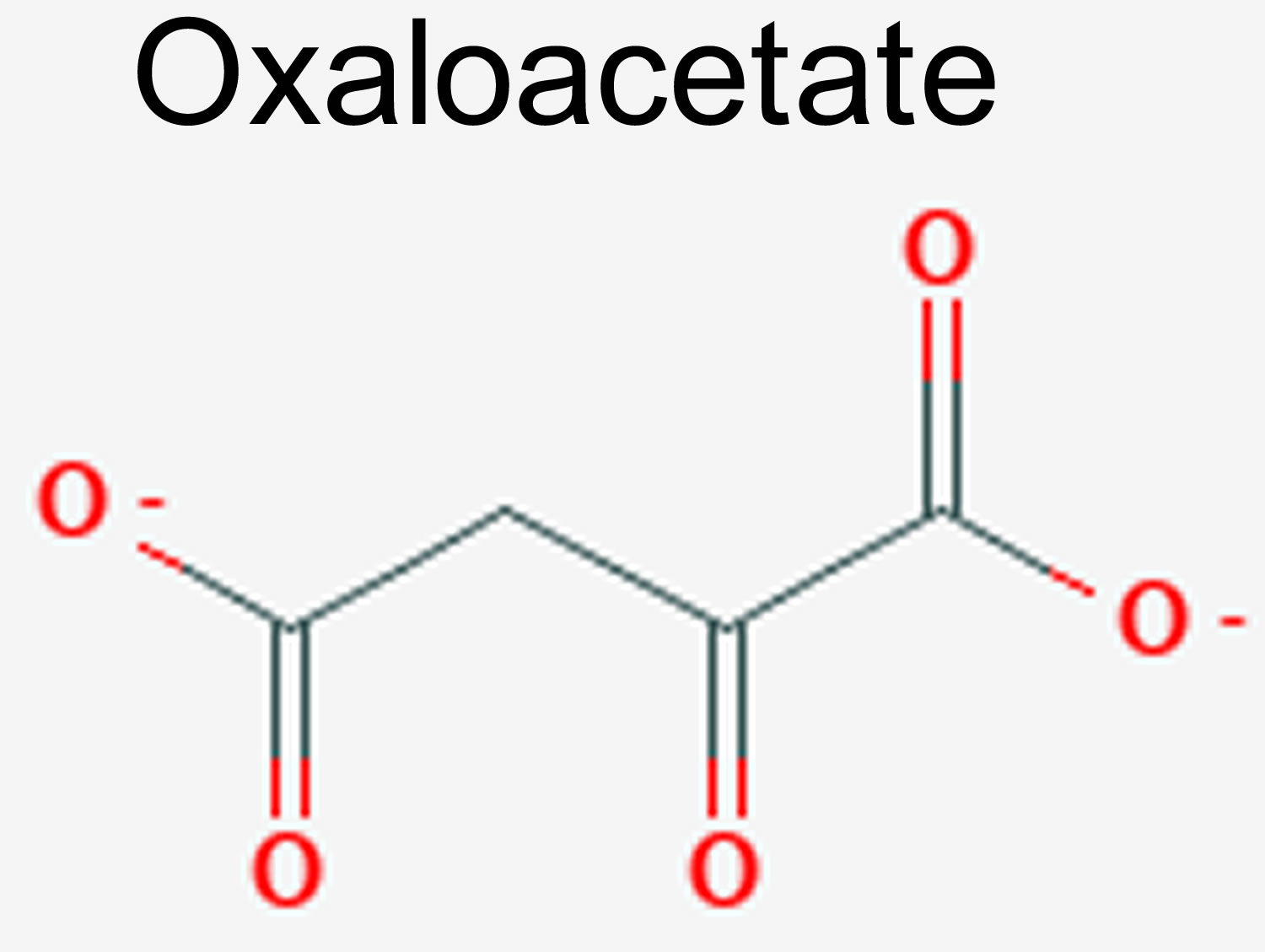
What is oxaloacetate
Oxaloacetate is a 4 carbon-dicarboxylate, which is an intermediate of the citric acid cycle (Krebs cycle or tricarboxylic acid cycle), is involved in energy production within the mitochondria 1. Oxaloacetate is also involved in many biochemical reactions including gluconeogenesis (glucose synthesis), the glyoxylate cycle, glyoxylate degradation, anaerobic respiration, the Krebs cycle (citric acid cycle), aspartate biosynthesis, and degradation of glutamate (see Figure 1) 1.
It is well established that glutamate acts as an important mediator of neuronal degeneration during cerebral ischemia 2. The systemic administration of oxaloacetate represents a novel neuroprotective strategy to minimize the deleterious effect of glutamate in the brain tissue after ischemic stroke 3. The neuroprotective effect of oxaloacetate is based on the capacity of this molecule to reduce the brain and blood glutamate levels as a result of the activation of the blood-resident enzyme glutamate-oxaloacetate transaminase (GOT) 4.
Oxaloacetic acid is a critical metabolic intermediate and is found within every mitochondrion. Oxaloacetic acid is a small molecule that is highly water soluble, and oral supplementation results in distribution of the compound throughout the body via the bloodstream 5. Once in the body, oxaloacetate (the water soluble ion) can react in several ways. A highly energy favorable reaction is the conversion of oxaloacetate to L-malate, catalyzed by the enzyme malate dehydrogenase. During the conversion of oxaloacetate to malate, NADH is also converted to NAD+, which greatly increases the NAD+/NADH ratio.
High doses of oxaloacetic acid have been shown to successfully protect mitochondrial DNA damage in brain tissues 6. Interestingly, the oxaloacetate was delivered by intraperitoneal injection, and the protection in the brain indicates that oxaloacetate is able to penetrate the blood-brain barrier (BBB). Mitochondrial damage has been implicated in premature aging of animals 7. It was hypothesized that an excess of oxaloacetate may effectively mimic calorie restriction by raising the NAD+/NADH ratio. Published studies on Caenorhabditis elegans (roundworm) have indicated that excess oxaloacetate can lead to increased life span through the AMPK/FOXO-dependent pathway 8.
In addition to protecting mitochondrial DNA, oxaloacetic acid has been shown to be protective of various whole tissues. Wood 9 showed that retinal pigmented epithelial (RPE) cells can be protected by combinations of zinc and oxaloacetate, which works much better than zinc alone. Damaged RPE cells underlie age-related macular degeneration, which causes the loss of central vision in 30% of those over 75 years old 10.
In addition to protection from zinc, oxaloacetate also provides protection of neurons from hydrogen peroxide 11 and from other free radicals 12. Supplementing the diet with anti-oxidants has been suggested as a method to reduce age related illness such as cardiovascular disease, the number one cause of death. Although a popular theory of anti-aging therapy involves the use of antioxidants, there is little support that antioxidants (in general) increase lifespan. Large epidemiologic studies of populations that ingest higher amounts of carotenoids, vitamin C and vitamin E have shown conflicting efficacy. Furthermore, clinical trials have failed to find significant protection provided by Vitamin C, Vitamin E and Carotenoids 13. Perhaps the unique abilities of oxaloacetic acid stem not from general anti-oxidant abilities, but from proper placement of the antioxidant into the mitochondria, via mitochondrial membrane leakage 14.
Figure 1. Citric acid cycle (shows the position of oxaloacetate within the cycle)
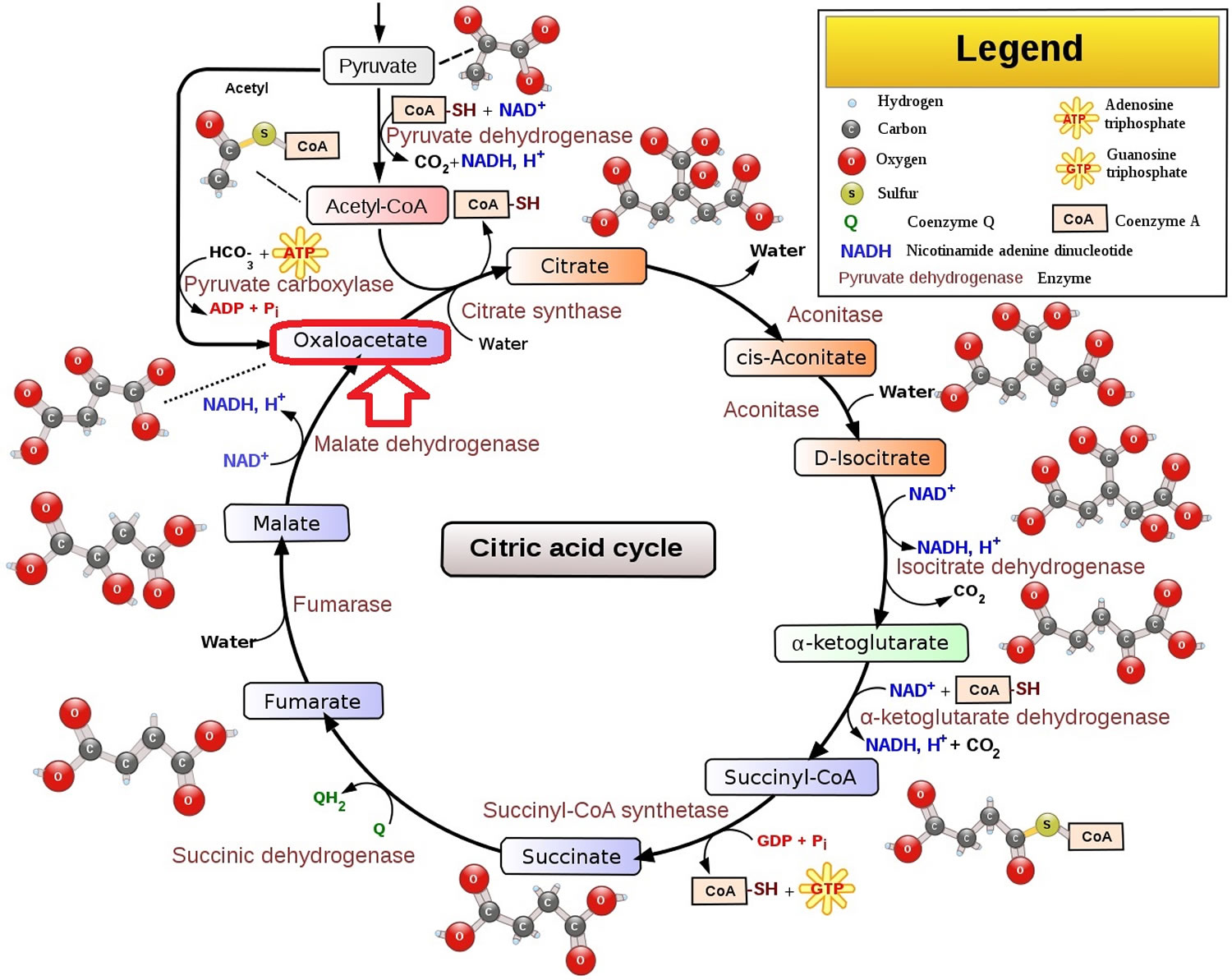
Footnote: The citric acid cycle begins with the condensation of an oxaloacetate (a four-carbon acceptor molecule) and the acetyl group of acetyl CoA (a two-carbon unit), to form a six-carbon molecule called citrate. Oxaloacetate reacts with acetyl CoA and water (H2O) to yield citrate and CoA 15. This reaction, which is an aldol condensation followed by a hydrolysis, is catalyzed by citrate synthase. Oxaloacetate first condenses with acetyl CoA to form citryl CoA, which is then hydrolyzed to citrate and CoA. The hydrolysis of citryl CoA, a high-energy thioester intermediate, drives the overall reaction far in the direction of the synthesis of citrate. In essence, the hydrolysis of the thioester powers the synthesis of a new molecule from two precursors. Because this reaction initiates the citric acid cycle, it is very important that side reactions be minimized. In the final stage of the citric acid cycle, oxaloacetate is regenerated by the oxidation of succinate.
- Two carbon atoms enter the citric acid cycle in the condensation of an acetyl unit (from acetyl CoA) with oxaloacetate. Two carbon atoms leave the cycle in the form of CO2 in the successive decarboxylations catalyzed by isocitrate dehydrogenase and alpha-ketoglutarate dehydrogenase. Interestingly, the results of isotope-labeling studies revealed that the two carbon atoms that enter each cycle are not the ones that leave.
- Four pairs of hydrogen atoms leave the cycle in four oxidation reactions. Two molecules of NAD+ are reduced in the oxidative decarboxylations of isocitrate and α-ketoglutarate, one molecule of FAD is reduced in the oxidation of succinate, and one molecule of NAD+ is reduced in the oxidation of malate.
- One compound with high phosphoryl transfer potential, usually GTP (guanosine triphosphate), is generated from the cleavage of the thioester linkage in succinyl CoA.
- Two molecules of water are consumed: one in the synthesis of citrate by the hydrolysis of citryl CoA and the other in the hydration of fumarate.
The net reaction of the citric acid cycle is: Acetyl CoA + 3NAD+ + FAD + GDP + Pi + 2H2O –> 2 CO2 + 3 NADH + FADH2 + GTP + 2 H+ + CoA
In a single turn of the citric acid cycle, two carbons enter from acetyl CoA and two molecules of carbon dioxide (CO2) are released; three molecules of NADH and one molecule of FADH2 are generated; and one molecule of ATP (adenosine triphosphate) or GTP (guanosine triphosphate) is produced.
GTP is similar to ATP, both serve as energy sources, and the two can be readily interconverted. Which of the two molecules is produced during the citric acid cycle depends on the organism and cell type. For example, ATP is made in human heart cells, but GTP is made in liver cells.
Where does oxaloacetate come from?
Oxaloacetate or oxaloacetic acid is central to metabolism. It is an intermediate of both gluconeogenesis and the citric acid cycle, and is found in every cell in your body. Oxalacetic acid can be found primarily in cellular cytoplasm, cerebrospinal fluid (CSF), and urine, as well as in human liver tissue.
Oxaloacetate forms in several ways in nature. A principal route is upon oxidation of malate, catalyzed by malate dehydrogenase, in the citric acid cycle (see Figure 1 above). Malate is also oxidized by succinate dehydrogenase in a slow reaction with the initial product being enol-oxaloacetate 16. Oxaloacetate also arises from the condensation of pyruvate with carbonic acid, driven by the hydrolysis of ATP. Oxaloacetate can also arise from trans- or de- amination of aspartic acid.
Oxaloacetate food sources
Oxalacetic acid can be found in a number of food items such as daikon radish, sacred lotus, cucurbita (gourd) and tarragon, which makes oxalacetic acid a potential biomarker for the consumption of these food products.
Oxaloacetate function
Oxaloacetate is an intermediate of the citric acid cycle, where it reacts with acetyl-CoA to form citrate, catalyzed by citrate synthase. It is also involved in gluconeogenesis, the urea cycle, the glyoxylate cycle, amino acid synthesis, and fatty acid synthesis. Oxaloacetate is also a potent inhibitor of complex II.
Oxaloacetate is required for the first step of gluconeogenesis, which produces fuel for the body during fasting. Certain tissues such as the heart and brain rely on the glucose produced by gluconeogenesis and without that glucose they are severely affected. One of the breakdown products of oxaloacetate is aspartic acid, which is required for the urea cycle. Reduced levels of aspartic acid leads to an increase in serum ammonia levels. The prognosis for patients with reduced levels of oxaloacetate is bleak, as progressive neurologic deterioration eventually leads to death within the first six months of life. Thus, a lack of oxaloacetate is not a preferred or healthy state 17.
Breakdown products of oxaloacetate in the body are well documented and include pyruvate, aspartic acid and malate, all of which are currently sold as or in, dietary supplements. In nature, there are many biochemical reactions that involve oxaloacetate, which include gluconeogenesis, the glyoxylate cycle, glyoxylate degradation, mixed acid fermentation, anaerobic respiration, aerobic respiration (Krebs cycle), aspartate biosynthesis and degradation and glutamate degradation.
Gluconeogenesis
Gluconeogenesis is an anabolic pathway that synthesizes glucose from nonglucose precursors (lactate, amino acids, and glycerol). Gluconeogenesis is the metabolic pathway consisting of 11 enzyme-catalyzed reactions by which D-glucose is generated from non-carbohydrate substrates 18. This metabolic pathway is important because the brain depends on glucose as its primary fuel and red blood cells use only glucose as a fuel. The daily glucose requirement of the brain in a typical adult human being is about 120 g, which accounts for most of the 160 g of glucose needed daily by the whole body 19. The amount of glucose present in body fluids is about 20 g and that readily available from glycogen, a storage form of glucose, is approximately 190 g. Thus, the direct glucose reserves are sufficient to meet glucose needs for about a day. During a longer period of starvation, glucose must be formed from noncarbohydrate sources.
The major site of gluconeogenesis is the liver, with a small amount also taking place in the kidney 19. Little gluconeogenesis takes place in the brain, skeletal muscle or heart muscle. Rather, gluconeogenesis in the liver and kidney helps to maintain the glucose level in your blood so that your brain and muscle can extract sufficient glucose from it to meet their metabolic demands.
The beginning of gluconeogenesis takes place in the mitochondrial matrix, where pyruvate molecules are found. A pyruvate molecule is carboxylated by a pyruvate carboxylase enzyme, activated by a molecule each of ATP and water. This reaction results in the formation of oxaloacetate. NADH reduces oxaloacetate to malate. This transformation is needed to transport the molecule out of the mitochondria. Once in the cytosol, malate is oxidized to oxaloacetate again using NAD+. Then oxaloacetate remains in the cytosol, where the rest of reactions will take place. Oxaloacetate is later decarboxylated and phosphorylated by phosphoenolpyruvate carboxykinase and becomes 2-phosphoenolpyruvate using guanosine triphosphate (GTP) as phosphate source. Glucose is obtained after further downstream processing.
Urea cycle
The urea cycle is a metabolic pathway that results in the formation of urea using two ammonium molecules and one bicarbonate molecule 18. This route commonly occurs in hepatocytes. The reactions related to the urea cycle produce NADH, and NADH can be produced in two different ways. One of these uses oxaloacetate. In the cytosol there are fumarate molecules. Fumarate can be transformed into malate by the actions of the enzyme fumarase. Malate is acted on by malate dehydrogenase to become oxaloacetate, producing a molecule of NADH. After that, oxaloacetate will be recycled to aspartate, as transaminases prefer these keto acids over the others. This recycling maintains the flow of nitrogen into the cell.
Amino acid synthesis
The amino acids that must be supplied in the diet are called essential amino acids (histidine, isoleucine, leucine, lysine, methionine, phenylalanine and threonine), whereas the others are termed nonessential amino acids. Six essential amino acids and three non-essential are synthesized from oxaloacetate and pyruvate 20. The nonessential amino acids are synthesized by quite simple reactions, whereas the pathways for the formation of the essential amino acids are quite complex. For example, the nonessential amino acids alanine and aspartate are synthesized in a single step from pyruvate and oxaloacetate, respectively, by transamination from glutamate 20. In contrast, the pathways for the essential amino acids require from 5 to 16 steps. Asparagine, methionine, lysine and threonine are synthesized by aspartate, therefore given importance to oxaloacetate as without it, no aspartate would be formed and the following other amino acids would neither be produced.
Fatty acid synthesis
Fatty acid synthesis starts with the carboxylation of acetyl CoA to malonyl CoA. This irreversible reaction is the committed step in fatty acid synthesis. Fatty acids are synthesized in the cytosol, whereas acetyl CoA is formed from pyruvate in mitochondria. Hence, acetyl CoA must be transferred from mitochondria to the cytosol. Mitochondria, however, are not readily permeable to acetyl CoA. The barrier to acetyl CoA is bypassed by citrate, which carries acetyl groups across the inner mitochondrial membrane. Citrate is formed in the mitochondrial matrix by the condensation of acetyl CoA with oxaloacetate. When present at high levels, citrate is transported to the cytosol, where it is cleaved by ATP-citrate lyase. Thus, acetyl CoA and oxaloacetate are transferred from mitochondria to the cytosol at the expense of the hydrolysis of a molecule of ATP.
Oxaloacetate formed in the transfer of acetyl groups to the cytosol must now be returned to the mitochondria. The inner mitochondrial membrane is impermeable to oxaloacetate. Hence, a series of bypass reactions are needed. Most important, these reactions generate much of the NADPH needed for fatty acid synthesis. First, oxaloacetate is reduced to malate by NADH. This reaction is catalyzed by a malate dehydrogenase in the cytosol. Second, malate is oxidatively decarboxylated by an NADP+-linked malate enzyme (also called malic enzyme). The pyruvate formed in this reaction readily enters mitochondria, where it is carboxylated to oxaloacetate by pyruvate carboxylase. Thus, one molecule of NADPH is generated for each molecule of acetyl CoA that is transferred from mitochondria to the cytosol.
Oxaloacetate benefits
There is little evidence that oxaloacetate will be beneficial for most diseases except possibly stroke or traumatic brain injury. There is little evidence that oxaloacetate will be beneficial for Alzheimer’s disease, except possibly at high doses. In a recent preclinical study, oxaloacetate in capsule form was administered to patients for the treatment of Alzheimer’s disease. The capsules contained 100 mg oxaloacetate and 150 mg of ascorbic acid. In addition to oxaloacetate’s proposed use in the treatment of Alzheimer’s disease and diabetes, recent preclinical research has also tested oxaloacetate which may be used for therapy of traumatic brain injury, stroke, and amyotrophic lateral sclerosis (ALS) 21, 22.
From the perspective of Alzheimer’s disease, there are two potential mechanisms by which oxaloacetate may be beneficial. As an intermediate of the citric acid cycle (Krebs cycle), it may increase mitochondrial respiration. Additionally, it may reduce glutamate toxicity through glutamate oxaloacetate transaminase 3. Glutamate oxaloacetate transaminase converts glutamate and oxaloacetate to L-aspartate and alpha-ketoglutarate. Therefore, by administering oxaloacetate, glutamate is reduced in the periphery and this reduction acts as a glutamate ‘sink’, pulling excess glutamate from the brain and preventing glutamate toxicity 3.
Oxaloacetate reportedly reduces hyperglycemia in type 2 diabetes 5 and extends longevity in Caenorhabditis elegans (roundworm) 8. Williams et al study in Caenorhabditis elegans worms suggested that oxaloacetate increased median lifespan by 25% and maximal lifespan by 13%. These increases required signaling through AMPK-FOXO 8. However, despite that Caenorhabditis elegans (roundworm) lifespan study 8, there is little evidence suggesting that oxaloacetate will be beneficial for age-related diseases. Oxaloacetate was also tested in the National Institute on Aging Interventions Testing Program and did not increase lifespan in mice 23. Oxaloacetate was ground into chow at an intended concentration of 2200 ppm. Oxaloacetate broke down in the food, and at different time points it was estimated the food did not contain more than 250-560 ppm of oxaloacetate during the lifespan study. Therefore, due to the steep reductions in expected dosing, conclusions cannot be drawn from this experiment 23.
Because oxaloacetate participates in the deamination of glutamate to alpha-ketoglutarate, some have tested oxaloacetate in seizure, stroke and traumatic brain injury rodent models and found therapeutic benefits 24. Wilkins et al 25 reported that two-week treatment with oxaloacetate increased markers of mitochondrial biogenesis (p-AMPK, PGC1αmRNA and nuclear translocation). There were no changes in cytochrome oxidase subunit 2 (COX2) or silent information regulator of transcription 1 (SIRT1); however, markers of inflammation (NF-kBand CCL11) decreased while neurogenesis increased. Oxaloacetate also increased phosphorylated mechanistic target of rapamycin (p-mTOR) and p-CREB. In vitro, oxaloacetate increased the NAD/NADH ratio and SIRT1 expression 26. In a rat model where either amyloid or TNFα were injected into the brain, peripheral treatment of oxaloacetate prevented the reduction of long-term potentiation 27. In rodent models of hemorrhagic and ischemic stroke, oxaloacetate improved neurologic performance, blood brain barrier integrity, long-term potentiation, and reduced lesion size 28. The hypothesized mechanism for neuroprotection using oxaloacetate is to reduce glutamate neurotoxicity through glutamate oxaloacetate transaminase (GOT). GOT converts glutamate and oxaloacetate to L-aspartate and alpha-ketoglutarate. Therefore, by administering oxaloacetate, glutamate is reduced in the periphery and this reduction acts as a glutamate ‘sink’, pulling excess glutamate from the brain.
Dementia
Swerdlow et al 29 treated 6 Alzheimer’s disease patients with 100mg of oxaloacetate twice per day for 28 days. On day one there was a transient (~30 minute) increase in plasma oxaloacetate levels with no increase 28 days later. The authors concluded that 100mg oxaloacetate twice per day was too low a dose to see an increase over background serum levels of oxaloacetate 29. The Trial of Oxaloacetate in Alzheimer’s Disease (TOAD) trial is an open label trial comparing the effects of 500mg oxaloacetate twice per day to 1000mg oxaloacetate twice per day in 21 Alzheimer’s disease patients over 28 days 30. An interim analysis found a greater increase in hippocampal fluorodeoxyglucose positron emission tomography (FDG PET) signal in the oxaloacetate 1000 mg twice per day group compared to the 500mg twice per day group from baseline (2.5% vs. 0.3% increase for the 1000 mg and 500 mg groups, respectively) 30. Over the course of the treatment month, there was an insignificant trend towards lower regional FDG PET‐determined glucose uptake in the 500 mg twice per day group and an insignificant trend towards higher regional FDG PET‐determined glucose uptake in the 1000 mg twice per day group. These trends were consistent across multiple regions, and in the default mode network, comparing the 500 to the 1000 mg twice per day dose showed a significant benefit for the 1000 mg twice per day dose 30. The oxaloacetate 1000 mg twice per day subjects, but not the 500 mg twice per day subjects, showed an increase in brain reduced glutathione (GSH) in the parietal and frontal‐parietal regions 30. The authors did not observe consistent changes in blood levels, which may reflect relatively high baseline concentrations of plasma oxaloacetate. Over a 1 month treatment period, 1000 mg twice per day oxaloacetate appears to engage brain energy metabolism. However, cognitive scores did not improve in either group 30. 1000 mg twice per day oxaloacetate, taken over 1 month in persons with Alzheimer’s disease, is safe and well‐tolerated.
Parkinson’s disease
One randomized controlled trial tested the effects of 100mg oxaloacetate plus 100mg ascorbic acid (vitamin C) per day versus placebo capsules that contain only 100 mg ascorbic acid (vitamin C), taken daily in 33 Parkinson’s disease patients over 4 months 31. There were no significant differences in any outcomes 31.
Cancer
One study reported that oxaloacetate reduced tumor size in glioma-implanted rodents and increased survival, especially when co-administered with a glioma chemotherapeutic 32. Similar to the neuroprotective studies above, the benefits of oxaloacetate treatment were thought to be due to a reduction in peripheral glutamate. Oxaloacetic acid supplementation has been shown to eliminate the ability of some cancer cell types to reproduce, while not affecting normal tissues 33. Micromolar levels of oxaloacetate in contact with human A549 lung cancer tissue in vitro resulted in differential massive debris within the cancer tissue, but not within normal tissue. The massive debris resulted in the inability of the cancer tissue to reproduce, even if the cancer tissue was moved away from the oxaloacetate solution for a period of six weeks 33. Interestingly, the herb Euonymus alatus (“winged Euonymus” or “burning bush”), the same herb from which oxaloacetic acid was extracted and identified for its anti-diabetic activity, also pre-vents the spread of some cancer types with low cytotoxicity 34. The mechanism of action appears to be inhibitory effects on matrix metalloproteinase (MMP)-9, which may be involved in tumor metastasis 34. Halting the cellular reproduction of human lung cancer tissues with low levels of oxaloacetate in vitro is an important finding, but one that needs to be verified in vivo.
Diabetes
One open label study suggested that oxaloacetate was effective in most patients with type 1 and type 2 diabetes 5. The Yoshikawa paper is from 1968 and the quality of the data is low (no specific outcome measures of oxaloacetate’s effects, just whether it was effective or ineffective) 5. Yoshikawa 5 showed that 100 to 1,000 mg of the sodium salt of oxaloacetic acid is effective in lowing the blood and urine glucose levels of diabetic patients. Yoshikawa initially investigated oxaloacetic acid for diabetes treatment after identifying the compound as the active ingredient extracted from the Asian mountain shrub “Euonymus alata” (“winged Euonymus” or “burning bush”), a traditional herb used for hundreds of years and still in use today to treat diabetes in Asian countries 35. Yoshikawa showed that oral consumption of sodium oxaloacetate entered the bloodstream within one hour of consumption and lowered fasting glucose levels in blood and urine to normal levels in the majority of diabetic patients without any noted side effects 5. In animal studies, Yoshikawa showed that sodium oxaloacetate increased the uptake of glucose by tissues 300% in diabetic animals, and 180% in normal animals.
Oxaloacetate dosage
Oxaloacetate is unstable and a thermally stabilized drug should be used. The ongoing Trial of Oxaloacetate in Alzheimer’s Disease study is using 500 or 1000 mg oxaloacetate 30.
Oxaloacetate supplement side effects
Oxaloacetate is probably safe, but there are few human studies. One small study in Parkinson’s disease patients suggested that oxaloacetate increased the risk of gastrointestinal symptoms and possible worsening of Parkinson’s symptoms and insomnia. However, the study is too small to draw conclusions 31. One small, open-label study reported no side effects, though the data was poor 5.
Drug interactions
There is little clinical work completed and drug interactions are unknown.
- Cash, A. (2009). Oxaloacetic Acid Supplementation as a Mimic of Calorie Restriction. Open Longevity Science (formerly ‘the Open Aging Journal’), 3, 22-27. https://benthamopen.com/contents/pdf/TOLSJ/TOLSJ-3-22.pdf[↩][↩]
- Campos F, Sobrino T, Ramos-Cabrer P, Castillo J. Oxaloacetate: a novel neuroprotective for acute ischemic stroke. Int J Biochem Cell Biol. 2012 Feb;44(2):262-5. doi: 10.1016/j.biocel.2011.11.003[↩]
- Zlotnik A., Sinelnikov I., Gruenbaum B.F., Gruenbaum S.E., Dubilet M., Dubilet E., Leibowitz A., Ohayon S., Regev A., Boyko M., et al. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. Anesthesiology. 2012;116:73–83. doi: 10.1097/ALN.0b013e31823d7731[↩][↩][↩]
- Campos F., Sobrino T., Ramos-Cabrer P., Castillo J. Oxaloacetate: A novel neuroprotective for acute ischemic stroke. Int. J. Biochem. Cell Biol. 2012;44:262–265. doi: 10.1016/j.biocel.2011.11.003[↩]
- Yoshikawa K. Studies on the anti-diabetic effect of sodium oxaloacetate. Tohoku J Exp Med. 1968 Oct;96(2):127-41. doi: 10.1620/tjem.96.127[↩][↩][↩][↩][↩][↩][↩]
- Yamamoto HA, Mohanan PV. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol Lett 2003; 143: 115-22.[↩]
- Navarro CL, Cau P, Levy N. Molecular bases of progeroid syndromes. Hum Mol Genet 2006; 15 Spec No 2: R151-61.[↩]
- Williams, D. S., Cash, A., Hamadani, L., & Diemer, T. (2009). Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging cell, 8(6), 765–768. https://doi.org/10.1111/j.1474-9726.2009.00527.x[↩][↩][↩][↩]
- Wood JP, Osborne NN. Zinc and energy requirements in induction of oxidative stress to retinal pigmented epithelial cells. Neurochem Res 2003; 28: 1525-33.[↩]
- Friedman DS, O’Colmain BJ, Munoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol 2004; 122: 564-72.[↩]
- Desagher S, Glowinski J, Premont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci 1997; 17: 9060-7.[↩]
- Puntel RL, Roos DH, Grotto D, Garcia SC, Nogueira CW, Rocha JB. Antioxidant properties of Krebs cycle intermediates against malonate pro-oxidant activity in vitro: a comparative study using the colorimetric method and HPLC analysis to determine malondialdehyde in rat brain homogenates. Life Sci 2007; 81: 51-62.[↩]
- Willcox BJ, Curb JD, Rodriguez BL. Antioxidants in cardiovascular health and disease: key lessons from epidemiologic studies. Am J Cardiol 2008; 101: 75D-86D.[↩]
- Haslam JM, Krebs HA. The permeability of mitochondria to oxaloacetate and malate. Biochem J 1968; 107: 659-67.[↩]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition. New York: W H Freeman; 2002. Section 17.1, The Citric Acid Cycle Oxidizes Two-Carbon Units. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22427[↩]
- Panchenko, M.V. and Vinogradov, A.D.(1991), Direct demonstration of enol-oxaloacetate as an immediate product of malate oxidation by the mammalian succinate dehydrogenase, FEBS Letters, 286, doi: 10.1016/0014-5793(91)80944-X[↩]
- Ahmad A, Kahler SG, Kishnani PS, et al. Treatment of pyruvate carboxylase deficiency with high doses of citrate and aspartate. Am J Med Genet 1999; 87: 331-8.[↩]
- Nelson, David L.; Cox, Michael M. (2005). Principles of Biochemistry (4th ed.). New York: W. H. Freeman. ISBN 0-7167-4339-6.[↩][↩]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition. New York: W H Freeman; 2002. Section 16.3, Glucose Can Be Synthesized from Noncarbohydrate Precursors. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22591[↩][↩]
- Berg JM, Tymoczko JL, Stryer L. Biochemistry. 5th edition. New York: W H Freeman; 2002. Section 24.2, Amino Acids Are Made from Intermediates of the Citric Acid Cycle and Other Major Pathways. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22459[↩][↩]
- Ruban A., Berkutzki T., Cooper I., Mohar B., Teichberg V.I. Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Investig. New Drugs. 2012;30:2226–2235. doi: 10.1007/s10637-012-9794-x[↩]
- Ruban A., Cohen-Kashi Malina K., Cooper I., Graubardt N., Babakin L., Jona G., Teichberg V.I. Combined Treatment of an Amyotrophic Lateral Sclerosis Rat Model with Recombinant GOT1 and Oxaloacetic Acid: A Novel Neuroprotective Treatment. Neurodegener. Dis. 2015;15:233–242. doi: 10.1159/000382034[↩]
- Strong, R., Miller, R. A., Astle, C. M., Baur, J. A., de Cabo, R., Fernandez, E., Guo, W., Javors, M., Kirkland, J. L., Nelson, J. F., Sinclair, D. A., Teter, B., Williams, D., Zaveri, N., Nadon, N. L., & Harrison, D. E. (2013). Evaluation of resveratrol, green tea extract, curcumin, oxaloacetic acid, and medium-chain triglyceride oil on life span of genetically heterogeneous mice. The journals of gerontology. Series A, Biological sciences and medical sciences, 68(1), 6–16. https://doi.org/10.1093/gerona/gls070[↩][↩]
- Campos, F., Sobrino, T., Ramos-Cabrer, P., Argibay, B., Agulla, J., Pérez-Mato, M., Rodríguez-González, R., Brea, D., & Castillo, J. (2011). Neuroprotection by glutamate oxaloacetate transaminase in ischemic stroke: an experimental study. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism, 31(6), 1378–1386. https://doi.org/10.1038/jcbfm.2011.3[↩]
- Wilkins, H. M., Harris, J. L., Carl, S. M., E, L., Lu, J., Eva Selfridge, J., Roy, N., Hutfles, L., Koppel, S., Morris, J., Burns, J. M., Michaelis, M. L., Michaelis, E. K., Brooks, W. M., & Swerdlow, R. H. (2014). Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Human molecular genetics, 23(24), 6528–6541. https://doi.org/10.1093/hmg/ddu371[↩]
- Wilkins, H. M., Koppel, S., Carl, S. M., Ramanujan, S., Weidling, I., Michaelis, M. L., Michaelis, E. K., & Swerdlow, R. H. (2016). Oxaloacetate enhances neuronal cell bioenergetic fluxes and infrastructure. Journal of neurochemistry, 137(1), 76–87. https://doi.org/10.1111/jnc.13545[↩]
- Zhang, D., Mably, A. J., Walsh, D. M., & Rowan, M. J. (2017). Peripheral Interventions Enhancing Brain Glutamate Homeostasis Relieve Amyloid β- and TNFα- Mediated Synaptic Plasticity Disruption in the Rat Hippocampus. Cerebral cortex (New York, N.Y. : 1991), 27(7), 3724–3735. https://doi.org/10.1093/cercor/bhw193[↩]
- Boyko, M., Melamed, I., Gruenbaum, B. F., Gruenbaum, S. E., Ohayon, S., Leibowitz, A., Brotfain, E., Shapira, Y., & Zlotnik, A. (2012). The effect of blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome in a rat model of subarachnoid hemorrhage. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics, 9(3), 649–657. https://doi.org/10.1007/s13311-012-0129-6[↩]
- Swerdlow, R. H., Bothwell, R., Hutfles, L., Burns, J. M., & Reed, G. A. (2016). Tolerability and pharmacokinetics of oxaloacetate 100 mg capsules in Alzheimer’s subjects. BBA clinical, 5, 120–123. https://doi.org/10.1016/j.bbacli.2016.03.005[↩][↩]
- Vidoni, E.D., Choi, I., Lee, P., Clutton, J., Becker, A.M., Sherry, E., Bothwell, R., Anderson, H., Mahnken, J.D., Wilkins, H.M., Brooks, W., Reed, G., Burns, J.M. and Swerdlow, R.H. (2019), P3‐008: TRIAL OF OXALOACETATE IN ALZHEIMER’S DISEASE (TOAD): FINAL RESULTS. Alzheimer’s & Dementia, 15: P927-P927. https://doi.org/10.1016/j.jalz.2019.06.3034[↩][↩][↩][↩][↩][↩]
- A Pilot Study of Oxaloacetate in Subjects With Treated Parkinson’s disease. https://clinicaltrials.gov/ct2/show/results/NCT01741701[↩][↩][↩]
- Ruban, A., Berkutzki, T., Cooper, I., Mohar, B., & Teichberg, V. I. (2012). Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Investigational new drugs, 30(6), 2226–2235. https://doi.org/10.1007/s10637-012-9799-5[↩]
- Farah IO. Differential modulation of intracellular energetics in A549 and MRC-5 cells. Biomed Sci Instrum 2007; 43: 110-5.[↩][↩]
- Cha BY, Park CJ, Lee DG, et al. Inhibitory effect of methanol extract of Euonymus alatus on matrix metalloproteinase-9. J Ethnopharmacol 2003; 85: 163-7. [↩][↩]
- Fang XK, Gao Y, Yang HY, et al. Alleviating effects of active fraction of Euonymus alatus abundant in flavonoids on diabetic mice. Am J Chin Med 2008; 36: 125-40.[↩]





{kind=link}