood pressure
- Confusion
- Reduced consciousness or delirium
- Low blood pressure
- High potassium (hyperkalemia) and low sodium (hyponatremia)
How is Addison’s disease diagnosed?
Your doctor will talk to you first about your medical history and your signs and symptoms. You may undergo some of the following tests:
- Blood test. Tests can measure your blood levels of sodium, potassium, cortisol and adrenocorticotropic hormone (ACTH), which stimulates the adrenal cortex to produce its hormones. A blood test can also measure antibodies associated with autoimmune Addison’s disease.
ACTH stimulation test. ACTH signals your adrenal glands to produce cortisol. This test measures the level of cortisol in your blood before and after an injection of synthetic ACTH. - Insulin-induced hypoglycemia test. You may be given this test if doctors think you may have adrenal insufficiency as a result of pituitary disease (secondary adrenal insufficiency). The test involves checking your blood sugar (blood glucose) and cortisol levels after an injection of insulin. In healthy people, glucose levels fall and cortisol levels increase. In certain situations doctors may do alternative tests for secondary adrenal insufficiency, such as a low-dose ACTH stimulation test, prolonged ACTH stimulation test or glucagon stimulation test.
- Imaging tests. You may undergo a computerized tomography (CT) scan of your abdomen to check the size of your adrenal glands and look for other abnormalities. You may also undergo an MRI scan of your pituitary gland if testing indicates you might have secondary adrenal insufficiency.
Addison’s disease treatment
Treatment for an addisonian crisis, which is a medical emergency, typically includes intravenous injections of:
- Corticosteroids
- Saline solution
- Sugar (dextrose)
Treating Addison’s disease usually involves taking prescription hormones. This can include hydrocortisone, prednisone, or cortisone acetate. If your body is not making enough of the hormone aldosterone, your doctor may prescribe fludrocortisone. These medicines are taken every day by mouth (in pill form).
- Hydrocortisone (Cortef), prednisone or methylprednisolone to replace cortisol. These hormones are given on a schedule to mimic the normal 24-hour fluctuation of cortisol levels.
- Fludrocortisone acetate to replace aldosterone.
Your doctor may also recommend that you take a medicine called dehydroepiandrosterone. Some women who have Addison’s disease find that taking this medicine improves their mood and sex drive.
If you are experiencing an Addisonian crisis, you need immediate medical care. The treatment typically consists of intravenous (IV) injections of hydrocortisone, saline (salt water), and dextrose (sugar). These injections help restore blood pressure, blood sugar, and potassium levels to normal.
Living with Addison’s disease
Living with Addison’s disease involves learning to live with the unpleasant symptoms. In addition, you need to prepare for the possibility of Addisonian crisis. This is a medical emergency that requires you to:
- Carry a medical alert card and bracelet at all times. This gives emergency medical workers information about your condition.
- Keep extra medicine with you in case you forget to take your medicine. Ask your doctor for an injectable form of corticosteroids for use in an emergency.
- Carry a glucocorticoid injection kit. The kit contains a needle, syringe and injectable form of corticosteroids to use in case of emergency.
- Tell your doctor if your symptoms change or if your medicines stop working the way they used to.
- Have annual checkups. See your doctor or an endocrinology specialist at least once a year. Your doctor may recommend annual screening for a number of autoimmune diseases.
Mineralocorticoid vs Glucocorticoid
Cortisol also known as hydrocortisone, is the major glucocorticoid and increases in response to stress which activates the hypothalamic-pituitary-adrenal (HPA) axis axis. Therefore, all of its functions can be thought of as allowing the body to function with increased stress. Upon engaging glucocorticoid receptors, cortisol increases the expression of genes that will regulate metabolism, the immune system, cardiovascular function, growth, and reproduction. Cortisol is essential for maintaining blood pressure because it increases the sensitivity of vascular smooth muscle to vasoconstrictors like catecholamines and suppresses the release of vasodilators like nitrous oxide 29. Cortisol suppresses the immune system, which is the basis for immunosuppressive drug therapy with glucocorticoids. Regarding metabolism, cortisol increases gluconeogenesis and decreases peripheral glucose uptake. These oppose the actions of insulin, and the net effect is an increase in serum glucose. Cortisol also activates lipolysis and stimulates adipocyte growth, which leads to fat deposition. Generally, growth is inhibited, leading to muscle atrophy, increased bone resorption, and thinning of the skin. Of note, glucocorticoids can act on mineralocorticoid receptors. However, aldosterone effects predominate in the kidney because the renal enzyme, 11-beta-hydroxysteroid dehydrogenase-2 converts cortisol to cortisone 30. The 11-beta-hydroxysteroid dehydrogenase-1 converts cortisone into cortisol. Hence, these enzymes add another layer of regulation to cortisol. Licorice toxicity inhibits 11-beta-hydroxysteroid dehydrogenase-2, causing hypertension and hypokalemic alkalosis with normal aldosterone levels. Also, there can be a loss of function mutations in 11-beta-hydroxysteroid dehydrogenase-2, resulting in hypertension with low aldosterone 31.
In contrast to loss of mineralocorticoids, failure to produce glucocorticoids is not acutely life-threatening. Nevertheless, loss or profound diminishment of glucocorticoid secretion leads to a state of deranged metabolism and an inability to deal with stressors which, if untreated, is fatal.
In addition to their physiologic importance, glucocorticoids are also among the most frequently used drugs, and often prescribed for their anti-inflammatory and immunosuppressive properties.
Physiologic effects of glucocorticoids
There seem to be no cells that lack glucocorticoid receptors and as a consequence, these steroid hormones have a huge number of effects on physiologic systems. That having been said, it can be stated that the best known and studied effects of glucocorticoids are on carbohydrate metabolism and immune function.
Effects on metabolism
The name glucocorticoid derives from early observations that these hormones were involved in glucose metabolism. In the fasted state, cortisol stimulates several processes that collectively serve to increase and maintain normal concentrations of glucose in blood. These effects include:
- Stimulation of gluconeogenesis, particularly in the liver: This pathway results in the synthesis of glucose from non-hexose substrates such as amino acids and lipids and is particularly important in carnivores and certain herbivores. Enhancing the expression of enzymes involved in gluconeogenesis is probably the best known metabolic function of glucocorticoids.
- Mobilization of amino acids from extrahepatic tissues: These serve as substrates for gluconeogenesis.
- Inhibition of glucose uptake in muscle and adipose tissue: A mechanism to conserve glucose.
- Stimulation of fat breakdown in adipose tissue: The fatty acids released by lipolysis are used for production of energy in tissues like muscle, and the released glycerol provide another substrate for gluconeogenesis.
Effects on inflammation and immune function
Glucocorticoids have potent anti-inflammatory and immunosuppressive properties. This is particularly evident when they administered at pharmacologic doses, but also is important in normal immune responses. As a consequence, glucocorticoids are widely used as drugs to treat inflammatory conditions such as arthritis or dermatitis, and as adjunction therapy for conditions such as autoimmune diseases.
Other effects of glucocorticoids
Glucocorticoids have multiple effects on fetal development. An important example is their role in promoting maturation of the lung and production of the surfactant necessary for extrauterine lung function. Mice with homozygous disruptions in the corticotropin-releasing hormone gene (see below) die at birth due to pulmonary immaturity.
Several aspects of cognitive function are known to both stimulate glucocorticoid secretion and be influenced by glucocorticoids. Fear provides an interesting example of this. Fear-inducing stimuli lead to secretion of glucocorticoids from the adrenal gland, and treatment of phobic individuals with glucocorticoids prior to a fear-inducing stimulus can blunt the fear response.
Excessive glucocorticoid levels resulting from administration as a drug or hyperadrenocorticism have effects on many systems. Some examples include inhibition of bone formation, suppression of calcium absorption and delayed wound healing. These observations suggest a multitide of less dramatic physiologic roles for glucocorticoids.
Control of cortisol secretion
Cortisol and other glucocorticoids are secreted in response to a single stimulator: adrenocorticotropic hormone (ACTH) from the anterior pituitary. ACTH is itself secreted under control of the hypothalamic peptide corticotropin-releasing hormone (CRH). The central nervous system is thus the commander and chief of glucocorticoid responses, providing an excellent example of close integration between the nervous and endocrine systems.
Virtually any type of physical or mental stress results in elevation of cortisol concentrations in blood due to enhanced secretion of CRH in the hypothalamus. This fact sometimes makes it very difficult to assess glucocorticoid levels, particularly in animals. Observing the approach of a phlebotomist, and especially being restrained for blood sampling, is enough stress to artificially elevate cortisol levels several fold!
Cortisol secretion is suppressed by classical negative feedback loops. When blood concentrations rise above a certain theshold, cortisol inhibits CRH secretion from the hypothalamus, which turns off ACTH secretion, which leads to a turning off of cortisol secretion from the adrenal. The combination of positive and negative control on corticotropin-releasing hormone (CRH) secretion results in pulsatile secretion of cortisol. Typically, pulse amplitude and frequency are highest in the morning and lowest at night.
ACTH binds to receptors in the plasma membrane of cells in the zona fasiculata and reticularis of the adrenal. Hormone-receptor engagement activates adenyl cyclase, leading to elevated intracellular levels of cyclic AMP which leads ultimately to activation of the enzyme systems involved in biosynthesis of cortisol from cholesterol.
Disease states
The most prevalent disorder involving glucocorticoids in man and animals is hyperadrenocorticism or Cushings disease. Excessive levels of glucocorticoids are seen in two situations:
- Excessive endogenous production of cortisol, which can result from a primary adrenal defect (ACTH-independent) or from excessive secretion of ACTH (ACTH-dependent).
- Administration of glucocorticoids for theraputic purposes. This is a common side-effect of these widely-used drugs.
Cushing’s disease has widespread effects on metabolism and organ function, which is not surprising considering the ubiquitous distribution of glucocorticoid receptors. A diverse set of clinical manifestations accompany this disorder, including hypertension, apparent obesity, muscle wasting, thin skin, and metabolic aberrations such as diabetes.
Insufficient production of cortisol, often accompanied by an aldosterone deficiency, is called hypoadrenocorticism or Addison’s disease. Most commonly, this disease is a result of infectious disease (e.g. tuberculosis in humans) or autoimmune destruction of the adrenal cortex. As with Cushing’s disease, numerous and diverse clincial signs accompany Addison’s disease, including cardiovascular disease, lethargy, diarrhea, and weakness. Aldosterone deficiency can be acutely life threatening due to disorders of electrolyte balance and cardiac function.
- Dutt M, Jialal I. Physiology, Adrenal Gland. [Updated 2019 Jan 2]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2018 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK537260[↩][↩]
- Baranowski ES, Arlt W, Idkowiak J. Monogenic Disorders of Adrenal Steroidogenesis. Horm Res Paediatr. 2018;89(5):292-310.[↩][↩]
- Wagner CA. Effect of mineralocorticoids on acid-base balance. Nephron Physiol. 2014;128(1-2):26-34.[↩]
- Arai K, Chrousos GP. Aldosterone Deficiency and Resistance. [Updated 2016 May 11]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279079[↩]
- Landau RL, Lugibihl K. Inhibition of the sodium-retaining influence of aldosterone by progesterone. J Clin Endocrunol Metab 18:1237-1245, 1958.[↩]
- Hudson JB, Chobanian AV, et al. Hypoaldosteronism. A clinical study of a patient with isolated mineralocorticoid deficiency, resulting in hypokalemia and Strokes-Adama attacks. N Engl J Med 257:529-536, 1957.[↩]
- Schambelan M, Stockight JR, Bigloreli EG. Isolated hypoaldosteronism in adults, a renin-deficiency syndrome. N Engl J Med 287:573-578, 1972.[↩]
- Sebastian A, Schambelan M, LindenfeldS, et al. Amelioration of metabolic acidosis with fludrocortisone therapy in hyperreninemic hypoaldosteronism. N Engl J Med 297:576-583, 1977[↩]
- Perez GO, Lespier L. Jacobi J, et al. Hyporeninemic hypoaldosteronism in diabetes mellitus. Arch Intern Med 137:852-855, 1977[↩]
- Schembelan M, Sebastian A. Hyporeninemic hypoaldosteronism. Adv Intern Med 24:385-405, 1979[↩]
- Shaked Y, Blau A, Shipiberg O, et al. Hyporeninemic hypoaldosteronism associated with multiple myeloma: 11 years follow up. Clin Nephrol 40:79-82, 1993.[↩]
- Masud T, Wincour P, Clarke F. Reversible hyporeninemic hypoaldosteronism and lifethreatening cardiac dysrhythmias: the interaction of non-steroidal anti-inflammatory drugs and autonomic dysfunction. Postgrad Med J 69:593-594, 1993.[↩]
- Schambelan M, Stockight JR, Bigloreli EG. Isolated hypoaldosteronism in adults, a renin-deficiency syndrome. N Engl J Med 287:573-578, 1972[↩]
- Sunderlin FS, Anderson GH, streeten DHP, et al. The renin-angiotensin-aldosterone system in diabetic patients with hyperkalemia. Diabetes 30:335-340, 1981.[↩]
- Perez GO, Lespier LE, Oster JR, et al. Effects of alteration of sodium intake in patients with hyporeninemic hypoaldosteronism. Nephron 18:249-265, 1977[↩]
- Tuck ML, Sambhi MP, Levin L. Hyporeninemic hypoaldosteronism in diabetes mellitus: studies of the autonomic nervous system’s control of renin release. Diabetes 28:237-241, 1979.[↩]
- Delevia A, Christlieb AR, Melby JC, et al. Big renin and biosynthetic defect of aldosterone in diabetes mellitus. N Engl J Med 295:639-43, 1976.[↩]
- Oster JA, Whorton AR, Gerkens JH, et al. The participation of prostaglandins in the control of renin release. Fed Proc 38:72-74, 1979[↩]
- Nadler JL, Lee FO, Hsuch W, et al. Evidence of prostacyclin deficiency in the syndrome of hyporeninemic hypoaldosteronism. N Engl J Med 314:1015-1020, 1986[↩]
- Fitzgerald GA, Hossman V, Himmerich W, et al. The renin-kallikrein-prostaglandin system: plasma active and inactive renin and urinary kallikrein during prostacyclin infusion in man. Prostaglandins Med 5:445-456, 1980[↩]
- Hambling C, Jung RT, Browning MC, et al. Primary hyperaldosteronism-evaluation of procedures for diagnosis and localization. Q J Med 86:383-392, 1993.[↩]
- Ulick S, Wang JZ, Morton DH. The biochemical phenotypes of two inborn errors in the biosynthesis of aldosterone. J Clin Endocrinol Metab 74:1415-1420, 1992[↩][↩]
- Kawamoto T, Mitsuuchi Y, Toda K, et al. Role of steroid 11b-hydoxylase and steroid 18-hydroxylase in the biosynthesis of glucocorticoids and mineralocorticoids in humans. Proc Natl AcadSci USA89:1458-1462,1992[↩]
- Geley S, Johrer K, Peter M, et al. Amino acid substitution R384P in aldosterone synthase causes corticosterone methyloxidase type I deficiency. J Clin Endocrinol Metab 80:424-429, 1995[↩]
- Ulick S. Correction of the nomenclature and mechanism of the aldosterone biosynthetic defects. J Clin Endocrinol Metab 81:1299-1300, 1996[↩][↩]
- White PC. Aldosterone synthase deficiency and related disorders. Mol Cell Endocrinol 217:81-87, 2004[↩][↩]
- Petr M, Partsch CJ, Sillel WG. Multisteroid analysis in children with terminal aldosterone biosynthesis defects. J Clin Endocrinol Metab 80:1622-1627, 1995.[↩]
- Ulick S. Editorial: Cortisol as mineralocorticoid. J Clin Endocrinol Metab 81:1307-1308, 1996.[↩]
- Burford NG, Webster NA, Cruz-Topete D. Hypothalamic-Pituitary-Adrenal Axis Modulation of Glucocorticoids in the Cardiovascular System. Int J Mol Sci. 2017 Oct 16;18, 10[↩]
- Miller WL. The Hypothalamic-Pituitary-Adrenal Axis: A Brief History. Horm Res Paediatr. 2018;89(4):212-223[↩]
- Vaidya A, Dluhy R. Hyperaldosteronism. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, Hershman JM, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Perreault L, Purnell J, Rebar R, Singer F, Trence DL, Vinik A, Wilson DP, editors. Endotext [Internet]. MDText.com, Inc.; South Dartmouth (MA): Oct 19, 2016.[↩]





{kind=link}