Nelson syndrome
Nelson syndrome refers to a spectrum of symptoms and signs in 8%–47% of patients as a complication following bilateral adrenalectomy for Cushing’s disease, with high circulating adrenocorticotropin (ACTH) levels, pigmentation due to the effects of the high serum concentrations of ACTH on the skin, the secondary loss of other pituitary hormones and is due to the development of an invasive secreting macroadenoma tumor of the corticotroph cells in the pituitary 1.
In 1958, Dr. Don Nelson et al. 2 reported a case in which a 33-year-old woman, who had undergone bilateral adrenalectomy for Cushing’s disease 3 years earlier, experienced skin hyperpigmentation, high plasma levels of adrenocorticotropic hormone (ACTH), and ultimately a pituitary tumor. By the 1960s, Nelson and other colleagues found that after bilateral adrenalectomy, ACTH-producing pituitary tumors appeared in several patients, thus leading to increased levels of ACTH and hyperpigmentation. Hence, these 3 symptoms have since become the clinical triad of Nelson’s syndrome 3.
Nelson syndrome occurs in up to 47% of patients with Cushing’s disease undergoing bilateral adrenalectomy 4 although progression of the size of a corticotroph tumor as assessed by MRI is more common and is detected in 50% of patients within 10 years of bilateral adrenalectomy for Cushing’s disease 5. The corticotroph tumor may be small in some cases but may also be extensive and locally invasive in others; patients can present with mass effects, headache, visual field defects, and external ophthalmoplegia 6. The hallmarks of the Nelson syndrome are skin hyperpigmentation and high plasma adrenocorticotropic hormone (ACTH) levels that reflect the activity of the tumor and are used for monitoring 7. Treatment of Nelson syndrome is restricted to pituitary surgery and radiotherapy only when there is an amenable anatomic target and the patient’s condition allows 8. In many patients with Nelson’s syndrome these conditions are not met; the levels of ACTH continue to rise, the symptoms persist and there are limited treatment options.
There is currently no medical therapy that can consistently reduce plasma ACTH levels and corticotroph tumor growth, and there is a real need for an effective medical management for Nelson’s syndrome. The anti-epileptic sodium valproate was frequently used in the past with disappointing or variable results 9 and dopamine agonists such as cabergoline only occasionally result in satisfactory response 10. Peroxisome proliferator-activated receptor gamma (PPARγ) agonists such as rosiglitazone have also been studied: one report showed biochemical response in two out of three patients, but one of these subsequently escaped 11. Munir et al 12 previously showed that even high doses of rosiglitazone (12 mg/day) do not reduce plasma ACTH levels. Temozolomide is a medical treatment for aggressive pituitary tumors, but is associated with significant toxicity limiting its use 13.
Pasireotide exerts its pharmacologic effects by binding and activating multiple somatostatin receptor subtypes (1, 2, 3, and 5). In vitro experiments have shown that pasireotide inhibits ACTH secretion in cultured corticotroph adenoma cells 14 and prospective clinical trials have proved effectiveness in lowering cortisol levels in patients with active Cushing’s disease 15. More recently, pasireotide LAR was used to treat a patient with an invasive corticotroph tumor resulting in clinical improvement, and reductions in tumor size and plasma ACTH levels 16.
Figure 1. Nelson syndrome
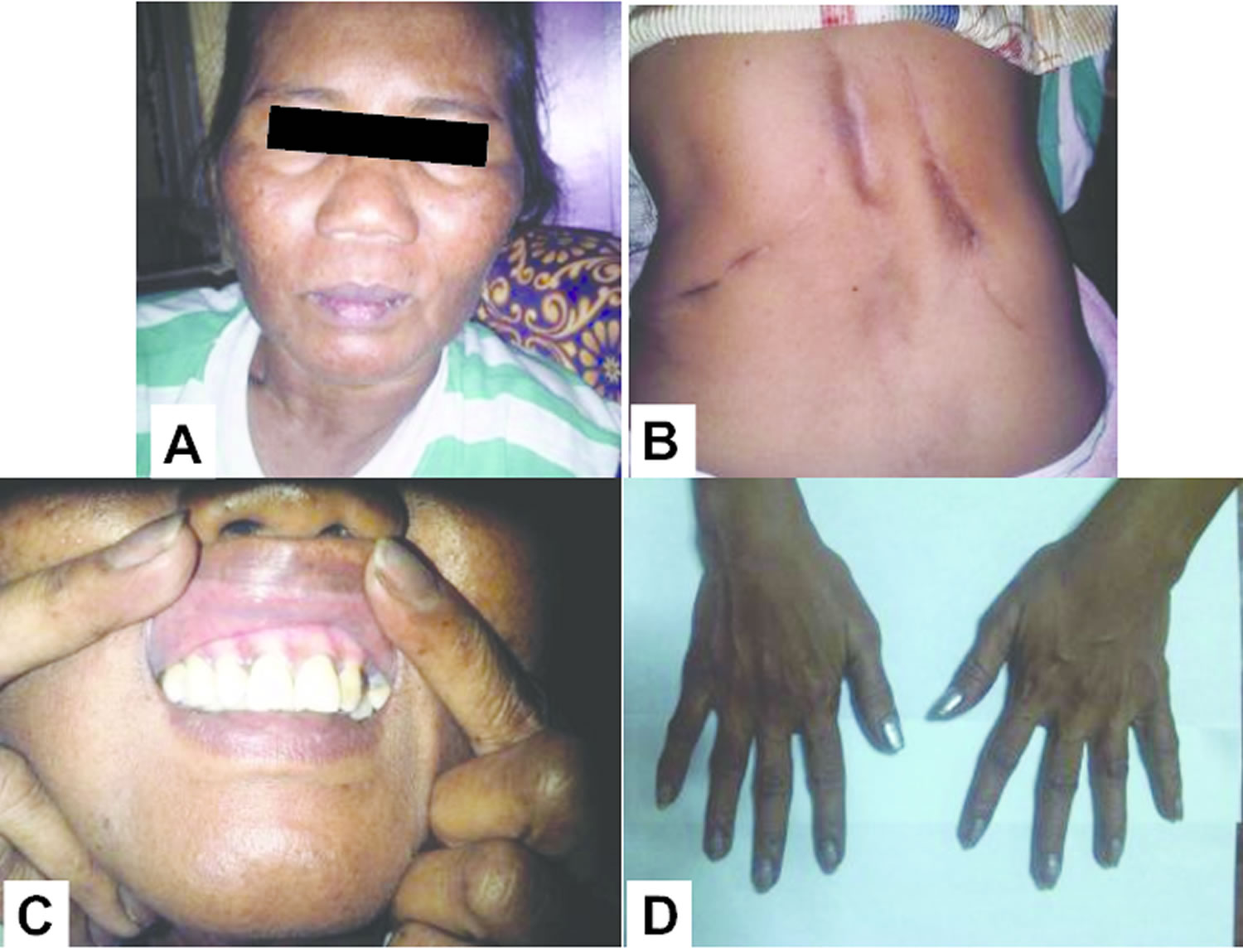
Footnote: A 53-year-old Filipino woman consulted her doctor with a 1½-month history of headache. Seh was seen in another center for amenorrhea, headache, polyuria, polydipsia, polyphagia and easy bruising. These were accompanied by the development of hypertension, moon facies, prominence of the dorsocervical fat pad, pitting edema of legs, and maculopapular rashes on the face, anterior neck and chest. She had no history of prolonged medication intake prior to the onset of symptoms. The patient had frequent emergency room visits due to recurrent symptoms of weakness, nausea and vomiting, but had always deferred admission. Generalized hyperpigmentation of the patient: face (A), bilateral adrenalectomy surgical scar (B), oral mucosa (C), and hands (D). Cranial MRI eventually revealed a pituitary macroadenoma (12.5 mm x 16.3 mm x 13.7 mm) with no evidence of invasion of the cavernous sinuses and abnormal changes in the brain parenchyma (see Figure 2).
[Source 17 ]Figure 2. Pituitary macroadenoma MRI scan
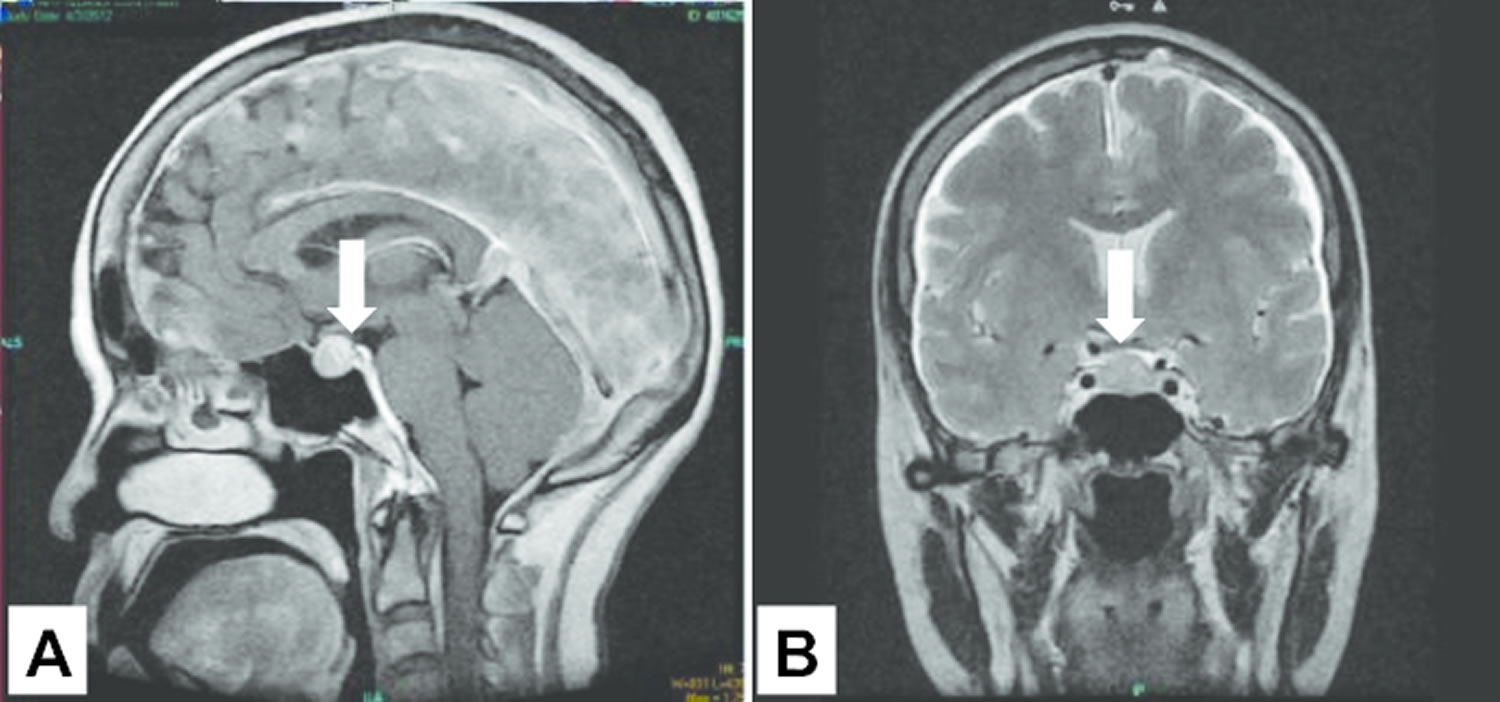
Footnote: Cranial pituitary MRI showing the presence of a pituitary macroadenoma of the patient in figure 1 above.
[Source 17 ]Figure 3. Nelson syndrome treatment options
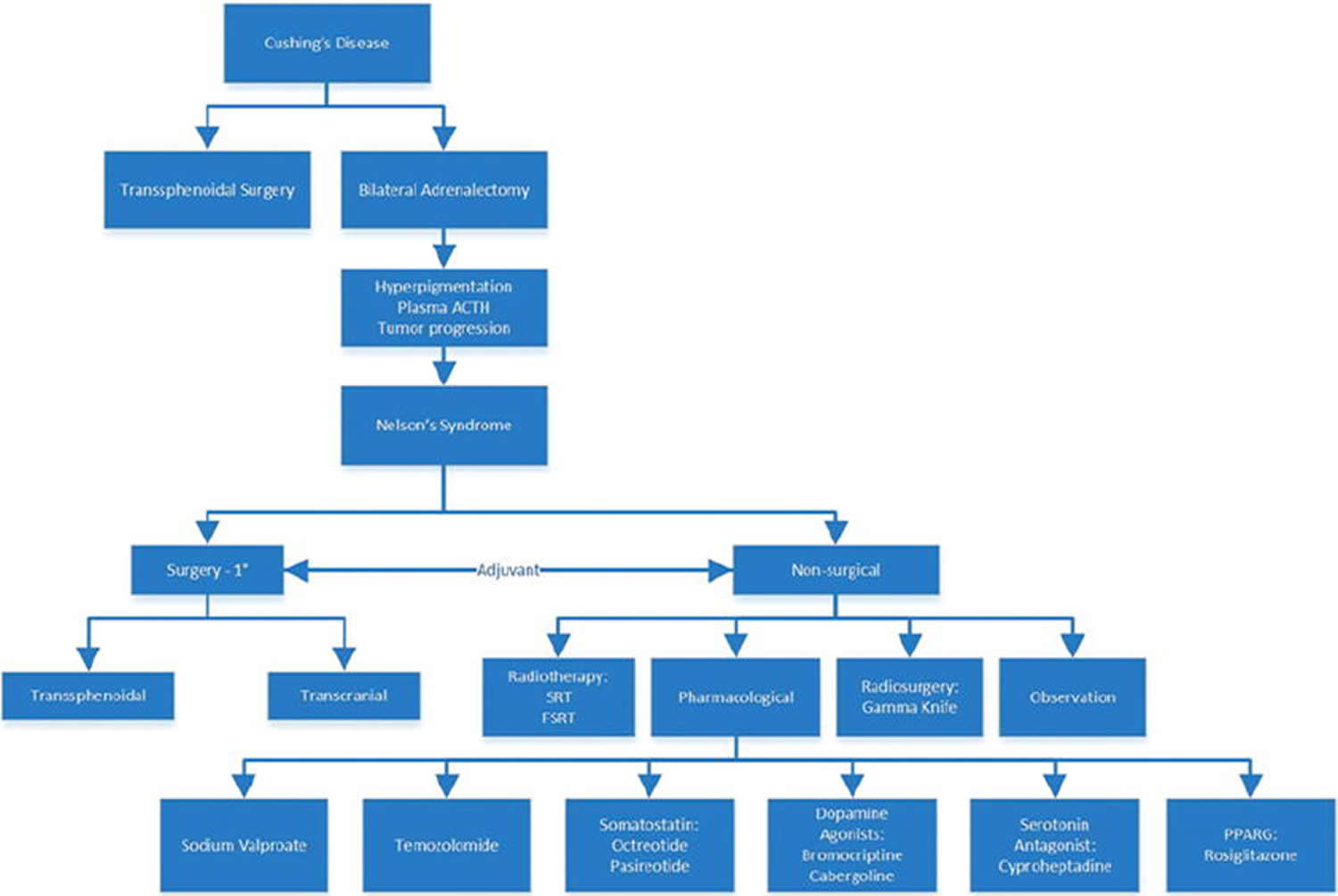
Footnote: Major treatment options for Cushing’s disease and Nelson’s syndrome (not all of the possible treatments for Cushing’s disease are included). Prophylactic radiotherapy at the time of bilateral adrenalectomy may have a highly protective effect in preventing Nelson’s syndrome. Transsphenoidal surgery remains the primary treatment for the management of Nelson’s syndrome. Nonsurgical treatments can either be stand-alone or used as adjuvants to surgery. Temozolomide, octreotide, and pasireotide show the most promise as pharmacotherapies, but they are often administered after transsphenoidal surgery. The usefulness of sodium valproate is still inconclusive. The usefulness of cabergoline has been shown in some studies, but that of other dopamine agonists is inconclusive. Rosiglitazone has shown some efficacy in murine models, but these results have not yet been found in human studies.
Abbreviations: PPARG = peroxisome proliferator-activated receptor gamma (peroxisome proliferator-activated receptor γ).
[Source 18 ]Nelson syndrome causes
Almost all cases of Nelson syndrome follow bilateral adrenalectomy in patients who have Cushing disease due to an adrenocorticotropin (ACTH)-secreting pituitary adenoma 19. Cortisol is a steroid hormone produced in the zona fasciculata of the adrenal cortex. In normal physiological systems, it provides negative feedback on the release of corticotropin-releasing hormone (CRH) produced by the hypothalamus. Bilateral adrenalectomy is meant to curb hypercortisolemia in patients with Cushing’s disease, thus releasing the system from the negative-feedback loop. Along the same line, without negative feedback, it is hypothesized that corticotropin-releasing hormone (CRH) levels increase, leading to increased production of proopiomelanocortin and its subsequent products adrenocorticotropin (ACTH) and melanocyte-stimulating hormone (MSH).
Recently, high-resolution MRI has allowed for detection of pituitary microadenomas at an early phase of Nelson syndrome. Most adenomatous corticotropes still retain their responsiveness to corticotropin-releasing hormone (CRH). Following bilateral adrenalectomy and normalization of cortisol levels that had suppressed hypothalamic corticotropin-releasing hormone (CRH) production, an increase in corticotropin-releasing hormone (CRH) occurs, which then has a trophic effect on the tumor, stimulating its growth. Regulatory gene mutations and mutations in the glucocorticoid receptor may also be important in determining tumor behavior.
Studies have demonstrated the difference in the mechanism of increased ACTH secretion in Nelson syndrome and untreated Cushing disease. Detailed analyses delineate marked ACTH secretory burst mass amplification and anomalous regularity of successive pulse size and timing in Nelson syndrome, compared with Cushing disease or controls 20. Authors of these studies speculate that these distinctions are due to unique tumoral secretory properties, concurrently required glucocorticoid replacement, and/or hypothalamic injury associated with prior radiotherapy in Nelson syndrome.
Nelson syndrome is a rare disorder, making accurately determining its incidence difficult. One review indicates that Nelson syndrome may be seen in anywhere from 8-44% of patients who have undergone bilateral adrenalectomy for Cushing disease 21. Fewer series were published in the 1990s than in the 1980s, suggesting that the syndrome is becoming increasingly less common. This decline in prevalence can be attributed to significant improvements in all aspects of the assessment and management of patients with Cushing syndrome in the last 10-20 years 22. These include introduction of the sensitive ACTH assay, the advent of high-resolution MRI, the availability in some centers of inferior petrosal sinus sampling, the refinement of the transsphenoidal pituitary surgery, and advances in pituitary radiation therapy, which have made bilateral adrenalectomy a less attractive therapy for Cushing disease.
Even in early series, only 20-40% of patients with a pituitary adenoma who had bilateral adrenalectomy developed Nelson syndrome. Younger age and pregnancy appeared to be risk factors. The former possibly represents a bias because younger patients have a longer time for tumor growth to occur and symptoms to become manifest.
Nelson syndrome symptoms
Corticotroph adenomas are observed predominantly in young and middle-aged women. The risk of developing Nelson syndrome appears to be higher in younger individuals than in older individuals. Corticotroph adenomas are observed predominantly in females; thus, Nelson syndrome is more common in women than men.
After a patient undergoes bilateral adrenalectomy for Cushing’s disease, the chance of Nelson’s syndrome development ranges from 8% through 47% 23.
The presentation of Nelson’s syndrome is variable and depends on the extent of adrenalectomy, length between surgery and presentation, and the source of hypercortisolemia (potentially ectopic sources) 24. When radiation therapy is used to treat an ACTH-secreting pituitary tumor after Nelson syndrome has developed, it is considered as a therapeutic measure for Nelson’s syndrome. However, when pituitary radiation therapy is undertaken before or immediately after bilateral adrenalectomy but before Nelson’s syndrome has developed, it is considered a prophylactic measure that can potentially prevent the development of Nelson’s syndrome 25. The signs and symptoms used to diagnose Nelson’s syndrome are elevated plasma ACTH levels, hyperpigmentation, and tumor progression. These tumors are capable of invading the cavernous sinus and compressing cranial nerves and optic pathways. In a recent case, bilateral oculomotor palsy was present in a patient with Nelson’s syndrome 26.
Hypopituitarism
Hypopituitarism occurs when the hypothalamic-pituitary portal system is disrupted or normal pituitary tissue is destroyed by the adenoma. Hypopituitarism may be partial and involves the adenohypophysis (anterior pituitary) more commonly than the neurohypophysis (posterior pituitary). Frequently, only partial hormone deficiencies occur. Information should be obtained about growth, the presence of symptoms of hypothyroidism, age of pubertal onset and its progression (in adolescents), presence of galactorrhea, and polyuria and polydipsia.
When no previous growth measurements are available, information should be obtained about the child’s rate of growth compared with friends or siblings. Information about growth may also be obtained indirectly by inquiring about the frequency with which larger clothes or shoes have been purchased for the child. Other diseases also need to be taken into account when obtaining a growth history. Patients with Cushing syndrome grow poorly because of hypercortisolemia.
Provided the epiphyses are open, growth should return to normal in these patients once adrenalectomy has been performed. If it does not, further investigation, including evaluation for possible hypothyroidism and growth hormone deficiency, is required.
Central hypothyroidism is usually mild and may be asymptomatic other than poor growth. Questions should focus on cold intolerance (whether the child feels the cold more than before or more than other family members), constipation, slowing of mentation, dry skin and coarse hair, and change in the shape of the face in addition to poor growth. Weight gain may occur but is not usually marked.
When looking for evidence of pubertal delay in girls, questions should establish the age at which breast development (thelarche) started (this may be difficult in the setting of obesity because of previous Cushing syndrome), with specific questioning about the age at which areolar enlargement began. A reduction in breast size or a noticeable softening in previously firm breasts is suggestive of hypoestrogenism.
In boys, questions should focus on when scrotal development and testicular enlargement commenced.
Pubarche (the development of pubic hair) is not a sensitive indicator of pubertal development in either sex because adrenal hyperandrogenemia commonly accompanies hypercortisolemia in Cushing syndrome. In addition, pubic hair may precede puberty in healthy girls and boys.
Hyperprolactinemia from interruption of the hypothalamic-pituitary portal axis may cause galactorrhea in pubertal or post-pubertal females.
Diabetes insipidus should be considered in patients with polyuria and polydipsia. Inadequate glucocorticoid replacement following adrenalectomy may mask these symptoms because glucocorticoids are required for normal water excretion. Inquiry should be made about the frequency, volume, and concentration of the urine being passed. During overnight sleep, the urine normally becomes concentrated because of a reduction in glomerular filtration rate and increased vasopressin secretion. The presence of nocturia or dilute urine on the first void in the morning is suggestive of diabetes insipidus.
Headache
Headaches are a common symptom in patients with pituitary masses and are probably the result of stretching of the diaphragma sellae.
Raised intracranial pressure due to obstruction of cerebrospinal fluid (CSF) flow is a late and uncommon sign because it requires a tumor large enough to extend into the third ventricle and obstruct the foramen of Monro.
Visual disturbance
Loss of vision can occur as a result of invasion or compression of the visual apparatus. This may be insidious and may not be noticed by the patient. The symptoms and signs vary depending upon where the optic apparatus is pressured by the pituitary lesion.
Prechiasmatic lesions usually result in symptoms affecting one eye only, while chiasmatic lesions result in the classic bitemporal hemianopia or quadrantanopsia. Postchiasmatic lesions can result in homonymous hemianopia.
Inquiry should be made about loss of peripheral vision (eg, bumping into walls or corners of tables, not seeing objects “out of the corner of the eye”), or visual loss in one eye or in one direction.
Physical examination
The physical examination of a patient in whom Nelson syndrome is suggested needs to include assessment of adequacy of steroid replacement, in addition to assessment of vision, cranial nerves, and general skin pigmentation.
Height
Height and growth velocity should be assessed as an index of growth hormone secretion, which may be affected by pituitary lesions.
Weight
Measurement of weight should be performed in older children when they are lightly clothed, in younger children when they are in their underclothes, and in infants when they are naked.
Vital signs
- Pulse: The pulse may be slow if significant hypothyroidism is present, although symptoms of central hypothyroidism are commonly mild. In acute adrenal insufficiency, reduced pulse volume with tachycardia may be present.
- Blood pressure: Blood pressure is frequently elevated in patients with hypercortisolism or if mineralocorticoid replacement is excessive. Hypotension or a postural fall in blood pressure may be present in patients with adrenal insufficiency as a result of inadequate replacement or because of acute adrenal crisis.
Eye examination
Loss of vision can occur as a result of invasion or compression of the optic apparatus.
Initial assessment should include assessment of visual acuity in each eye, confrontation testing to look for visual field defects, and examination of the optic fundi to look for papilledema or optic nerve atrophy.
Formal ophthalmologic assessment is necessary.
Thyroid examination
The thyroid gland should be examined to look for enlargement due to autoimmune hypothyroidism or hyperthyroidism that may accompany autoimmune adrenal insufficiency, one of the differential diagnoses in the hyperpigmented child.
Abdominal examination
In a hyperpigmented child, the abdominal examination should exclude hepatomegaly and splenomegaly (ie, possible liver disease), although the appearance is typically different in jaundice (yellow color with scleral involvement) or in hemochromatosis (bronze color). Inspection should also include a review of scars, which may indicate prior adrenalectomies. Scars may be hyperpigmented.
Liver disease is suggested if hepatomegaly, small liver, splenomegaly, or peripheral signs of liver disease are present.
Pubertal staging
All children of pubertal age should undergo an assessment of pubertal stage, looking for evidence of incomplete pubertal development or hypogonadism.
Premature appearance of pubic hair may occur in either sex because of excessive adrenal androgens in patients with Cushing syndrome. Other features observed are sex specific.
In females, secondary hypoestrogenism results in soft breasts and an unestrogenized vaginal mucosa.
In males, loss or softening of androgen-dependent body hair, small soft testes, increased upper–to–lower segment ratio, and gynecoid fat distribution are possible. Adrenal rest tissue may cause testicular enlargement that may be painful and can be unequal.
Neurologic examination
Hyporeflexia and delayed relaxation time of reflexes are signs that may be present in patients with hypothyroidism.
Cranial nerve involvement in a patient with Nelson syndrome can occur if tumor invasion of the ipsilateral cavernous sinus occurs. Examination should include assessment of sensation on the forehead (first division of trigeminal nerve) and ocular movements (III, IV, VI).
Visual examination should include assessment of visual acuity in each eye, visual field assessment, and examination of the optic nerve for evidence of papilledema or optic nerve atrophy.
Skin examination
Hyperpigmentation of the skin is usually obvious and is not limited to sun-exposed areas. The degree of pigmentation varies depending on the racial origin of the child and the serum concentrations of adrenocorticotropin (ACTH) 27.
Patients usually appear hyperpigmented with a linea nigra (pigmentation extending up the midline from the pubis to the umbilicus) and pigmentation of scars, gingivae, scrotum and areolae. Distinguishing this type of pigmentation from that of hemochromatosis, which is more of a bronze color, and jaundice, which also affects the sclera, is not usually difficult.
Nelson syndrome diagnosis
Visual field assessment
All children with large masses in the region of the pituitary or optic nerves should be referred for formal visual field assessment by a pediatric ophthalmologist. Patients, especially children, do not always report visual symptoms.
Laboratory studies
Adrenocorticotropin (ACTH) measurement
ACTH levels are markedly elevated in Nelson syndrome, usually in the thousands of picograms per milliliter. Levels of other derivatives of the precursor peptide, proopiomelanocortin (POMC), are also elevated, although their measurement is not required for diagnosis. Patients with Nelson syndrome often have an exaggerated ACTH response to corticotropin-releasing hormone (CRH). This test is not required for diagnostic purposes.
Thyroid function tests
Central hypothyroidism, which is not always clinically detectable, may be present. Free thyroxine (T4) levels are commonly just below the lower limit of the reference range, and thyroid-stimulating hormone (TSH) levels may be low, normal, or even mildly elevated.
Prolactin measurement
Any lesion that disrupts the pituitary stalk or hypothalamic-pituitary portal system results in mild hyperprolactinemia (< 100 ng/dL) because of loss of dopaminergic inhibition. Patients with prolactin-secreting pituitary adenomas typically have levels that exceed 150-200 ng/dL, with levels being many times greater in patients with macroadenomas.
Growth hormone measurement
Insulinlike growth factor–1 (IGF-1) and insulinlike growth factor–binding protein-3 (IGF-BP3) measurement is a useful means of screening for growth hormone deficiency in children older than 3 years. These results must be interpreted according to age-appropriate, sex-appropriate, and pubertal stage–appropriate reference range values. If these are low, more formal testing should be performed.
Gonadotropin measurement
Measure gonadotropin levels in adolescents in whom pubertal arrest or delay is suggested. Hyperprolactinemia causes hypogonadotropic hypogonadism. If androgenization with testicular enlargement, symptoms of pituitary or visual disturbance, and suppressed gonadotropins are present, human chorionic gonadotropin (hCG) should be measured to exclude a germ cell tumor.
Urine osmolality or specific gravity tests
Central diabetes insipidus occurs if the tumor has destroyed the posterior pituitary gland or disrupted the stalk. This is a rare occurrence because vasopressin secretion can occur more proximally if impingement on the neurohypophysis is gradual.
If an history of polyuria and polydipsia is noted, an early morning urine specimen should be collected. In healthy children, the urine osmolality should be greater than 600-700 mOsm/kg or the specific gravity should be greater than 1.010 in the morning. If the early morning urine is dilute, a water deprivation test should be performed.
Imaging studies
MRI of the pituitary and parasellar region
A gadolinium-enhanced MRI of the pituitary and parasellar region with 3-mm cuts through the pituitary adequately demonstrates the tumor and provides evidence of compression or invasion of surrounding structures.
In young children and patients with claustrophobia, sedation or anesthesia is required to obtain good quality images.
Nelson syndrome treatment
Nelson syndrome treatment options include observation, surgery, radiation therapy, and pharmacotherapy.
Surgical treatment
The first line of treatment for Nelson’s syndrome is resection, which is performed primarily via a transsphenoidal approach or, less commonly, via a transcranial approach 18. One of the first reports of pituitary surgery for Nelson’s syndrome was by Espinoza et al. 28, who described 3 patients who had undergone transsphenoidal surgery for the removal of the ACTH-secreting pituitary adenomas. Surgical treatment enables potentially curative resection of the expanding corticotroph tumor and decompression of the optic chiasm, if needed. Unfortunately, considering the rarity of Nelson’s syndrome, not many long-term case series for this surgery have been studied, and with the current advancements in nonsurgical therapies, the likelihood of finding any such series will decrease. Kelly et al. 29 attempted total hypophysectomy on 13 patients with Nelson’s syndrome; 9 patients underwent transsphenoidal resection and 4 underwent transcranial resection (2 by subfrontal approach and 2 by pterional approach). Tumor recurred in 2 patients, both of whom had undergone transsphenoidal resection; however, no regrowth occurred in patients who had undergone transcranial resection. De Tommasi et al. 30 reported on 6 patients with Nelson’s syndrome who had undergone transsphenoidal surgery. Two patients had extrasellar extension. In all
6 patients, the adenoma persisted after the first transsphenoidal surgery; half of these patients subsequently underwent repeat transsphenoidal surgery and half underwent Gamma Knife radiosurgery. Remission occurred in only 1 patient; disease persisted for the remainder. The range of efficacy of resection for treatment of Nelson syndrome is 10%–70%. Complications can include CSF leaks, cranial nerve deficits, meningitis, and visual field defects 31.
Radiotherapy and Radiosurgery
Radiotherapy can be considered as an option for patients in whom surgery for Nelson’s syndrome was unsuccessful or patients who are not optimal surgical candidates. One pitfall of radiotherapy is that it does not immediately reduce and normalize ACTH levels. Achieving normal ACTH levels can take weeks to months, depending on the size of the tumor; in the interim, control of excessive ACTH must be achieved by another manner such as medical therapy. Potential complications of radiation therapy include radiation-induced optic neuropathy, hypopituitarism, radiation necrosis, cerebral edema, and vasculopathy 32. Radiation therapy can also be used as an adjunctive treatment 29. More conformal techniques such as fractionated stereotactic radiotherapy or proton beam therapy can help reduce the adverse effects of disseminated radiation. For prevention of optic nerve injury, brain necrosis, and damage to surrounding tissues, the typical radiation dose (approximately 45–50 Gy) is given at 1.8- to 2.0-Gy fractions 31. Some investigators suggest that neoadjuvant radiation therapy after bilateral adrenalectomy may protect and possibly delay development of Nelson’s syndrome 29. According to a review of 39 patients followed up over 15 years after bilateral adrenalectomy, Nelson’s syndrome did not develop in any patients who had received neoadjuvant radiation therapy but did develop in 50% of those who did not.21 Although there is currently no consensus as to the role of neoadjuvant prophylactic radiation therapy, it has been the practice at the University of Oxford to administer it to patients with residual pituitary tissue who will undergo bilateral adrenalectomy 23. However, further definitive follow-up data are pending.
Stereotactic radiosurgery directs several beams of low-powered radiation to a specific target in the brain by a 3D coordination system. Although the low-powered beams prevent collateral radiation injury to the brain, a strong radiation dose is delivered to the point of convergence. One of the more common radiosurgical techniques used to manage Cushing’s disease is gamma knife radiosurgery 33. Vik-Mo et al. 34 published a study in which 10 patients with Nelson’s syndrome underwent gamma knife treatment. On average, tumor volume decreased by 32% for 9 patients but not at all for 1 patient. Of these 10 patients, ACTH levels decreased for 8 (range 2.0–278 pmol/L) and reached normal range for only 1 patient. However, the authors do not define their normal range for ACTH nor reveal the plasma ACTH levels for this patient. Recently, Marek et al. reported a study of 14 patients with Nelson’s syndrome treated by gamma knife radiosurgery in which the average
follow-up period was 10.4 years 35. Of these 14 patients, plasma ACTH levels decreased gradually in 12 and achieved normal levels 13–14 years later in 2. At approximately 2–5 years postirradiation, adenoma volume had decreased in 7 patients, remained unchanged in 6 patients, and completely disappeared in 1 patient. At 5–10 years after irradiation, among 11 patients reassessed, tumor volume decreased in 5 patients, remained unchanged in 3 patients, completely disappeared in 2 patients, and regrew in 1 patient. Most recently, Wilson et al. 36 published a report about use of a linear accelerator for stereotactic radiosurgery (5 patients) and fractionated stereotactic radiotherapy (2 patients). After stereotactic radiosurgery, tumor volume decreased for 2 patients, increased in another 2 patients, and did not change in 1 patient. After fractionated stereotactic radiotherapy, tumor volume decreased for 1 patient and increased after 15 months for the other. Unfortunately, no data concerning posttreatment ACTH levels were available. For patients in whom residual tumor is adjacent or in close approximation to the optic apparatus (< 2–3 mm), it might be safer to use fractionated stereotactic radiotherapy rather than stereotactic radiosurgery to avoid radiation-induced optic nerve injury.
Medical therapy
Medicinal therapy aims to control ACTH levels and curb tumor growth; however, no consistent series have confirmed its efficacy. Pharmacotherapies include sodium valproate, temozolomide, selective somatostatin analogs, dopamine agonists, and peroxisome proliferator-activated receptor γ.
Sodium valproate works as a therapeutic agent for Nelson syndrome by inhibiting reuptake of g-aminobutyric acid (GABA), which would decrease corticotropin-releasing hormone (CRH) release by the hypothalamus. In a study by Dornhorst et al. 37, 10 patients with Nelson’s syndrome received 600–1200 mg of sodium valproate per day for 5–32 weeks. Although initial treatment successfully decreased plasma ACTH levels, when treatment was discontinued, ACTH returned to pretreatment levels within 5–12 weeks. In another study 38, 11 patients with Nelson’s syndrome were given 600 mg of sodium valproate per day. At 6 weeks, plasma ACTH levels had decreased slightly. Of these 11 patients, sodium valproate was continued for 6 patients (600 mg/day for 5 patients and 1200 mg/day for 1 patient). At 1 year, ACTH levels had not changed significantly from pretreatment or 6-week levels for the 6 patients taking sodium valproate or the 5 patients not taking sodium valproate. Even when other reports are taken into consideration, effectiveness of sodium valproate for Nelson’s syndrome remains inconclusive 39.
Temozolomide is an orally administered alkylating agent capable of crossing the blood-brain barrier. Its active form is methyl-triazeno-imidazole-carboxamide, which methylates DNA at the O6 position of guanine. Methylation leads to mispairing with thymine, and continual mispairing eventually leads to apoptosis of the affected cell. The standard dosage of temozolomide is 150–200 mg/m² for 5 days in a 28-day cycle. Temozolomide was first used as a treatment for a prolactinomas (prolactin-secreting pituitary adenomas) 40. Temozolomide has been used in over 30 cases of
pituitary adenomas; for patients whose tumors respond to this treatment, hormone levels decrease almost immediately 41. Recently, Moyes et al. 42 described a case in which a 64-year-old woman with Nelson’s syndrome was given 200 mg/m2 of temozolomide orally for 5 days in a 28-day cycle. By the end of the fourth cycle, MR images showed markedly decreased tumor volume and plasma ACTH levels had fallen from 2472 to 389 pmol/L. However, the patient experienced complications of CSF leakage from the ears and nostrils from tumor shrinkage, which abated after about of bacterial meningitis. Although temozolomide seems to be an effective and well-tolerated treatment for Nelson’s syndrome, further studies with larger series and longer-term follow-up are warranted.
Somatostatin analogs such as octreotide have been used in the treatment of Nelson’s syndrome 43. ACTH-secreting pituitary adenomas primarily express somatostatin receptor (sst) 5, sst1, and sst2 44. Octreotide has a strong binding affinity to sst2 and only a moderate binding affinity to sst5 45. One of the earliest reports was made in 1975, when Tyrrell et al. 46 described a progressive decline in plasma ACTH levels (40%–71% of pretreatment basal values) in 5 patients who had been given somatostatin. After infusion cessation, ACTH values rose back to baseline levels. In a separate study 47, octreotide was given to a 27-year-old Nelson’s syndrome patient. Plasma ACTH levels fell 54% from pretreatment values (from 66.7pmol/L to 36pmol/L), and tumor diameter decreased by 3 mm. In a very recent quality of life assessment, the effect of long-acting repeatable octreotide was assessed in a 59-year-old woman with Nelson’s syndrome 48. Pretreatment plasma ACTH levels of 235 pM decreased to 116 and 71 pM at 18 and 21 months, respectively, after the patient received long-acting repeatable octreotide. Hyperpigmentation also decreased. Even after 3 months without receiving octreotide, the patient remained stable but ACTH levels began to increase (266 pM), thus corroborating the effects of octreotide.
In contrast to octreotide, pasireotide is a multireceptor ligand with high binding affinity to sst1, sst2, sst3, and sst5. In 2012, the US Food and Drug Administration (FDA) approved the use of pasireotide for the treatment of Cushing’s disease 49. In 2013, Katznelson 50 reported favorable biochemical and clinical responses in a patient with Nelson’s syndrome treated with pasireotide. The patient was a 55-year-old woman with an aggressive and persistent pituitary corticotroph adenoma previously treated with multiple transsphenoidal surgeries and fractionated radiotherapy. The patient received an intramuscular injection of 60 mg of pasireotide every 28 days. The pretreatment baseline plasma ACTH level of 42,710 pg/ml (reference range 5–27 pg/ml) declined significantly, almost a thousand-fold, to 4272 pg/ml, a month after treatment and remained around this level for approximately 19 months. In addition, the patient’s skin hyperpigmentation decreased markedly. Follow-up MR imaging at 9 months showed a reduced size of the suprasellar component of the adenoma. Perhaps the greater binding affinity of pasireotide to sst5 makes it more effective than octreotide for treatment of Nelson’s syndrome. Additional studies are warranted to corroborate the existing data for pasireotide as a treatment for Nelson’s syndrome, and studies comparing octreotide with pasireotide should be considered.
Dopamine agonists such as bromocriptine and cabergoline have also been used in the medical management of patients with Nelson’s syndrome. One study 51 assessed the effects of bromocriptine, cyproheptadine (serotonin antagonist), and valproic acid on ACTH levels in 6 female patients with Nelson’s syndrome. Only bromocriptine at 2.5 mg caused a significant (52%) decrease in plasma ACTH levels. In a different study, bromocriptine was ineffective at reducing plasma ACTH levels in a 26-year-old female Nelson’s syndrome patient, but cabergoline successfully reduced levels from 247 pg/ml to 64 pg/ml 52. According to
MRI analysis, cabergoline also led to complete remission of the pituitary adenoma. The single and combined effects of cyproheptadine and bromocriptine were also tested in 12 patients who had raised plasma ACTH levels but no pituitary macroadenoma after bilateral adrenalectomy for Cushing’s disease; plasma ACTH levels did not change significantly after treatment with this combination 53.
Rosiglitazone, a thiazolidinedione, is a selective ligand of peroxisome proliferator-activated receptor γ. These receptors are abundantly expressed in human ACTH secreting pituitary adenomas. Heaney et al. 54 showed that rosiglitazone effectively prevented corticotroph tumors and suppressed ACTH secretion in murine models (150 mg/kg/day). In in vitro studies, rosiglitazone induced G0/G1 cell-cycle arrest and apoptosis in human and murine corticotroph, somatolactotroph, and gonadotroph pituitary tumor cells and further suppressed hormone secretion. However, the favorable effects of rosiglitazone have not been successfully replicated in human studies. Mullan et al. 55 presented a series of 7 patients with Nelson’s syndrome for whom 8 mg of rosiglitazone orally, once daily for 12 weeks, led to no statistically significant decrease in plasma ACTH levels. Of note, the US Food and Drug Administration limits the maximal dose of rosiglitazone in humans to 8 mg, but the murine models tested by Heaney and colleagues received 150 mg.
Observation
Although some clinicians may consider initial observation with repeat imaging for Nelson syndrome patients harboring smaller, stable pitutary tumors that have not grown or that have demonstrated limited progression, observation is generally not the first line of treatment 18. If left untreated,
most of these pitutary tumors will probably progress and warrant treatment. In a study by Kemink et al. 56, observation was the treatment modality for 8 of 15 patients. In these 8 patients, ACTH levels increased between the time of diagnosis and the time of the last follow-up visit, and in all 8 patients, the tumor progressed with parasellar extension or suprasellar extension. Of these 8 patients, 6 underwent elective pituitary surgery.
- Nelson DH, Meakin JW, Thorn GW. ACTH-producing pituitary tumors following adrenalectomy for Cushing’s syndrome. Ann Intern Med. 1960;52:560–569. doi: 10.7326/0003-4819-52-3-560.[↩]
- Nelson DH, Meakin JW, Dealy JB Jr, Matson DD, Emerson K Jr, Thorn GW: ACTH-producing tumor of the pituitary gland. N Engl J Med 259:161–164, 1958[↩]
- Nelson DH, Sprunt JG, Mims RB: Plasma ACTH determinations in 58 patients before or after adrenalectomy for Cushing’s syndrome. J Clin Endocrinol Metab 26:722–728, 1966[↩]
- Jenkins PJ, Trainer PJ, Plowman PN, Shand WS, Grossman AB, Wass JA, Besser GM. The long-term outcome after adrenalectomy and prophylactic pituitary radiotherapy in adrenocorticotropin-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 1995;80(1):165–171.[↩]
- Graffeo CS, Perry A, Carlstrom LP, Meyer FB, Atkinson JL, Erickson D, Nippoldt TB, Young WF, Jr., Pollock BE, Van Gompel JJ. Characterizing and predicting the Nelson–Salassa syndrome. J Neurosurg. 2017[↩]
- Munir A, Song F, Ince P, Walters SJ, Ross R, Newell-Price J. Ineffectiveness of rosiglitazone therapy in Nelson’s syndrome. J Clin Endocrinol Metab. 2007;92(5):1758–1763. doi: 10.1210/jc.2006-2005.[↩]
- Assie G, Bahurel H, Bertherat J, Kujas M, Legmann P, Bertagna X. The Nelson’s syndrome… revisited. Pituitary. 2004;7(4):209–215. doi: 10.1007/s11102-005-1403-y[↩]
- Marek J, Jezkova J, Hana V, Krsek M, Liscak R, Vladyka V, Pecen L. Gamma knife radiosurgery for Cushing’s disease and Nelson’s syndrome. Pituitary. 2015;18(3):376–384. doi: 10.1007/s11102-014-0584-7[↩]
- Reincke M, Allolio B, Kaulen D, Jaursch-Hancke C, Winkelmann W. The effect of sodium valproate in Cushing’s disease, Nelson’s syndrome and Addison’s disease. Klin Wochenschr. 1988;66(15):686–689. doi: 10.1007/BF01726927[↩]
- Shraga-Slutzky I, Shimon I, Weinshtein R. Clinical and biochemical stabilization of Nelson’s syndrome with long-term low-dose cabergoline treatment. Pituitary. 2006;9(2):151–154. doi: 10.1007/s11102-006-9290-4[↩]
- Andreassen M, Kristensen LO. Rosiglitazone for prevention or adjuvant treatment of Nelson’s syndrome after bilateral adrenalectomy. Eur J Endocrinol. 2005;153(4):503–505. doi: 10.1530/eje.1.01994[↩]
- Munir A, Song F, Ince P, Walters SJ, Ross R, Newell-Price J. Ineffectiveness of rosiglitazone therapy in Nelson’s syndrome. J Clin Endocrinol Metab. 2007;92(5):1758–1763. doi: 10.1210/jc.2006-2005[↩]
- Moyes VJ, Alusi G, Sabin HI, Evanson J, Berney DM, Kovacs K, Monson JP, Plowman PN, Drake WM. Treatment of Nelson’s syndrome with temozolomide. Eur J Endocrinol. 2009;160(1):115–119. doi: 10.1530/EJE-08-0557[↩]
- Hofland LJ, van der Hoek J, Feelders R, van Aken MO, van Koetsveld PM, Waaijers M, Sprij-Mooij D, Bruns C, Weckbecker G, de Herder WW, Beckers A, Lamberts SW. The multi-ligand somatostatin analogue SOM230 inhibits ACTH secretion by cultured human corticotroph adenomas via somatostatin receptor type 5. Eur J Endocrinol. 2005;152(4):645–654. doi: 10.1530/eje.1.01876[↩]
- Pivonello R, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, Trovato A, Hughes G, Salgado LR, Lacroix A, Schopohl J, Biller BM, Pasireotide BSG. Pasireotide treatment significantly improves clinical signs and symptoms in patients with Cushing’s disease: results from a Phase III study. Clin Endocrinol (Oxf) 2014;81(3):408–417. doi: 10.1111/cen.12431[↩]
- Katznelson L. Sustained improvements in plasma ACTH and clinical status in a patient with Nelson’s syndrome treated with pasireotide LAR, a multireceptor somatostatin analog. J Clin Endocrinol Metab. 2013;98(5):1803–1807. doi: 10.1210/jc.2013-1497[↩]
- Darmawan, Guntur & Pagsisihan, Daveric & Andag-Silva, Aime. (2012). Tan Without the Sun: A Case of Nelson’s Syndrome. Journal of the ASEAN Federation of Endocrine Societies. 27. 214-216. 10.15605/jafes.027.02.14.[↩][↩]
- Nelson’s syndrome: a review of the clinical manifestations, pathophysiology, and treatment strategies. Neurosurg Focus 38 (2):E14, 2015 https://doi.org/10.3171/2014.10.FOCUS14681[↩][↩][↩]
- Nelson syndrome. https://emedicine.medscape.com/article/923425-overview[↩]
- van Aken MO, Pereira AM, van den Berg G, Romijn JA, Veldhuis JD, Roelfsema F. Profound amplification of secretory-burst mass and anomalous regularity of ACTH secretory process in patients with Nelson’s syndrome compared with Cushing’s disease. Clin Endocrinol (Oxf). 2004 Jun. 60(6):765-72.[↩]
- Tiyadatah BN, Kalavampara SV, Sukumar S, Mathew G, Pooleri GK, Prasanna AT, et al. Bilateral Simultaneous Laparoscopic Adrenalectomy in Cushing’s Syndrome: Safe, Effective, and Curative. J Endourol. 2012 Feb. 26(2):157-63.[↩]
- Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008 Jul. 93(7):2454-62.[↩]
- Barber TM, Adams E, Ansorge O, Byrne JV, Karavitaki N, Wass JA: Nelson ‘s syndrome. Eur J Endocrinol 163:495–507, 2010[↩][↩]
- Hornyak M, Weiss MH, Nelson DH, Couldwell WT: Nelson syndrome: historical perspectives and current concepts. Neurosurg Focus 23:3 E12, 2007[↩]
- Gil-Cárdenas A, Herrera MF, Díaz-Polanco A, Rios JM, Pantoja JP: Nelson’s syndrome after bilateral adrenalectomy for Cushing’s disease. Surgery 141:147–152, 2007[↩]
- Gundgurthi A, Kharb S,Garg MK, Brar KS, Bharwaj R, Pathak HC: Nelson ‘s syndrome presenting as bilateral oculomotor palsy. Indian J Endocrinol Metab 17:1114–1116, 2013[↩]
- Barabash R, Moreno-Suárez FG, Rodríguez L, Molina AM, Conejo-Mir J. [Nelson syndrome: a rare cause of generalized hyperpigmentation of the skin]. Actas Dermosifiliogr. 2010 Jan-Feb. 101(1):76-80.[↩]
- Espinoza A, Nowakowski H, Kautzky R, Lüdecke D: ACTH determinations before and after selective removal of pituitary adenomas in Nelson ‘s syndrome. Acta Endocrinol Suppl (Copenh) 173:34:1973[↩]
- Kelly PA, Samandouras GGrossman AB, Afshar F, Besser GM, Jenkins PJ: Neurosurgical treatment of Nelson’s syndrome. J Clin Endocrinol Metab 87:5465–5469, 2002[↩][↩][↩]
- De Tommasi C, Vance ML, Okonkwo DO, Diallo A, Laws ER Jr: Surgical management of adrenocorticotropic hormone-secreting macroadenomas: outcome and challenges in patients with Cushing’s disease or Nelson ‘s syndrome. J Neurosurg 103:825–830, 2005[↩]
- Banasiak MJ, Malek AR: Nelson syndrome: comprehensive review of pathophysiology, diagnosis, and management. Neurosurg Focus 23:3E13, 2007[↩][↩]
- Vance ML: Cushing’s disease: radiation therapy. Pituitary 12:11–14, 2009[↩]
- Brada M, Ajithkumar TV, Minniti G: Radiosurgery for pituitary adenomas. Clin Endocrinol (Oxf) 61:531–543, 2004[↩]
- Vik-Mo EO, Øksnes M, Pedersen PH, Wentzel-Larsen T, Rødahl E, Thorsen F: Gamma knife stereotactic radiosurgery of Nelson syndrome. Eur J Endocrinol 160:143–148, 2009[↩]
- Marek JJežková J, Hána V, Kršek M, Liščák R, Vladyka V: Gamma knife radiosurgery for Cushing’s disease and Nelson’s syndrome. Pituitary [epub ahead of print], 2014[↩]
- Wilson PJ, Williams JR, Smee RI: Cushing’s disease: a single centre’s experience using the linear accelerator (LINAC) for stereotactic radiosurgery and fractionated stereotactic radiotherapy. J Clin Neurosci 21:100–106, 2014[↩]
- Dornhorst A, Jenkins JS, Lamberts SW, Abraham RR, Wynn V, Beckford U: The evaluation of sodium valproate in the treatment of Nelson ‘s syndrome. J Clin Endocrinol Metab 56:985–991, 1983[↩]
- Kelly WAdams JE, Laing I, Longson D, Davies D: Long-term treatment of Nelson’s syndrome with sodium valproate. Clin Endocrinol (Oxf) 28:195–204, 1988[↩]
- Kasperlik-Załuska AA, Zgliczyński W, Jeske WZdunowski P: ACTH responses to somatostatin, valproic acid and dexamethasone in Nelson’s syndrome. Neuroendocrinol Lett 26:709–712, 2005[↩]
- Kovacs K, Horvath E, Syro LV, Uribe H, Penagos LC, Ortiz LD: Temozolomide therapy in a man with an aggressive prolactin-secreting pituitary neoplasm: morphological findings. Hum Pathol 38:185–189, 2007[↩]
- Ortiz LD, Syro LV, Scheithauer BW, Rotondo F, Uribe H, Fadul CE: Temozolomide in aggressive pituitary adenomas and carcinomas. Clinics (Sao Paulo) 67:Suppl 1119–123, 2012[↩]
- Moyes VJAlusi G, Sabin HI, Evanson J, Berney DM, Kovacs K: Treatment of Nelson’s syndrome with temozolomide. Eur J Endocrinol 160:115–119, 2009[↩]
- Petrini L, Gasperi M, Pilosu R, Marcello A, Martino E: Long-term treatment of Nelson’s syndrome by octreotide: a case report. J Endocrinol Invest 17:135–139, 1994[↩]
- Hofland LJ, Lamberts SW: The pathophysiological consequences of somatostatin receptor internalization and resistance. Endocr Rev 24:28–47, 2003[↩]
- Bruns CLewis IBriner U, Meno-Tetang G, Weckbecker G: SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol 146:707–716, 2002[↩]
- Tyrrell JB, Lorenzi M, Gerich JE, Forsham PH: Inhibition by somatostatin of ACTH secretion in Nelson’s syndrome. J Clin Endocrinol Metab 40:1125–1127, 1975[↩]
- Kelestimur F, Utas C, Ozbakir O, Selçuklu A, Kandemir O, Ozcan N: The effects of octreotide in a patient with Nelson’s syndrome. Postgrad Med J 72:53–54, 1996[↩]
- Arregger AL, Cardoso EM, Sandoval OB, Monardes Tumilasci EG, Sanchez RContreras LN: Hormonal secretion and quality of life in Nelson syndrome and Cushing disease after long acting repeatable octreotide: a short series and update. Am J Ther 21:e110–e116, 2014[↩]
- Schmid HA, Schoeffter P: Functional activity of the multi-ligand analog SOM230 at human recombinant somatostatin receptor subtypes supports its usefulness in neuroendocrine tumors. Neuroendocrinology 80:Suppl 147–50, 2004[↩]
- Katznelson L: Sustained improvements in plasma ACTH and clinical status in a patient with Nelson’s syndrome treated with pasireotide LAR, a multireceptor somatostatin analog. J Clin Endocrinol Metab 98:1803–1807, 2013[↩]
- Mercado-Asis LB, Yanovski JA, Tracer HL, Chik CL, Cutler GB Jr: Acute effects of bromocriptine, cyproheptadine, and valproic acid on plasma adrenocorticotropin secretion in Nelson’s syndrome. J Clin Endocrinol Metab 82:514–517, 1997[↩]
- Casulari LA, Naves LA, Mello PA, Pereira Neto A, Papadia C: Nelson ‘s syndrome: complete remission with cabergoline but not with bromocriptine or cyproheptadine treatment. Horm Res 62:300–305, 2004[↩]
- Whitehead HM, Beacom R, Sheridan B, Atkinson AB: The effect of cyproheptadine and/or bromocriptine on plasma ACTH levels in patients cured of Cushing’s disease by bilateral adrenalectomy. Clin Endocrinol (Oxf) 32:193–201, 1990[↩]
- Heaney AP: Novel pituitary ligands: peroxisome proliferator activating receptor-gamma. Pituitary 6:153–159, 2003[↩]
- Mullan KR, Leslie H, McCance DR, Sheridan B, Atkinson AB: The PPAR-gamma activator rosiglitazone fails to lower plasma ACTH levels in patients with Nelson’s syndrome. Clin Endocrinol (Oxf) 64:519–522, 2006[↩]
- Kemink SA, Grotenhuis JA, De Vries J, Pieters GF, Hermus AR, Smals AG: Management of Nelson’s syndrome: observations in fifteen patients. Clin Endocrinol (Oxf) 54:45–52, 2001[↩]





{kind=link}