Pelger-Huet anomaly
Pelger-Huet anomaly is an inherited blood condition in which the nuclei of several types of white blood cells (neutrophils and eosinophils) have unusual shape (bilobed, peanut or dumbbell-shaped instead of the normal trilobed shape) and unusual structure (coarse and lumpy). Pelger-Huet anomaly is considered to be a benign disorder in most instances, as individuals with Pelger-Huet anomaly are typically healthy 1. Pelger-Huet anomaly is caused by mutations in the lamin B receptor (LBR) gene. It is suspected that mutations within the LBR gene are responsible for a spectrum of disorders including isolated Pelger-Huet anomaly; Pelger-Huet anomaly with mild skeletal symptoms; and hydrops-ectopic calcification-moth-eaten skeletal dysplasia (HEM dysplasia or Greenberg skeletal dysplasia). Pelger-Huet anomaly was previously thought to be inherited in an autosomal dominant manner; however, co-dominant inheritance has been suggested as well 2. It is important to distinguish Pelger-Huet anomaly from acquired or pseudo-Pelger-Huet anomaly, which may be found in individuals with certain types of leukemia or myelodysplastic syndromes. Diagnosis is made based on characteristic appearance of white blood cell nuclei identified by a blood smear. Most individuals with Pelger-Huet anomaly do not require treatment as they do not have symptoms 1.
Pelger-Huet anomaly prevalence rate in the United States is estimated as 1 case in 5000 population. The prevalence rate of heterozygous Pelger-Huet anomaly is 1 case in 6000 population in the United Kingdom. The highest described incidence is in the Gelenau region of Germany (1.01%) and the Vasterbotten region of Sweden (0.6%) 3.
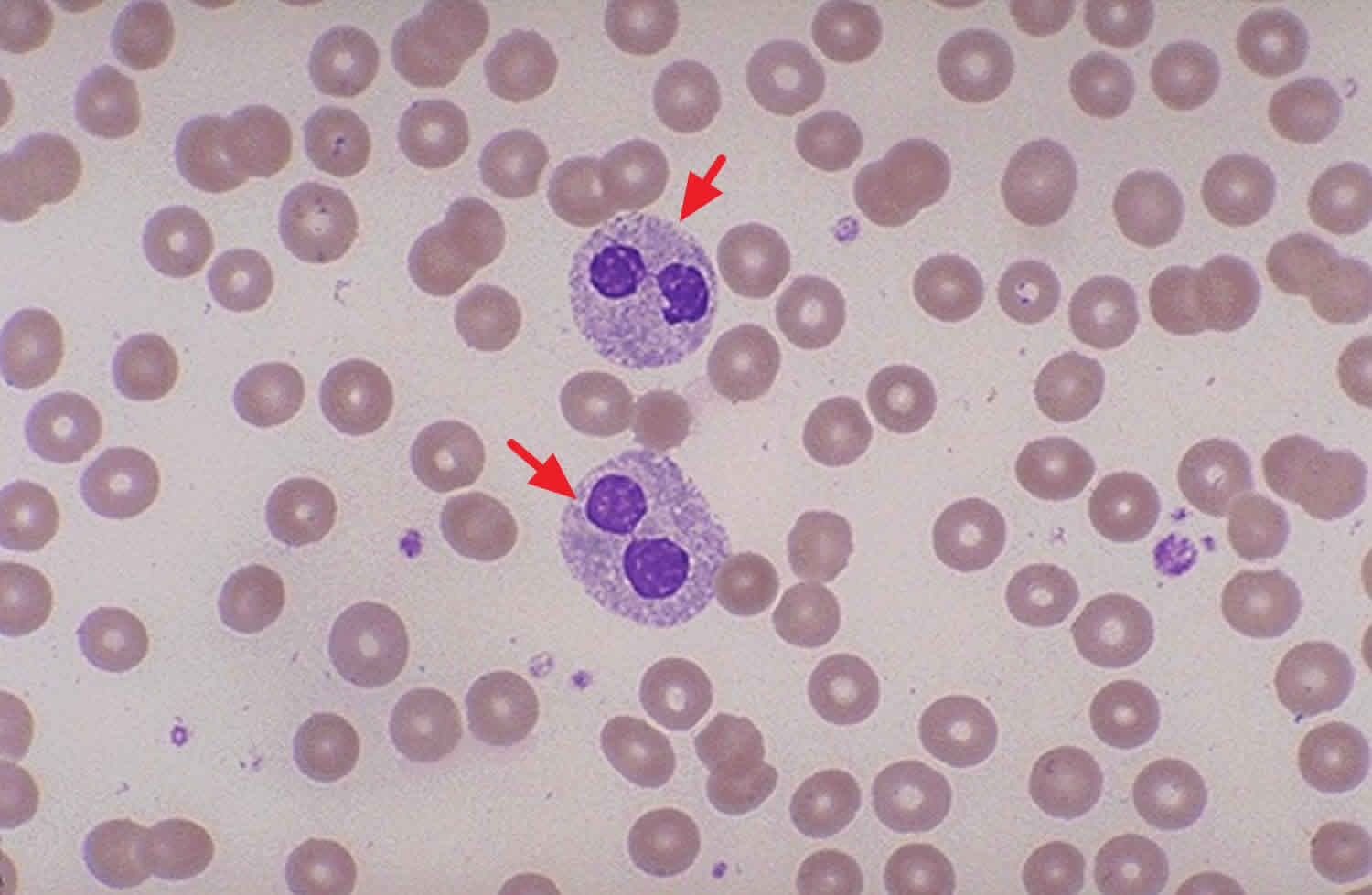
Figure 1. Pelger-Huet anomaly blood smear test
Pelger-Huet anomaly causes
Pelger-Huet anomaly is caused by a mutation of the lamin B receptor (LBR) gene located on subband 1q42.1 4. Pelger-Huet anomaly is inherited in a highly penetrant, autosomal dominant pattern.
The LBR gene product is essential for maintaining nuclear membrane structure, and heterozygotes have at least half of their neutrophils with bilobed, dumbbell-shaped nuclei, also described as pince-nez (ie, looking like pinched-nose spectacles) 5.
Lamin B receptor (LBR) also interacts with HP-1 heterochromatin proteins; this is hypothesized to account for the excessive coarse clumping of nuclear chromatin that is observed 6. Lamin B receptor (LBR) abnormalities do not affect neutrophil function, and Pelger-Huet cells survive normally in circulation and can phagocytize and kill microorganisms 7.
Research also suggests that Pelger-Huet anomaly and HEM skeletal dysplasia (Greenberg dysplasia) are related to a defect in cholesterol synthesis, resulting from LBR point mutations that cause a loss of sterol C14 reductase activity associated with the LBR protein. This loss occurs when the point mutations lower LBR’s affinity for the reducing agent nicotinamide adenine dinucleotide phosphate (NADPH) 8.
Homozygous LBR mutations are rare, with only 11 individuals described; neutrophils have a single, round nucleus with clumped chromatin, and basophils, eosinophils, and megakaryocytes also show rounded nuclear lobes and dense nuclear chromatin 9. Co-inherited LBR gene nonsense mutations can also result in lethal hydrops, ectopic calcification, moth-eaten (HEM) skeletal dysplasia/Greenberg skeletal dysplasia, and there continues to be debate as to how Pelger-Huet anomaly and HEM skeletal dysplasia overlap. Some patients with HEM skeletal dysplasia (Greenberg skeletal dysplasia) have neutrophils with features of Pelger-Huet anomaly, and some patients with Pelger-Huet anomaly have mild skeletal anomalies 10.
Pelger-Huet anomaly inheritance pattern
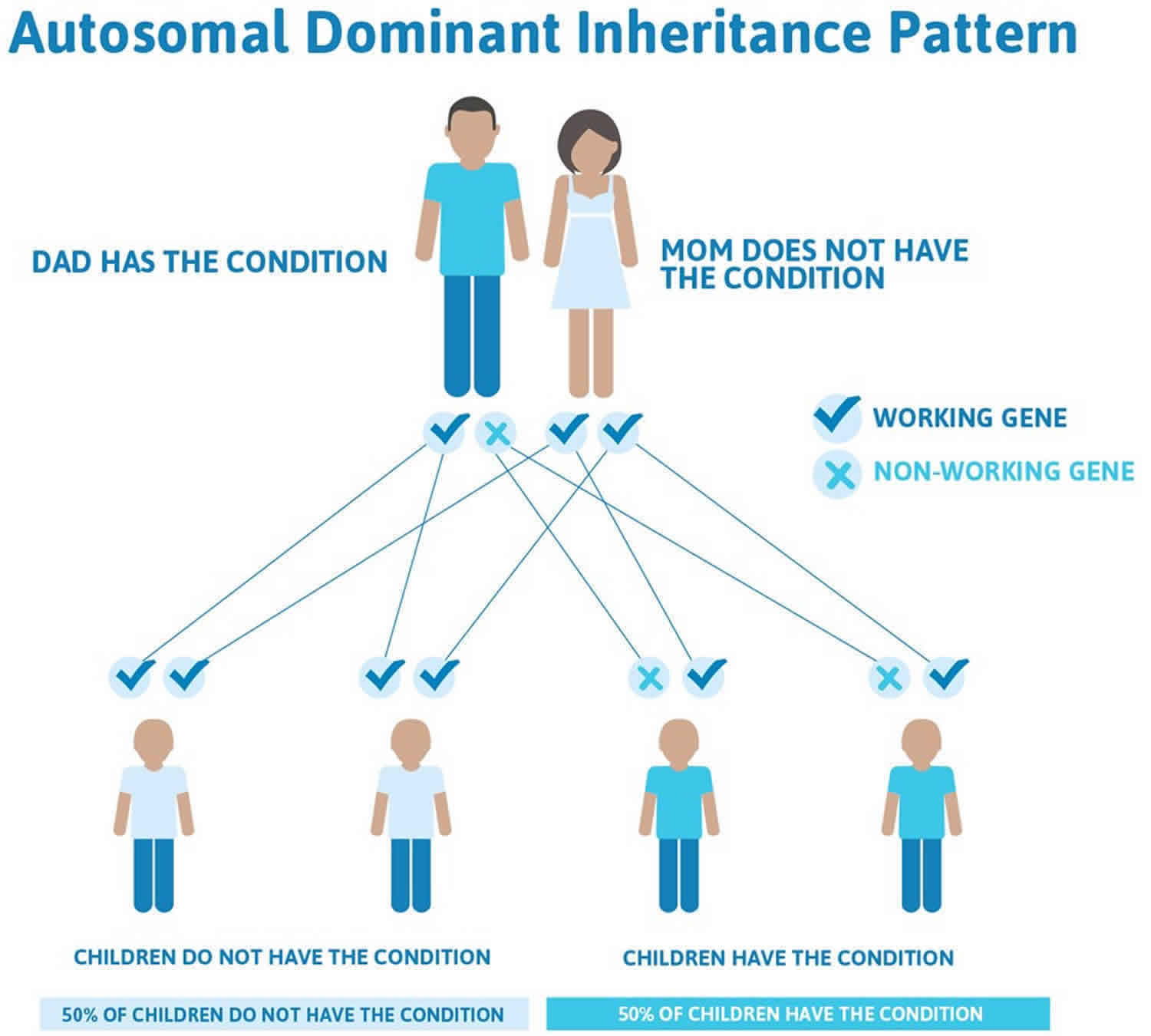
Pelger-Huet anomaly is inherited in a highly penetrant, autosomal dominant pattern. Often autosomal dominant conditions can be seen in multiple generations within the family. If one looks back through their family history they notice their mother, grandfather, aunt/uncle, etc., all had the same condition. In cases where the autosomal dominant condition does run in the family, the chance for an affected person to have a child with the same condition is 50% regardless of whether it is a boy or a girl. These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
- When one parent has the abnormal gene, they will pass on either their normal gene or their abnormal gene to their child. Each of their children therefore has a 50% (1 in 2) chance of inheriting the changed gene and being affected by the condition.
- There is also a 50% (1 in 2) chance that a child will inherit the normal copy of the gene. If this happens the child will not be affected by the disorder and cannot pass it on to any of his or her children.
There are cases of autosomal dominant gene changes, or mutations, where no one in the family has it before and it appears to be a new thing in the family. This is called a de novo mutation. For the individual with the condition, the chance of their children inheriting it will be 50%. However, other family members are generally not likely to be at increased risk.
Figure 2 illustrates autosomal dominant inheritance. The example below shows what happens when dad has the condition, but the chances of having a child with the condition would be the same if mom had the condition.
Figure 2. Pelger-Huet anomaly autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Pelger-Huet anomaly symptoms
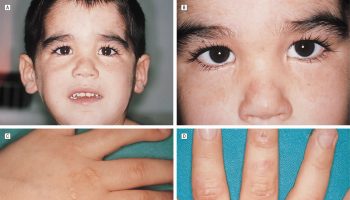
Neutrophil function is normal. Individuals with Pelger-Huet anomaly are in good health, and their natural resistance to infection is unimpaired. Individuals with the rare homozygous LBR mutations, Pelger-Huët anomaly may have skeletal anomalies 10. Affected individuals have extremely short bones in the arms and legs and abnormally flat vertebrae (platyspondyly). Other skeletal abnormalities may include short ribs and extra fingers (polydactyly). In addition, affected fetuses have extensive swelling of the body caused by fluid accumulation (hydrops fetalis).
Pelger-Huet anomaly diagnosis
Pelger-Huet anomaly is typically diagnosed by completing a type of blood test called a blood smear to examine the appearance of the nuclei of several types of white blood cells, including neutrophils. Normally the nuclei of these cells have a trilobed shape. In Pelger-Huet anomaly, they are bilobed, peanut, or dumb-bell shaped. They might additionally appear to not have any lobes. The structure is additionally abnormal and appear to be coarse or lumpy.
When Pelger-Huet anomaly is suspected, it is important to rule out other acquired causes of Pelger-Huet anomaly (known as pseudo-Pelger-Huet anomaly) such as medications (valproic acid, ibuprofen, docetaxel), chemical ingestion/use (benzene), hematologic disorders (leukemia, Fanconi anemia), and non-hematologic disorders (malaria, flu, lupus) 2. In pseudo–Pelger-Huet anomaly, cells may appear morphologically similar to Pelger-Huet anomaly, but absence of these findings in other family members, a low percentage of affected cells (usually 5-20%), and involvement of other cell lines (e.g., anemia or thrombocytopenia) suggest an acquired anomaly. Pseudo-Pelger-Huet anomaly may be predictive of the clinical onset of myelodysplastic disorders, myeloid leukemias, or myelofibrosis, and bone-marrow aspiration and biopsy may be warranted. A molecular technique that extracts and analyzes the nuclear skeleton can also be used to differentiate Pelger-Huet anomaly from pseudo-Pelger-Huet anomaly with a sensitivity and specificity of over 80% but is not in routine use 11.
Pelger-Huet anomaly treatment
No treatment is needed in individuals with Pelger-Huet anomaly 12. Individuals with Pelger-Huet anomaly have good health, and their natural resistance to infection is unimpaired.
- Pelger-Huet Anomaly. https://emedicine.medscape.com/article/957277-overview[↩][↩]
- Lior Borovik, Peggy Modaff,Hans R. Waterham, Anthony D. Krentz,Richard M. Pauli. Pelger-Huet Anomaly and a Mild Skeletal Phenotype Secondary to Mutations in LBR. American Journal of Medical Genetics. August 2013; 161A(8):2066-2073. http://www.ncbi.nlm.nih.gov/pubmed/23824842[↩][↩]
- Hoffmann K, Dreger CK, Olins AL, et al. Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huët anomaly). Nat Genet. 2002 Aug. 31(4):410-4.[↩]
- Nikolakaki E, Mylonis I, Giannakouros T. Lamin B Receptor: Interplay between Structure, Function and Localization. Cells. 2017 Aug 31. 6 (3).[↩]
- Hoffmann K, Sperling K, Olins AL, Olins DE. The granulocyte nucleus and lamin B receptor: avoiding the ovoid. Chromosoma. 2007 Jun. 116(3):227-35.[↩]
- Lukasova E, Kovarik A, Kozubek S. Consequences of Lamin B1 and Lamin B Receptor Downregulation in Senescence. Cells. 2018 Feb 6. 7 (2).[↩]
- Cohen TV, Klarmann KD, Sakchaisri K, Cooper JP, Kuhns D, Anver M, et al. The lamin B receptor under transcriptional control of C/EBPepsilon is required for morphological but not functional maturation of neutrophils. Hum Mol Genet. 2008 Oct 1. 17(19):2921-33.[↩]
- Turner EM, Schlieker C. Pelger-Huet anomaly and Greenberg skeletal dysplasia: LBR-associated diseases of cholesterol metabolism. Rare Dis. 2016. 4 (1):e1241363.[↩]
- Erice JG, Perez JM, Pericas FS. Homozygous form of the Pelger-Huët anomaly. Haematologica. 1999 Aug. 84(8):748.[↩]
- Borovik L, Modaff P, Waterham HR, Krentz AD, Pauli RM. Pelger-huet anomaly and a mild skeletal phenotype secondary to mutations in LBR. Am J Med Genet A. 2013 Aug. 161A(8):2066-73.[↩][↩]
- Sun M, Yang S, Jiang J, Wang Q. Detection of Pelger-Huet anomaly based on augmented fast marching method and speeded up robust features. Biomed Mater Eng. 2015. 26 Suppl 1:S1241-8.[↩]
- Pelger-Huet Anomaly Treatment & Management. https://emedicine.medscape.com/article/957277-treatment[↩]




{kind=link}