What is MitoQ
MitoQ also known as mitoquinone mesylate, mitochondrial-targeted coenzyme Q or 10-(4,5-Dimethoxy-2-methyl-3,6-dioxo-1,4-cyclohexadien-1-yl)decyl triphenyl phosphonium methanesulfonate, is a mitochondrial targeted anti‐oxidant composed of ubiquinone, a component of the electron transport chain (ETC), covalently bound via a ten carbon chain to triphenylphosphonium (TPP+), a lipophilic cation that targets the ubiquinone moiety to the inner mitochondrial membrane driven by the high electrochemical potential across that membrane 1. MitoQ is an orally active antioxidant developed by the pharmaceutical industry to potentially treat several diseases. MitoQ retains the antioxidant activity of coenzyme Q10 and the triphenylphosphonium cation (TPP+) substituent directs this agent to mitochondria 2. MitoQ or mitoquinone is a derivative of coenzyme Q, is produced by covalently binding ubiquinone, an endogenous antioxidant and component of the mitochondrial electron transport chain to a triphenylphosphonium (TPP+) cation. MitoQ rapidly crosses the blood–brain barrier (BBB) and neuronal membranes and concentrates several hundred-fold in mitochondria driven by the high membrane potential across the inner mitochondrial membrane 3. The ubiquinone moiety is delivered to the matrix side of the inner mitochondrial membrane, placing it at the site of most cellular reactive oxygen species (ROS) generation and was proven to be effective in reducing reactive oxygen species (ROS) generated in mitochondria 4. The triphenylphosphonium(TPP+) moiety adsorbs to the matrix side of the inner mitochondrial membrane and the ubiquinone penetrates into the membrane in which it is reduced to the active antioxidant ubiquinol by respiratory complex II 5. The ubiquinol acts as an antioxidant when oxidized to ubiquinone by reactive oxygen species. Complex II then reduces the ubiquinone to ubiquinol. MitoQ is a poor substrate for complex I and is negligibly oxidized by complex III. Therefore, it cannot substitute for endogenous ubiquinone in the electron transport chain (ETC) but primarily acts as an antioxidant capable of continuous regeneration by complex II 6. Because MitoQ is a poor substrate for complex I and III, it cannot substitute for endogenous ubiquinone and thus does not take part in mitochondrial respiration 7. It instead acts as a renewable antioxidant 8.
Mitochondrial dysfunction is associated with a diverse array of diseases ranging from dystrophy and heart failure to obesity and hepatosteatosis 9. One of the major biochemical consequences of impaired mitochondrial function is an accumulation of mitochondrial superoxide (O2–) or reactive oxygen species (ROS). Excessive ROS can be detrimental to cellular health and is proposed to underpin many mitochondrial diseases. Accordingly, much research has been committed to understanding ways to therapeutically prevent and reduce ROS accumulation. MitoQ (mitoquinone) was designed by Kelso et al. 10 as a potential therapeutic to target mitochondrial ROS. A large proportion of ROS is generated by the mitochondrial electron transport chain (ETC), which produces superoxide via the reduction of oxygen 11.
Reactive oxygen species (ROS) damage cellular structures and cause oxidative stress, but they also function as signaling molecules that regulate biological and pathological processes 12. Mitochondria produce a substantial portion of cellular ROS, in particular superoxide (O2–), as a byproduct of oxidative phosphorylation in complexes I, II, and III of the electron transport chain (ETC) 13. Superoxide (O2–) is then rapidly converted to form the ROS, hydrogen peroxide (H2O2), by the enzymatic activity of superoxide dismutases (superoxide dismutase 1, 2, and 3 [SOD1, SOD2, and SOD3]) located in the mitochondrial matrix 14. An accumulation of both superoxide (O2–) and hydrogen peroxide (H2O2) causes oxidative damage, which can lead to cellular dysfunction, and death 15, often via the formation of reactive lipid species formed by lipid peroxidation 16. The mitochondria-associated H2O2 can in turn stimulate intracellular signaling pathways by reversibly oxidizing cysteine residues in key proteins 17. Indeed, mitochondria-associated ROS can move into the cytosol and subsequently into the extracellular environment, where they can trigger different signaling cascades and pathways 12. Furthermore, mitochondrial ROS can target and mutate mitochondrial DNA and thereby contribute to carcinogenesis 18. However, several reports show conflicting results on the efficiency of MitoQ to limit tumor growth and metastasis 19. In this study in mice 20, the authors found that MitoQ administration had no impact on the number of primary tumors and lymph metastases in mice with BRAF-induced malignant melanoma and no impact on tumor burden in mice with KRAS-induced lung cancer. These results question the rationale of using these mitochondria-targeted antioxidants in anti-cancer therapy. Furthermore, manipulating mitochondrial ROS has been an area of therapeutic interest for conditions such as obesity, multiple sclerosis (MS) and type 2 diabetes, albeit with little success 9.
Previous lab studies and animal experiments have revealed mitoQ exerted its protective effects on diseases like traumatic brain injury 21, cardiovascular disease 22, inflammation 23, neurodegenerative disorders 24, intestinal ischemia reperfusion 25, diabetes 26 and diabetic kidney disease 27 through regulation of NF-E2-related factor 2 (Nrf2). In db/db mice, mitoQ partially rectified mitochondrial dysfunctions, such as defective mitophagy, mitochondrial reactive oxygen species (mtROS) overexpression and mitochondrial fragmentation in tubular cells and inhibited tubular cells apoptosis 27. In in vitro study mitoQ decreased deficiency of mitophagy, mitochondrial dysfunction and apoptosis in high glucose induced HK-2 cells via Nrf2/PINK pathway 27.
NF-E2-related factor 2 (Nrf2) is retained in the cytosol bounding to Kelch-like ECH-associated protein 1 (Keap1), which represses activity of Nrf2 by promoting the proteasomal degradation of Nrf2 28. Under quiescent conditions, the transcription factor Nrf2 interacts with the actin-anchored protein Keap1, largely localized in the cytoplasm. This quenching interaction maintains low basal expression of Nrf2-regulated genes. However, upon recognition of chemical signals imparted by oxidative and electrophilic molecules, Nrf2 is released from Keap1, escapes proteasomal degradation, translocates to the nucleus, and transactivates the expression of several dozen cytoprotective genes that enhance cell survival 29. In the presence of reactive oxygen species (ROS), Nrf2 will dissociate from Keap1, translocate to the nucleus, dimerize with a small protein Maf, namely MafF, MafG and MafK, and initiate transcription of many cytoprotective genes at antioxidant response elements (AREs) loci, including glutathione peroxidase (GPX), GSH-S-transferase, heme oxygenase (HO)-1, NADPH quinine oxidoreductases (NQO)-1, etc. 29.
Activation of Nrf2 and antioxidant response element (ARE) signaling is a crucial process in protecting cells from oxidative insults 30. However, there remain controversies in the mechanism how Nrf2 is exactly activated. Several studies reported that the gene expression of Nrf2 might be determined by complex I activity via an ERK5-myocyte enhancer factor 2 (MEF2) signaling pathway independent of ROS 31. While other studies reported changes in mitochondrial reactive oxygen species (mtROS) generation actually altered the cellular response, where changes of mtROS actually influenced the activation of Nrf2 through unknown mediators or signaling cascades 32. In a recent study from Ping Wang et al. 33 the stability of Nrf2 was modulated by kinases Mst1 and Mst2 (Mst1/2) which can sense site-specific ROS release and maintain cellular redox homeostasis. Activated Mst1/Mst2 by mtROS could phosphorylate Keap1 at four Ser/Thr residues in the N-terminus, which inactivated Keap1 and decreased the degradation of Nrf2. Thus, these results suggested that mtROS activated Nrf2 mainly by indirect and kinase-dependent way 33. Accumulating evidence also suggests that the Nrf2 and antioxidant response element (ARE) pathway reduces apoptosis and alleviates the inflammatory response, resulting in a significant improvement in tissue retention and beneficial role in coordination 34. It is also reported that oxidative signaling and Nrf2 were tightly linked with cellular responses 35.
Figure 1. MitoQ (chemical structure of triphenylphosphonium-substituted coenzyme Q)
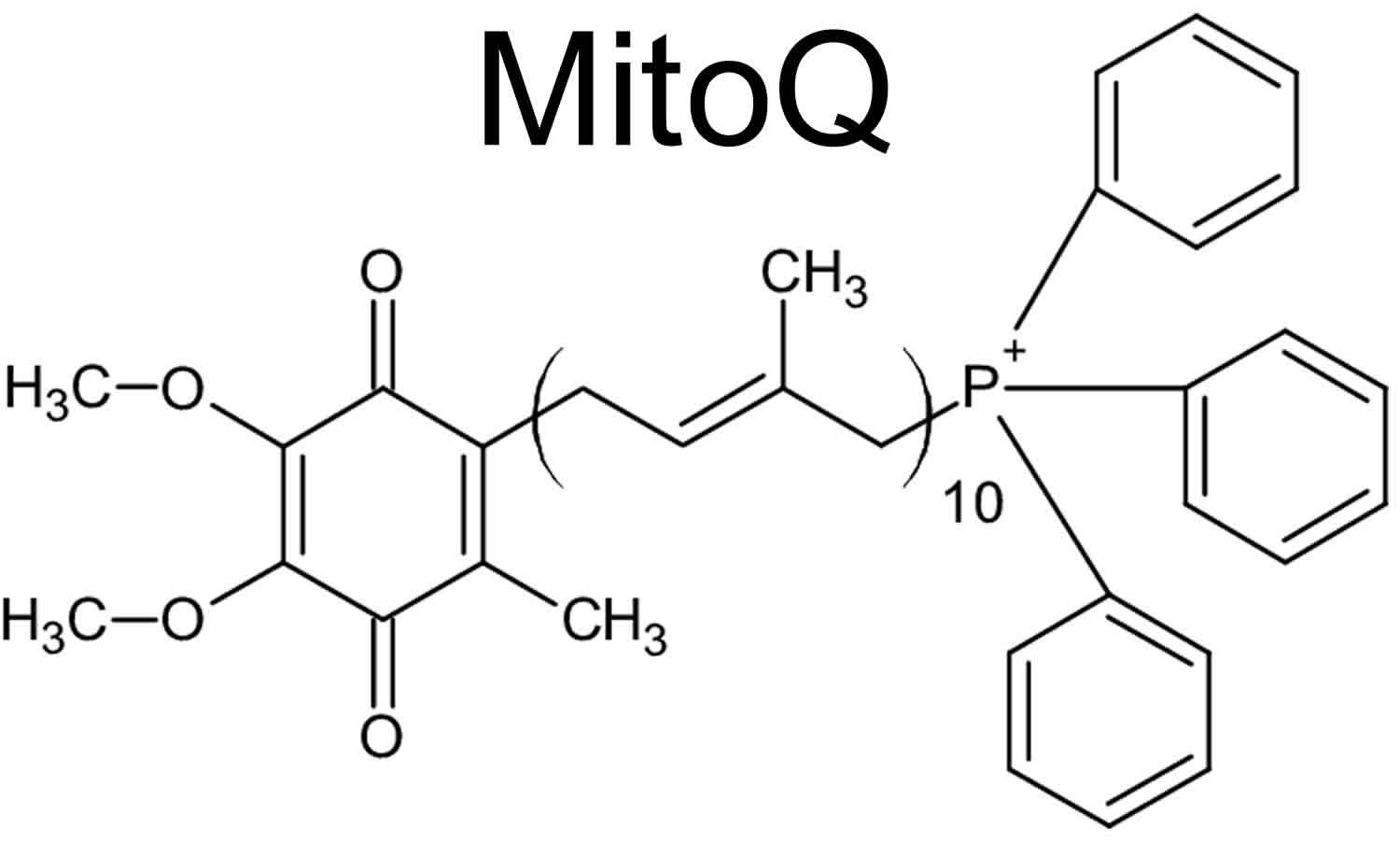
Footnote: MitoQ (mitoquinone or mitochondrial-targeted coenzyme Q), is a mitochondrial targeting antioxidant with an antioxidant ubiquinone, which is identical to the active antioxidant in Coenzyme Q10, attached to a triphenylphosphonium (TPP+) cation
MitoQ health benefits
MitoQ is an orally active antioxidant developed by the pharmaceutical industry to potentially treat several diseases. MitoQ retains the antioxidant activity of coenzyme Q10 and the triphenylphosphonium cation (TPP+) substituent directs this agent to mitochondria 2. Mitochondria are the major resource of reactive oxygen species (ROS) and mitoQ is a mitochondria targeted antioxidant composed of lipophilic triphenylphosphonium (TPP+) cation which can easily move through phospholipid bilayers driven by mitochondrial membrane potential and an ubiquinol antioxidant 36. Once accumulating within mitochondria, mitoQ will be adsorbed to the surface of the inner membrane where it will be continually transformed to the antioxidant quinol form in the respiratory chain by complex II 37, which may react directly with superoxide (O2–) 38. In cultured cells MitoQ was able to accumulate into mitochondria and act against oxidative stress 39. It was also demonstrated a protective effect of MitoQ in a sepsis model by decreasing the oxidative stress and protecting mitochondria against damage as well as by suppressing proinflammatory cytokine release 40. It was proposed that the antioxidant action of MitoQ may be useful in the treatment of diseases associated to the impairment of mitochondrial Complex I 41. On the other hand, it was recently showed that MitoQ may be prooxidant and present proapoptotic action due its quinone group that may participates in redox cycling and superoxide production 42. It is also shown that mitoQ is able to protect cells against peroxynitrite damage but it can hardly eliminate hydrogen peroxide (H2O2) like other quinols 43.
Growing evidence shows that MitoQ exerts beneficial effects on a wide range of diseases including Parkinson’s disease 44, diabetes 27 and cardiac ischemia-reperfusion injury 45 where mitochondrial oxidative damage plays a crucial role in the pathology. In addition, in some inflammatory diseases, such as sepsis, mitoQ is also proved to be effective both in vitro and in vivo 46. Previously it was reported that mitoQ improved neurological deficits, attenuated brain edema and decreased cortical neuronal apoptosis through activating the NF-E2-related factor 2 (Nrf2) and antioxidant response element (ARE) pathway 21. Keap1-Nrf2-ARE signaling plays a significant role in protecting cells from endogenous and exogenous stresses 29.
Alzheimer’s disease
MitoQ may reduce the burden of oxidative stress in Alzheimer’s disease, though there is not strong evidence it would change clinical outcomes in patients.
In vitro, MitoQ prevented amyloid β (Aβ)-induced neuronal death, ROS production and change in mitochondrial membrane depolarization. In Alzheimer’s mice treated with MitoQ from a young age for five months, treatment improved cognition, reduced oxidative stress, increased synaptic markers, reduced gliosis, and reduced levels of amyloid 47. In older Alzheimer’s mice (beginning treatment at 12 months of age), MitoQ treatment for five months improved cognitive performance, increased markers of synapses, reduced oxidative stress, reduced astro-and micro-gliosis, and reduced tau and ptau levels. MitoQ also increased lifespan of Alzheimer’s mice to a similar extent as control mice –although the study was not long enough to test the full extent of an increase in lifespan 48. In vitro studies suggest that MitoQ can increase neurite outgrowth in neurons from an Alzheimer’s mouse model and protect against Aβ toxicity in cells 49.
In a Caenorhabditis elegans (roundworm) model of Alzheimer’s disease, MitoQ extended lifespan by 15% and delayed paralysis (a marker of healthspan in these worms) 50. MitoQ had no effect on oxidative stress, mitochondrial DNA (mtDNA) damage, or oxygen consumption rate and ATP levels (measures of mitochondrial metabolism). However, MitoQ increased mitochondrial complex I and IV activity and increased the level of certain cardiolipin species 50.
Amyotrophic lateral sclerosis (ALS)
In a mouse model of amyotrophic lateral sclerosis (ALS), treatment with MitoQ early in the disease improved mitochondrial function in the muscle and spinal cord, reduced levels of 3-nitrotyrosine and gliosis in the spinal cord, improved motor unit integrity at the neuromuscular junction, and increased lifespan 51.
Multiple sclerosis
In a mouse model of multiple sclerosis (experimental autoimmune encephalomyelitis injection), pretreatment with MitoQ delayed the onset of neurological symptoms and treatment with MitoQ after experimental autoimmune encephalomyelitis injection reduced the severity of neurological symptoms 52. In the spinal cord, MitoQ reduced microgliosis, IL-6 expression, and neurodegeneration 52.
Traumatic brain injury
When administered 30 minutes after a traumatic brain injury in mice, MitoQ (4mg/kg and 8mg/kg, but not 2mg/kg) improved neurological outcomes and reduced brain edema 53. It increased the activity of antioxidant enzymes (superoxide dismutase –SOD, and glutathione peroxidate) and reduced a marker of oxidative stress (malondialdehyde). In addition, it reduced neuronal apoptosis and increased nuclear Nrf2 levels and the expression of proteins downstream of Nrf2 53.
Subarachnoid hemorrhage
In a rat model of subarachnoid hemorrhage, administration of MitoQ 1 hour after hemorrhage improved neurological outcomes andreduced brain edema (in the 3mg/kg but not 1mg/kg or 9mg/kg groups) 54. This was accompanied by an increase in Nrf2 expression, improved mitochondrial morphology, and improved blood brain barrier integrity (measured by increased claudin-5 expression and reduced brain albumin levels) 54.
Angelman syndrome
In a mouse model of Angelman Syndrome, MitoQ increased synaptic plasticicy and improved memory 55.
Heart failure
In a model of pressure overload-induced heart failure, MitoQ (100μM in drinking water) failed to improve cardiac functioning 56. MitoQ had a few minor benefits in some measures of mitochondrial function, but not all 56. In aged mice, 4-week treatment with MitoQ (250μM) reduced aortic stiffness (aortic pulse-wave velocity) and increased elastin expression, but had no effect on collagen expression or the expression of pro-inflammatory cytokines (e.g. IL-6, IL-10, IFN-γ, IL-1β) in the aorta 57. Another study from the same group showed that 4-week treatment with MitoQ increased endothelium-dependent (but not independent) dilation, reduced mitochondria-specific superoxide production, and reduced 3-nitrotyrosine levels. It also increased the aortic expression of PGC-1α, superoxide dismutase (MnSOD), and reduced expression of COX-IV 58.
MitoQ shows consistent antioxidant properties, though whether it would have beneficial effects on clinicaloutcomes in humans is not known.
In aged rats, 5-week treatment with MitoQ (500μM in drinking water) reduced markers of oxidative damage (malondialdehyde, protein carbonylation, and nitrosative stress), improved mitochondrial complex I and IV activity, increased the mitochondrial membrane potential, and increased mitochondrial ATP levels 59.
In a meta-analysis of aging-related biomarkers from preclinical studies, Braakhuis et al 60 reported that three biomarkers were included in enough studies to conduct a meta-analysis (3-nitrotyrosine, 8 studies; mitochondrial membrane potential, 4 studies; and protein carbonyls, 8 studies). MitoQ significantly reduced 3-nitrotyrosine levels, increased mitochondria membrane potential, and non-significantly reduced protein carbonyl levels 60.
Kidney ischemia-reperfusion injury
In a mouse model of ischemia-reperfusion of the kidney, administration of MitoQ 15 minutes prior to ischemia reduced kidney damage (as measured by creatine levels) and reduced oxidative damage (as measured by protein carbonyl content and mtDNA damage) 61. In a model of hemorrhagic shock with reperfusion, administration of MitoQ had mixed effects with a reduction in morbidity, no effect on liver necrosis, a trend toward increased lipid peroxidation, elevated glutathione peroxidase (GPx) activity, reduced catalase activity, and a reduction in inflammation (hepatic IL-6 and TNFα) 62.
Diabetes and diabetic kidney disease
In an ex vivo study of leukocytes from patients with type 2 diabetes, MitoQ reduced mitochondrial ROS levels, leukocyte adhesion properties, and reduced the expression of inflammatory markers (NFkB and TNFα) 63. In a mouse model of diabetic kidney disease, MitoQ (0.6mg/kg/day) over 12 weeks had no effects on plasma glucose or insulin levels. Beneficial effects on renal function were similar to an ACE inhibitor (ramipril), but there were no additive effects when given together 64. In a mouse model of metabolic disease, 14-week treatment with MitoQ prevented an increase in fat, improved energy expenditure, reduced liver lipid content, reduced serum liver enzymes, and reduced serum lipid markers (cholesterol, triglycerides, and LDL [low density lipoprotein or “bad” cholesterol]). Although it had no effect on atherosclerotic plaque size, it did reduce the proliferation of plaque-associated macrophages. It reduced mitochondrial DNA (mtDNA) damage in the liver but had no effect on protein carbonyl content 65.
MitoQ clinical trials
One pilot study of MitoQ in Alzheimer’s disease patients is underway. It is a 2-day cross-over trial in 12 patients looking at carotid artery blood flow, oxidative stress, cerebrovascular oxygenation, EEG, and endothelial function 66. Other clinical trials are ongoing including fatigue in multiple sclerosis, cardiovascular function, peripheral artery disease, and chronic kidney disease (https://clinicaltrials.gov/ct2/results?cond=&term=mitoq).
Parkinson’s disease
In a mouse model of Parkinson’s disease, 14-day treatment with MitoQ increased the number of dopaminergic neurons and increased the expression of PGC-1α (a gene involved in mitochondrial fusion) 67. However, in 128 patients with newly diagnosed Parkinson’s disease, MitoQ (40mg or 80mg/day) had no effect on clinical outcome measures [Unified Parkinson’s Disease Rating Scale (UPDRS)] over 12 months. More patients discontinued in the MitoQ group (13 in 80 mg, 2 in 40 mg, and 1 in placebo), mostly due to gastrointestinal side effects 68. Reasons suggested for the failed trial include treating patients too late in the disease progression and insufficient brain penetration of MitoQ.
In 20 healthy older adults (average age 68) with impaired endothelial function (brachial artery flow-mediated dilation <60%), 6-week cross-over treatment with MitoQ (20mg/day) improved endothelial-dependent flow-mediated dilation by 42% (but had no effect on endothelial-independent flow-mediated dilation) and reduced aortic stiffness (carotid-femoral pulse wave velocity) in patients with elevated aortic stiffness 69. MitoQ reduced oxidized-LDL (low density lipoprotein or “bad” cholesterol) but had no effect on circulating inflammatory markers (CRP or IL-6) 69.
MitoQ supplement dosage
MitoQ is being developed by Antipodean Pharmaceuticals. It is also sold as a supplement by MitoQ. Recommended doses are 10mg/day with therapeutic doses in the Parkinson’s disease trial at 40mg or 80mg/day.
MitoQ side effects
Some evidence from clinical studies suggest there are gastrointestinal side effects of MitoQ at higher doses, though no large, extended clinical trials have been conducted.
MitoQ has been tested in 128 Parkinson’s patients over 12 months (40mg or 80mg/day) 68, 20 healthy patients over 3 weeks (10mg/day) 70, in 20 healthy older adults with endothelial dysfunction over 6 weeks (20mg/day) 69 and in 30 patients with chronic hepatitis C over 28 days (40mg or 80mg/day) 71. The only adverse effects so far reported are gastrointestinal side effects (at 40mg and 80mg doses).
Drug interactions
Not currently known. Although, theoretically, MitoQ may interact with other mitochondria-targeted antioxidants (e.g Skq1 andSS-31).
MitoQ summary
Consistent evidence suggests that MitoQ can reduce oxidative stress in mitochondria; though whether this will impact clinical outcomes in humans is unknown.
- Smith RA, Porteous CM, Gane AM, Murphy MP. Delivery of bioactive molecules to mitochondria in vivo. Proc Natl Acad Sci U S A. 2003 Apr 29;100(9):5407-12. doi: 10.1073/pnas.0931245100[↩]
- Tauskela, J.S. (2007). MitoQ – a mitochondria-targeted antioxidant. IDrugs, Vol.10, No. 6, (June 2007), pp. (399-412), ISSN 2040-3410[↩][↩]
- Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629-56. doi: 10.1146/annurev.pharmtox.47.120505.105110[↩]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Ed 4. Oxford, UK: Oxford UP; 2007.[↩]
- The Mitochondria-Targeted Antioxidant MitoQ Prevents Loss of Spatial Memory Retention and Early Neuropathology in a Transgenic Mouse Model of Alzheimer’s Disease. J Neurosci. 2011 Nov 2; 31(44): 15703–15715. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3334845[↩]
- Rodriguez-Cuenca S, Cochemé HM, Logan A, Abakumova I, Prime TA, Rose C, Vidal-Puig A, Smith AC, Rubinsztein DC, Fearnley IM, Jones BA, Pope S, Heales SJ, Lam BY, Neogi SG, McFarlane I, James AM, Smith RA, Murphy MP. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic Biol Med. 2010 Jan 1;48(1):161-72. doi: 10.1016/j.freeradbiomed.2009.10.039[↩]
- James AM, Cochemé HM, Smith RA, Murphy MP. Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools. J Biol Chem. 2005 Jun 3;280(22):21295-312. doi: 10.1074/jbc.M501527200[↩]
- Young, M. L., & Franklin, J. L. (2019). The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Molecular and cellular neurosciences, 101, 103409. https://doi.org/10.1016/j.mcn.2019.103409[↩]
- The Antioxidant Moiety of MitoQ Imparts Minimal Metabolic Effects in Adipose Tissue of High Fat Fed Mice. Front. Physiol., 08 May 2019. https://doi.org/10.3389/fphys.2019.00543[↩][↩]
- Kelso, G. F., Porteous, C. M., Coulter, C. V., Hughes, G., Porteous, W. K., Ledgerwood, E. C., et al. (2001). Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 276, 4588–4596. doi: 10.1074/jbc.M009093200[↩]
- Murphy, M. P. (2009). How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. doi: 10.1042/bj20081386[↩]
- Holmstrom K.M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801[↩][↩]
- Murphy M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009;417:1–13. doi: 10.1042/BJ20081386[↩]
- Fridovich I. Superoxide anion radical (O2−●), superoxide dismutases, and related matters. J. Biol. Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515.[↩]
- Pandey, K. B., and Rizvi, S. I. (2010). Markers of oxidative stress in erythrocytes and plasma during aging in humans. Oxid. Med. Cell. Longev. 3, 2–12. doi: 10.4161/oxim.3.1.10476[↩]
- Higdon, A., Diers, A. R., Oh, J. Y., Landar, A., and Darley-Usmar, V. M. (2012). Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem. J. 442, 453–464. doi: 10.1042/bj20111752[↩]
- Finkel T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095[↩]
- Chen E.I. Mitochondrial dysfunction and cancer metastasis. J. Bioenerg. Biomembr. 2012;44:619–622. doi: 10.1007/s10863-012-9465-9[↩]
- Shetty S., Kumar R., Bharati S. Mito-TEMPO, a mitochondria-targeted antioxidant, prevents N-nitrosodiethylamine-induced hepatocarcinogenesis in mice. Free Radic. Biol. Med. 2019;136:76–86. doi: 10.1016/j.freeradbiomed.2019.03.037[↩]
- Le Gal, K., Wiel, C., Ibrahim, M. X., Henricsson, M., Sayin, V. I., & Bergo, M. O. (2021). Mitochondria-Targeted Antioxidants MitoQ and MitoTEMPO Do Not Influence BRAF-Driven Malignant Melanoma and KRAS-Driven Lung Cancer Progression in Mice. Antioxidants (Basel, Switzerland), 10(2), 163. https://doi.org/10.3390/antiox10020163[↩]
- Zhou J., Wang H., Shen R., Fang J., Yang Y., Dai W., Zhu Y., Zhou M. Mitochondrial-targeted antioxidant mitoq provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018;10:1887–1899.[↩][↩]
- Gioscia-Ryan R.A., LaRocca T.J., Sindler A.L., Zigler M.C., Murphy M.P., Seals D.R. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J. Physiol. 2014;592:2549–2561. doi: 10.1113/jphysiol.2013.268680[↩]
- Formentini L., Santacatterina F., Nunez de Arenas C., Stamatakis K., Lopez-Martinez D., Logan A., Fresno M., Smits R., Murphy M.P., Cuezva J.M. Mitochondrial ROS Production Protects the Intestine from Inflammation through Functional M2 Macrophage Polarization. Cell Rep. 2017;19:1202–1213. doi: 10.1016/j.celrep.2017.04.036[↩]
- Manabe T., Matsumura A., Yokokawa K., Saito T., Fujikura M., Iwahara N., Matsushita T., Suzuki S., Hisahara S., Kawamata J., et al. Evaluation of Mitochondrial Oxidative Stress in the Brain of a Transgenic Mouse Model of Alzheimer’s Disease by in vitro Electron Paramagnetic Resonance Spectroscopy. J. Alzheimers Dis. 2019;67:1079–1087. doi: 10.3233/JAD-180985[↩]
- Hu Q., Ren J., Li G., Wu J., Wu X., Wang G., Gu G., Ren H., Hong Z., Li J. The mitochondrially targeted antioxidant MitoQ protects the intestinal barrier by ameliorating mitochondrial DNA damage via the Nrf2/ARE signaling pathway. Cell Death Dis. 2018;9 doi: 10.1038/s41419-018-0436-x[↩]
- Lian K., Wang Q., Zhao S., Yang M., Chen G., Chen Y., Li C., Gao H., Li C. Pretreatment of Diabetic Adipose-derived Stem Cells with mitoTEMPO Reverses their Defective Proangiogenic Function in Diabetic Mice with Critical Limb Ischemia. Cell Transpl. 2019;28:1652–1663. doi: 10.1177/0963689719885076[↩]
- Xiao L., Xu X., Zhang F., Wang M., Xu Y., Tang D., Wang J., Qin Y., Liu Y., Tang C., He L., Greka A., Zhou Z., Liu F., Dong Z., Sun L. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022[↩][↩][↩][↩]
- Dinkova-Kostova A.T., Holtzclaw W.D., Cole R.N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. Unit. States Am. 2002;99:11908–11913. doi: 10.1073/pnas.172398899[↩]
- Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89-116. doi: 10.1146/annurev.pharmtox.46.120604.141046[↩][↩][↩]
- Peake B.F., Nicholson C.K., Lambert J.P., Hood R.L., Amin H., Amin S., Calvert J.W. Hydrogen sulfide preconditions the db/db diabetic mouse heart against ischemia-reperfusion injury by activating Nrf2 signaling in an Erk-dependent manner. Am. J. Physiol. Heart Circ. Physiol. 2013;304:1215–1224. doi: 10.1152/ajpheart.00796.2012[↩]
- Tsushima M., Liu J., Hirao W., Yamazaki H., Tomita H., Itoh K. Emerging evidence for crosstalk between Nrf2 and mitochondria in physiological homeostasis and in heart disease. Arch. Pharm. Res. 2020;43:286–296. doi: 10.1007/s12272-019-01188-z[↩]
- Robb E.L., Gawel J.M., Aksentijević D., Cochemé H.M., Stewart T.S., Shchepinova M.M., Qiang H., Prime T.A., Bright T.P., James A.M., Shattock M.J., Senn H.M., Hartley R.C., Murphy M.P. Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 2015;89:883–894. doi: 10.1016/j.freeradbiomed.2015.08.021[↩]
- Wang P., Geng J., Gao J., Zhao H., Li J., Shi Y., Yang B., Xiao C., Linghu Y., Sun X., Chen X., Hong L., Qin F., Li X., Yu J.S., You H., Yuan Z., Zhou D., Johnson R.L., Chen L. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019;10 doi: 10.1038/s41467-019-08680-6[↩][↩]
- Slocum S.L., Kensler T.W. Nrf2: control of sensitivity to carcinogens. Arch. Toxicol. 2011;85:273–284. doi: 10.1007/s00204-011-0675-4[↩]
- Moritani C., Kawakami K., Shimoda H., Hatanaka T., Suzaki E., Tsuboi S. Protective effects of rice peptide oryza peptide-P60 against oxidative injury through activation of Nrf2 signaling pathway in vitro and in vivo. ACS. Omega. 2020;5:13096–13107. doi: 10.1021/acsomega.0c01016[↩]
- Murphy M.P., Smith R.A.J. Drug delivery to mitochondria: the key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000;41:235–250. doi: 10.1016/S0169-409X(99)00069-1[↩]
- Kelso G.F., Porteous C.M., Coulter C.V., Hughes G., Porteous W.K., Ledgerwood E.C., Smith R.A.J., Murphy M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties. J. Biol. Chem. 2001;276:4588–4596. doi: 10.1074/jbc.M009093200[↩]
- Maroz A., Anderson R.F., Smith R.A.J., Murphy M.P. Reactivity of ubiquinone and ubiquinol with superoxide and the hydroperoxyl radical: implications for in vivo antioxidant activity. Free Radic. Biol. Med. 2009;46:105–109. doi: 10.1016/j.freeradbiomed.2008.09.033[↩]
- Murphy, MP &.Smith, RA. (2007). Targeting Antioxidants to Mitochondria by Conjugation to Lipophilic Cations. Annual. Review of Pharmacology and Toxicology, Vol. 47, (February, 2007), pp. (629–656), ISSN 0362-1642[↩]
- Lowes, D.A., Thottakam, B.M., Webster, N.R., Murphy, M.P., & Galley, H.F. (2008). The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide-peptidoglycan model of sepsis. Free Radic Biol Med, Vol.45, No. 11, (December 2008), pp. (1559-1565), 0891-5849[↩]
- Plecitá-Hlavatá, L; Jezek, J & Jezek, P. (2009). Pro-oxidant Mitochondrial Matrix-Targeted Ubiquinone MitoQ10 Acts as Anti-oxidant at Retarded Electron Transport or Proton Pumping Within Complex I. The International Journal of Biochemistry and Cell Biology, Vol. 41, No. 8-9, (August-September, 2009), pp. (1697-1707), ISSN 1357-2725[↩]
- Doughan, A.K., & Dikalov, S.I. (2007). Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid Redox Signal, Vol. 9, No. 11, (November 2007), pp. (1825-1836), ISSN 1557-7716[↩]
- James A.M., Sharpley M.S., Manas A.-R.B., Frerman F.E., Hirst J., Smith R.A.J., Murphy M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007;282:14708–14718. doi: 10.1074/jbc.M611463200[↩]
- Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O’Sullivan J.D., Fung V., Smith R.A.J., Murphy M.P., Taylor K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148[↩]
- Adlam V.J., Harrison J.C., Porteous C.M., James A.M., Smith R.A.J., Murphy M.P., Sammut I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia‐reperfusion injury. FASEB. J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com[↩]
- Lowes D.A., Thottakam B.M.V., Webster N.R., Murphy M.P., Galley H.F. The mitochondria-targeted antioxidant MitoQ protects against organ damage in a lipopolysaccharide–peptidoglycan model of sepsis. Free Radic. Biol. Med. 2008;45:1559–1565. doi: 10.1016/j.freeradbiomed.2008.09.003[↩]
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J Neurosci. 2011 Nov 2;31(44):15703-15. doi: 10.1523/JNEUROSCI.0552-11.2011[↩]
- Young ML, Franklin JL. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol Cell Neurosci. 2019 Dec;101:103409. doi: 10.1016/j.mcn.2019.103409[↩]
- Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20 Suppl 2(Suppl 2):S609-31. doi: 10.3233/JAD-2010-100564[↩]
- Ng LF, Gruber J, Cheah IK, Goo CK, Cheong WF, Shui G, Sit KP, Wenk MR, Halliwell B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic Biol Med. 2014 Jun;71:390-401. doi: 10.1016/j.freeradbiomed.2014.03.003[↩][↩]
- Miquel E, Cassina A, Martínez-Palma L, Souza JM, Bolatto C, Rodríguez-Bottero S, Logan A, Smith RA, Murphy MP, Barbeito L, Radi R, Cassina P. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic Biol Med. 2014 May;70:204-13. doi: 10.1016/j.freeradbiomed.2014.02.019[↩]
- Mao P, Manczak M, Shirendeb UP, Reddy PH. MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim Biophys Acta. 2013 Dec;1832(12):2322-31. doi: 10.1016/j.bbadis.2013.09.005[↩][↩]
- Zhou, J., Wang, H., Shen, R., Fang, J., Yang, Y., Dai, W., Zhu, Y., & Zhou, M. (2018). Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. American journal of translational research, 10(6), 1887–1899.[↩][↩]
- Zhang T, Xu S, Wu P, Zhou K, Wu L, Xie Z, Xu W, Luo X, Li P, Ocak U, Ocak PE, Travis ZD, Tang J, Shi H, Zhang JH. Mitoquinone attenuates blood-brain barrier disruption through Nrf2/PHB2/OPA1 pathway after subarachnoid hemorrhage in rats. Exp Neurol. 2019 Jul;317:1-9. doi: 10.1016/j.expneurol.2019.02.009[↩][↩]
- Santini E, Turner KL, Ramaraj AB, Murphy MP, Klann E, Kaphzan H. Mitochondrial Superoxide Contributes to Hippocampal Synaptic Dysfunction and Memory Deficits in Angelman Syndrome Model Mice. J Neurosci. 2015 Dec 9;35(49):16213-20. doi: 10.1523/JNEUROSCI.2246-15.2015[↩]
- Ribeiro Junior RF, Dabkowski ER, Shekar KC, O Connell KA, Hecker PA, Murphy MP. MitoQ improves mitochondrial dysfunction in heart failure induced by pressure overload. Free Radic Biol Med. 2018 Mar;117:18-29. doi: 10.1016/j.freeradbiomed.2018.01.012[↩][↩]
- Gioscia-Ryan RA, Battson ML, Cuevas LM, Eng JS, Murphy MP, Seals DR. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J Appl Physiol (1985). 2018 May 1;124(5):1194-1202. doi: 10.1152/japplphysiol.00670.2017[↩]
- Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014 Jun 15;592(12):2549-61. doi: 10.1113/jphysiol.2013.268680[↩]
- Maiti AK, Spoorthi BC, Saha NC, Panigrahi AK. Mitigating peroxynitrite mediated mitochondrial dysfunction in aged rat brain by mitochondria-targeted antioxidant MitoQ. Biogerontology. 2018 Jul;19(3-4):271-286. doi: 10.1007/s10522-018-9756-6[↩]
- Braakhuis, A. J., Nagulan, R., & Somerville, V. (2018). The Effect of MitoQ on Aging-Related Biomarkers: A Systematic Review and Meta-Analysis. Oxidative medicine and cellular longevity, 2018, 8575263. https://doi.org/10.1155/2018/8575263[↩][↩]
- Dare AJ, Bolton EA, Pettigrew GJ, Bradley JA, Saeb-Parsy K, Murphy MP. Protection against renal ischemia-reperfusion injury in vivo by the mitochondria targeted antioxidant MitoQ. Redox Biol. 2015 Aug;5:163-168. doi: 10.1016/j.redox.2015.04.008[↩]
- Powell RD, Swet JH, Kennedy KL, Huynh TT, Murphy MP, Mckillop IH, Evans SL. MitoQ modulates oxidative stress and decreases inflammation following hemorrhage. J Trauma Acute Care Surg. 2015 Mar;78(3):573-9. doi: 10.1097/TA.0000000000000533[↩]
- Escribano-Lopez I, Diaz-Morales N, Rovira-Llopis S, de Marañon AM, Orden S, Alvarez A, Bañuls C, Rocha M, Murphy MP, Hernandez-Mijares A, Victor VM. The mitochondria-targeted antioxidant MitoQ modulates oxidative stress, inflammation and leukocyte-endothelium interactions in leukocytes isolated from type 2 diabetic patients. Redox Biol. 2016 Dec;10:200-205. doi: 10.1016/j.redox.2016.10.017[↩]
- Ward MS, Flemming NB, Gallo LA, Fotheringham AK, McCarthy DA, Zhuang A, Tang PH, Borg DJ, Shaw H, Harvie B, Briskey DR, Roberts LA, Plan MR, Murphy MP, Hodson MP, Forbes JM. Targeted mitochondrial therapy using MitoQ shows equivalent renoprotection to angiotensin converting enzyme inhibition but no combined synergy in diabetes. Sci Rep. 2017 Nov 9;7(1):15190. doi: 10.1038/s41598-017-15589-x[↩]
- Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant MitoQ decreases features of the metabolic syndrome in ATM+/-/ApoE-/- mice. Free Radic Biol Med. 2012 Mar 1;52(5):841-9. doi: 10.1016/j.freeradbiomed.2011.11.026[↩]
- Effects of Mitochondrial-targeted Antioxidant on Mild Cognitive Impairment (MCI) Patients. https://clinicaltrials.gov/ct2/show/NCT03514875[↩]
- Xi Y, Feng D, Tao K, Wang R, Shi Y, Qin H, Murphy MP, Yang Q, Zhao G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1α. Biochim Biophys Acta Mol Basis Dis. 2018 Sep;1864(9 Pt B):2859-2870. doi: 10.1016/j.bbadis.2018.05.018[↩]
- Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O’Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM; Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010 Aug 15;25(11):1670-4. doi: 10.1002/mds.23148[↩][↩]
- Rossman MJ, Santos-Parker JR, Steward CAC, Bispham NZ, Cuevas LM, Rosenberg HL, Woodward KA, Chonchol M, Gioscia-Ryan RA, Murphy MP, Seals DR. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension. 2018 Jun;71(6):1056-1063. doi: 10.1161/HYPERTENSIONAHA.117.10787[↩][↩][↩]
- Shill DD, Southern WM, Willingham TB, Lansford KA, McCully KK, Jenkins NT. Mitochondria-specific antioxidant supplementation does not influence endurance exercise training-induced adaptations in circulating angiogenic cells, skeletal muscle oxidative capacity or maximal oxygen uptake. J Physiol. 2016 Dec 1;594(23):7005-7014. doi: 10.1113/JP272491[↩]
- Gane EJ, Weilert F, Orr DW, Keogh GF, Gibson M, Lockhart MM, Frampton CM, Taylor KM, Smith RA, Murphy MP. The mitochondria-targeted anti-oxidant mitoquinone decreases liver damage in a phase II study of hepatitis C patients. Liver Int. 2010 Aug;30(7):1019-26. doi: 10.1111/j.1478-3231.2010.02250.x[↩]




{kind=link}