What is retinitis pigmentosa
Retinitis pigmentosa is a group of inherited eye diseases that affect the light-sensitive part of the eye (retina). Retinitis pigmentosa causes cells in the retina to die, causing progressive vision loss. The first sign of retinitis pigmentosa usually is night blindness (nyctalopia), which becomes apparent in childhood. Problems with night vision can make it difficult to navigate in low light. Later, as the condition progresses affected individuals develop blind spots in the side (peripheral) vision. Over time, these blind spots merge to produce tunnel vision. The disease progresses over years or decades to affect central vision, which is needed for detailed tasks such as reading, driving, and recognizing faces. In adulthood, many people with retinitis pigmentosa become legally blind.
The signs and symptoms of retinitis pigmentosa are most often limited to vision loss. When retinitis pigmentosa occurs by itself, it is described as nonsyndromic retinitis pigmentosa. Researchers have identified several major types of nonsyndromic retinitis pigmentosa, which are usually distinguished by their pattern of inheritance: autosomal dominant, autosomal recessive, or X-linked.
Less commonly, retinitis pigmentosa occurs as part of syndromes that affect other organs and tissues in the body. These forms of retinitis pigmentosa are described as syndromic retinitis pigmentosa. The most common form of syndromic retinitis pigmentosa is Usher syndrome 1, which is characterized by the combination of vision loss and hearing loss beginning early in life. Retinitis pigmentosa is also a feature of several other genetic syndromes, including Bardet-Biedl syndrome; Refsum disease; and neuropathy, ataxia, and retinitis pigmentosa.
Retinitis pigmentosa may be caused by mutations in any of at least 60 genes. Association with RPE65 is important as there is genetic therapy available which is effective for young patients 2. Inheritance can be autosomal dominant, autosomal recessive, or X-linked 3. In 10 to 40 percent of all cases of retinitis pigmentosa, only one person in a family is affected. In these families, the disorder is described as simplex. It can be difficult to determine the inheritance pattern of simplex cases because affected individuals may have no affected relatives or may be unaware of other family members with the disease. Simplex cases can also result from a new gene mutation that is not present in other family members 4. Treatment options to slow the progression of vision loss include light avoidance, use of low-vision aids, and vitamin A supplementation. Researchers are working to develop new treatment options for the future such as gene therapy, stem cell transplantation and prosthetic implants 5.
The prevalence of retinitis pigmentosa in the United States and Europe is estimated to be between 1 in 3,500 to 1 in 4,000 individuals 3.
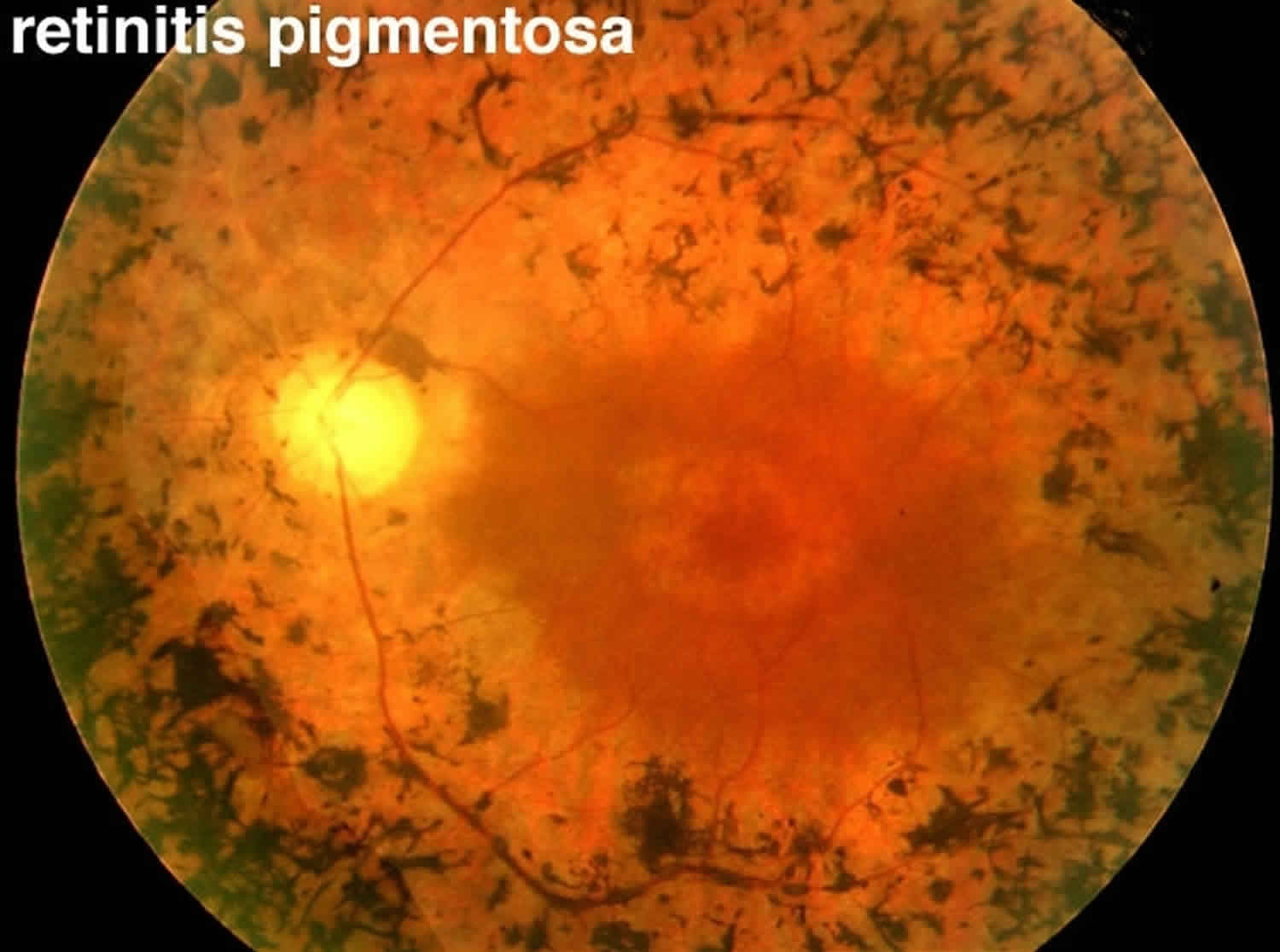
Figure 1. Retinitis pigmentosa
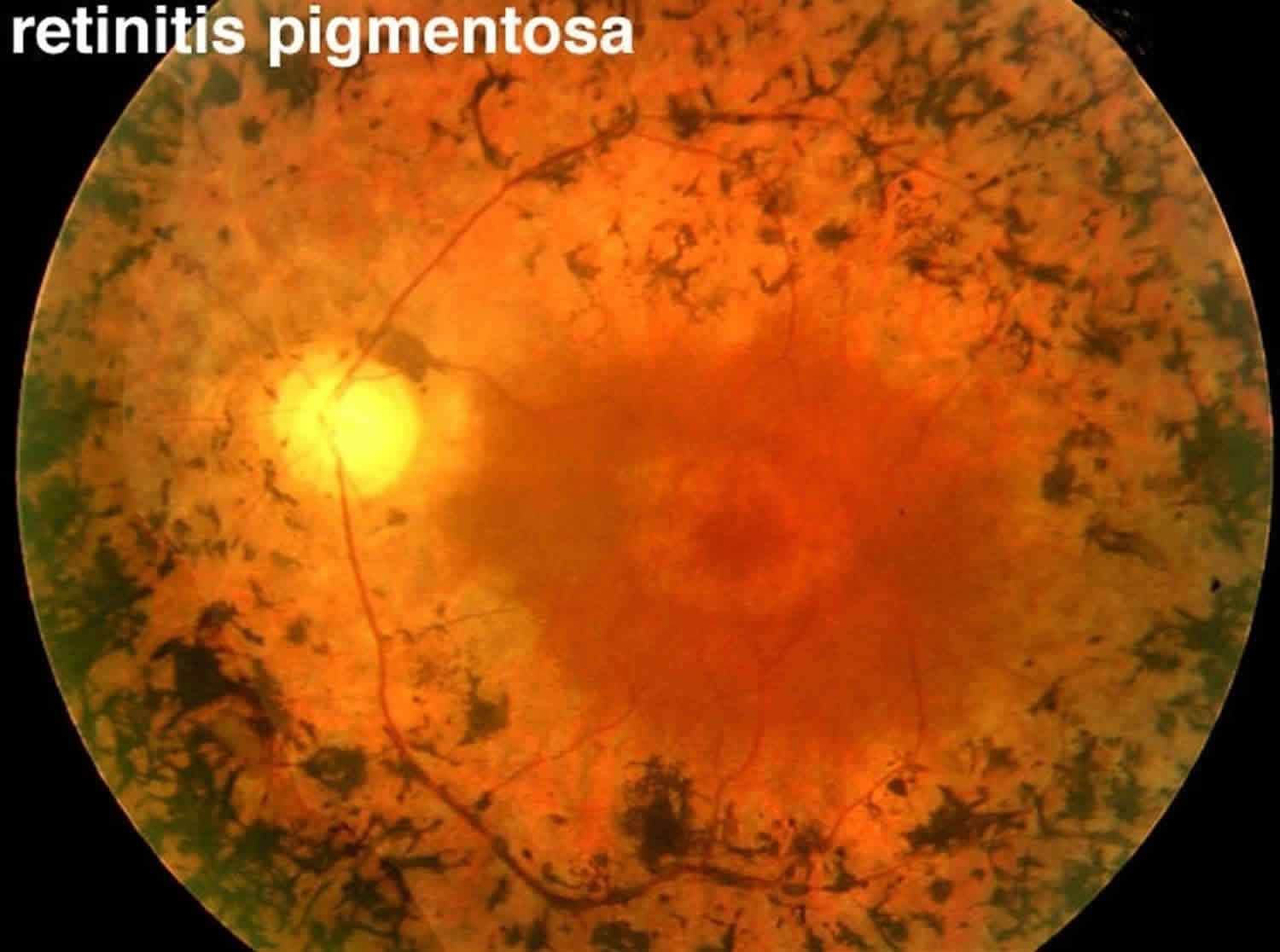
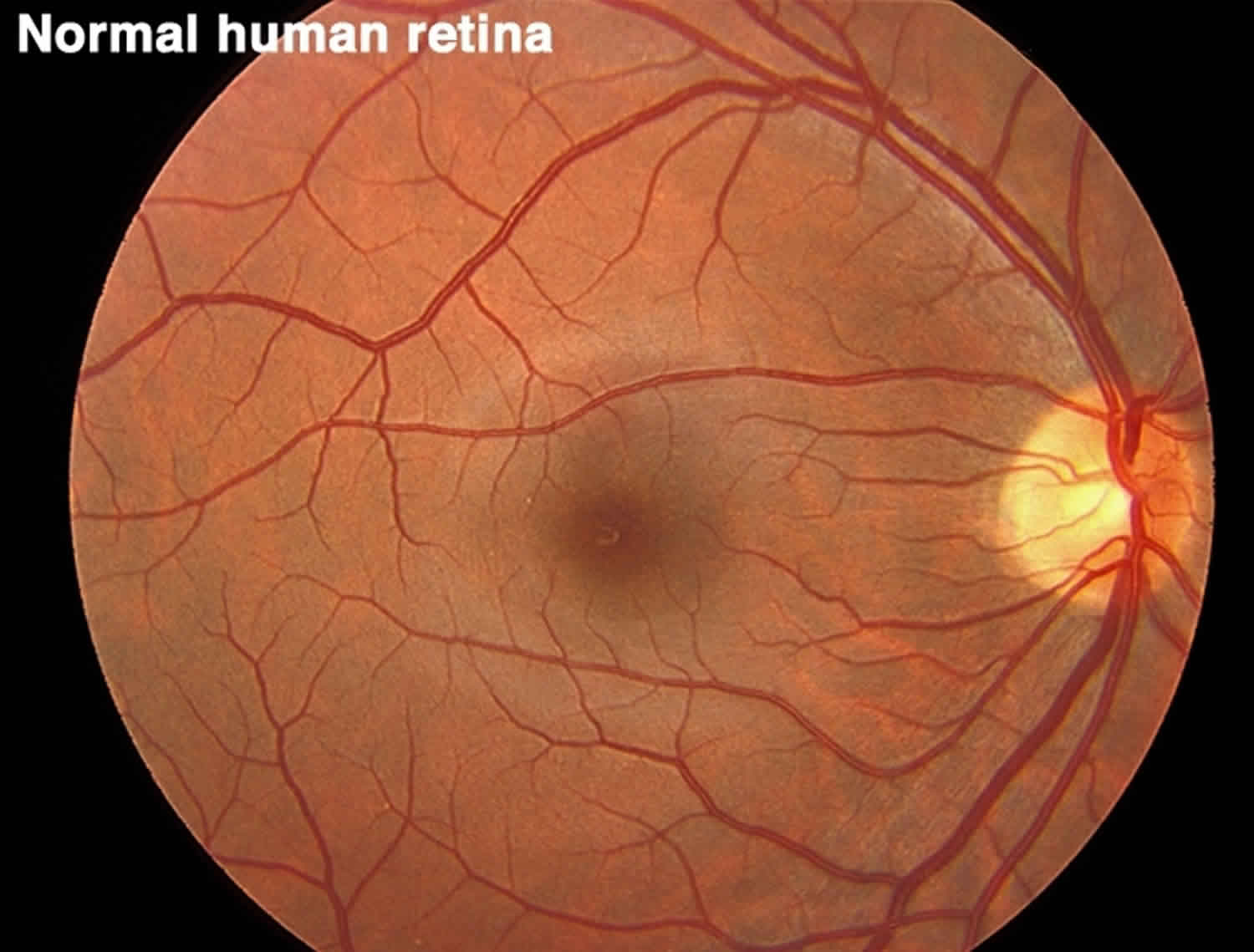
Figure 2. Normal human retina
Can retinitis pigmentosa be cured?
There is currently no cure for retinitis pigmentosa 6. Available treatments aim to slow the progression of the disease and primarily include light avoidance and the use of low-vision aids. Some practitioners also consider vitamin A as a possible treatment option. However, taking too much vitamin A can be toxic and the effects of vitamin A on the disease appear to be relatively weak 7. Studies have explored potential treatment with docosahexaenoic acid (DHA), an omega-3 fatty acid naturally found in fish. While DHA (docosahexaenoic acid) is known to play a structural role in retinal cells, more research is needed to determine whether supplements should be recommended 8.
Current research is focused on the development of new treatments including gene therapy, retinal transplantation, and the use of a retinal prosthesis. Stem cell transplantation would involve the injection and integration of stem cells into the retina, in hopes these cells will replace dead cells and provide the missing enzymes and chemicals needed for sight. Gene therapy could potentially be used when the disease-causing mutation is known and would aim to restore production of the missing or abnormal protein. Studies with retinal prosthetics have tested devices that transform light into electrical signals that can be sent directly to the inner retina and brain, avoiding the diseased part of the outer retina. Though challenges remain, preliminary research into these technologies has been promising 5, 8.
Retinitis pigmentosa causes
Mutations in more than 60 genes are known to cause nonsyndromic retinitis pigmentosa 2. More than 20 of these genes are associated with the autosomal dominant form of the disorder. Mutations in the RHO gene are the most common cause of autosomal dominant retinitis pigmentosa, accounting for 20 to 30 percent of all cases. At least 35 genes have been associated with the autosomal recessive form of the disorder. The most common of these is USH2A; mutations in this gene are responsible for 10 to 15 percent of all cases of autosomal recessive retinitis pigmentosa. Changes in at least six genes are thought to cause the X-linked form of the disorder. Together, mutations in the RPGR and RP2 genes account for most cases of X-linked retinitis pigmentosa.
The genes associated with retinitis pigmentosa play essential roles in the structure and function of specialized light receptor cells (photoreceptors) in the retina. The retina contains two types of photoreceptors, rods and cones. Rods are responsible for vision in low light, while cones provide vision in bright light, including color vision.
Mutations in any of the genes responsible for retinitis pigmentosa lead to a gradual loss of rods and cones in the retina. The progressive degeneration of these cells causes the characteristic pattern of vision loss that occurs in people with retinitis pigmentosa. Rods typically break down before cones, which is why night vision impairment is usually the first sign of the disorder. Daytime vision is disrupted later, as both rods and cones are lost.
Some of the genes associated with retinitis pigmentosa are also associated with other eye diseases, including a condition called cone-rod dystrophy. Cone-rod dystrophy has signs and symptoms similar to those of retinitis pigmentosa. However, cone-rod dystrophy is characterized by deterioration of the cones first, followed by the rods, so daylight and color vision are affected before night vision.
General Pathology
Histopathologic studies suggest that retinitis pigmentosa results from a primary defect in the rod and cone photoreceptors 9. Pathologic findings of an enucleated eye in a patient with autosomal recessive retinitis pigmentosa showed that the rod and cone outer segments were shortened and disorganized in the patient’s best field of vision, while in the area of visual loss; there was total loss of outer segments and a decrease in photoreceptors number 10. Two types of pigmented cells were found invading the retina: typical RPE cells that were migrating away from the retinal pigment epithelial layer, and macrophage-like cells that contained melanin. These changes were thought to be a reactive response to photoreceptor damage, since the RPE appeared relatively normal morphologically in areas of early photoreceptor involvement. A recent review described histopathologic findings in 10 patients with autosomal dominant retinitis pigmentosa, including poorly organized, shortened or absent outer segments with shortened inner segments. Inclusion bodies and/or perinuclear cytoplasmic membranous swirls were found in three cases 11.
Pathophysiology
The pathophysiology of retinitis pigmentosa has been studied in several animal models 12. In the rat, retinal degeneration caused by failure of retinal pigment epithelium to phagocytose the shed rod outer segment discs, leading to accumulation of rod outer segement debris. In mice homozygous recessive for retinal degeneration mutation, rod photoreceptors stop to develop and undergo degeneration before cellular maturation completes. A defect in cGMP-phosphodiesterase, which leads to toxic level of cyclic guanosine monophosphate, has also been documented. This is also found to be true in some autosomal recessive models of the dog. It is unknown whether the defect in these animal retinal degenerations is the pathophysiologic mechanism of human retinitis pigmentosa.
Molecular genetics of retinitis pigmentosa
More than 100 gene loci that cause retinitis pigmentosa have been mapped or identified 13. Genes that cause retinitis pigmentosa can be categorized into those that affect the phototransduction cascade, the retinoid cycle, photoreceptor structure, or other biological function of photoreceptor and retinal pigment epithelium. The most frequent known causes are mutations in the rhodopsin (phototransduction cascade), USH2A (photoreceptor structure), or RPGR (maintenance of cilia or ciliated cells with possible role in trafficking) genes. Patients with the same gene defect can have variable severity of disease at a given age. Despite recent advances, about 50% of cases still have an unknown molecular genetic basis. There is genetic treatment for RPE 65 defects in children.
Retinitis pigmentosa prevention
Since retinitis pigmentosa is a genetic disorder, there is currently no intervention that would prevent manifestations of retinitis pigmentosa 2.
Retinitis pigmentosa genetics
Mutations in more than 60 genes are known to cause nonsyndromic retinitis pigmentosa.
Genetics counseling
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Retinitis pigmentosa inheritance
Retinitis pigmentosa can be inherited in an autosomal dominant, autosomal recessive, or X-linked manner. The mode of inheritance in a particular family is determined by evaluating the family history and, in some instances, by molecular genetic testing. There are many potential complications in interpreting the family history, so in some cases, identifying the responsible gene with genetic testing is needed.
In 10 to 40 percent of all cases of retinitis pigmentosa, only one person in a family is affected. In these families, the disorder is described as simplex. It can be difficult to determine the inheritance pattern of simplex cases because affected individuals may have no affected relatives or may be unaware of other family members with the disease. Simplex cases can also result from a new gene mutation that is not present in other family members.
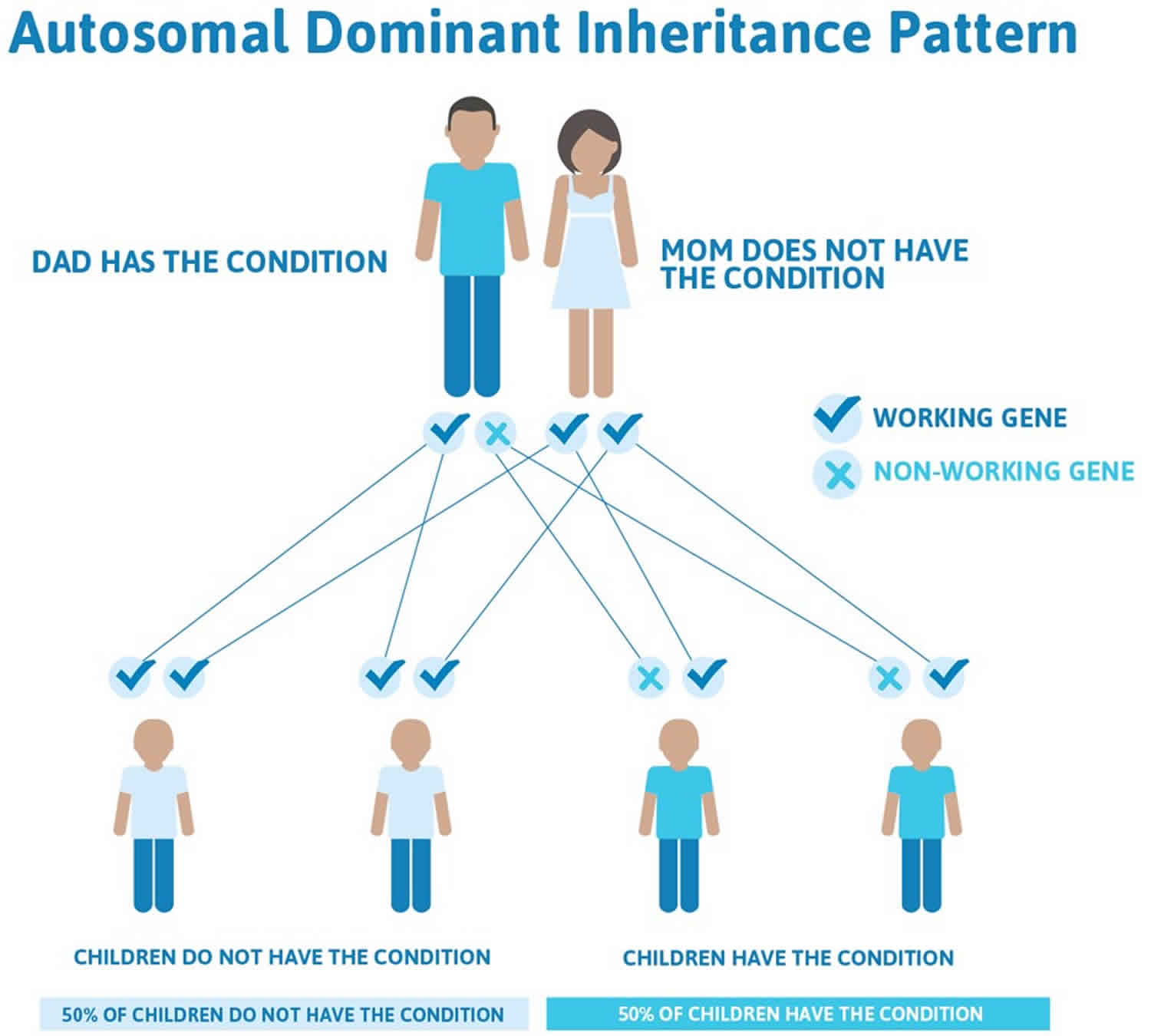
Retinitis pigmentosa autosomal dominant inheritance
Autosomal dominant inheritance means that having a change (mutation) in only one copy of the responsible gene in each cell is enough to cause features of the condition. In some cases, an affected person inherits the mutated gene from an affected parent. In other cases, the mutation occurs for the first time in a person with no family history of the condition. When a person with a mutation that causes an autosomal dominant condition has children, each child has a 50% chance to inherit that mutation.
Figure 3. Retinitis pigmentosa autosomal dominant inheritance
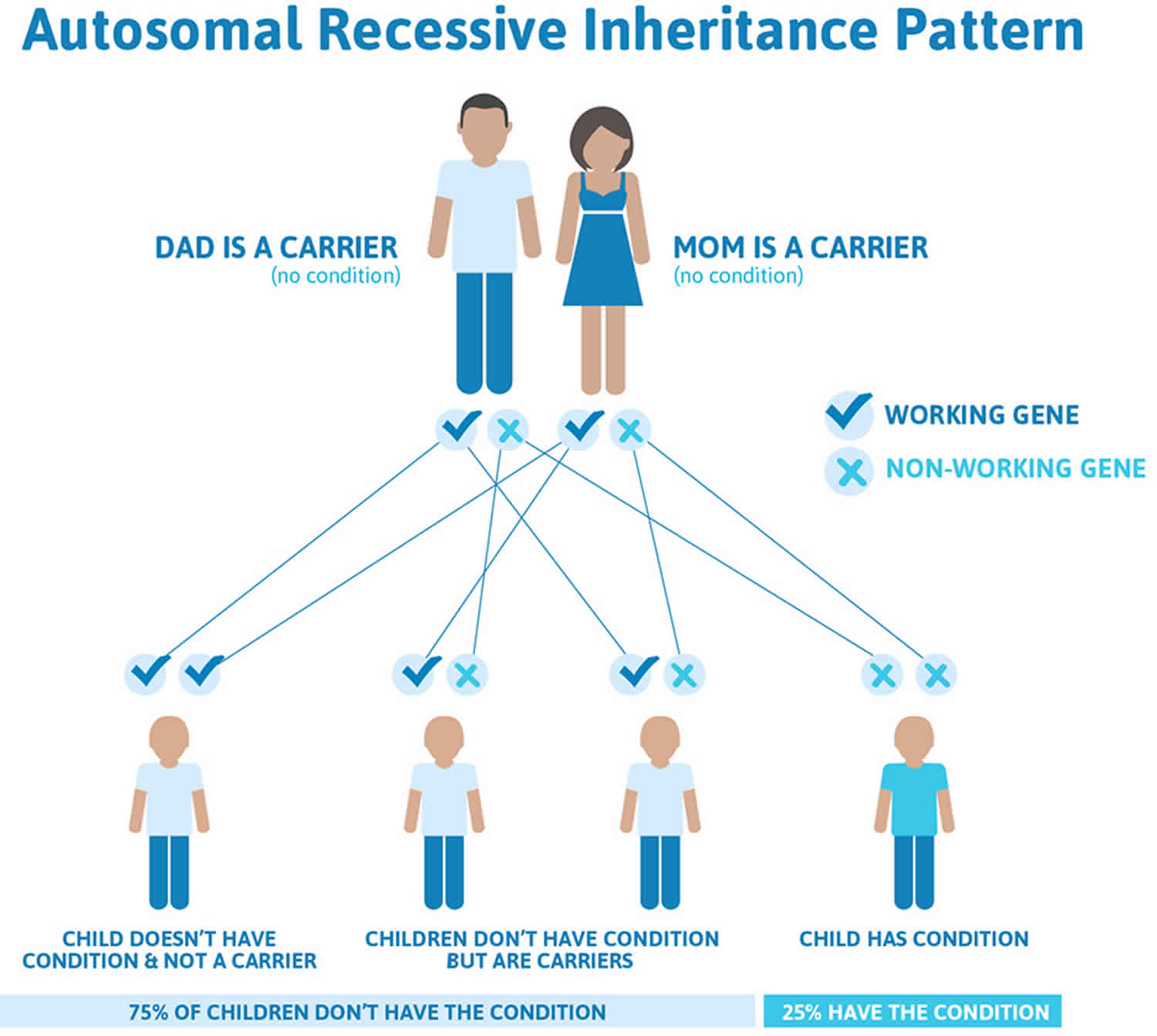
Retinitis pigmentosa autosomal recessive inheritance
Autosomal recessive inheritance means that to be affected, a person must have a mutation in both copies of the responsible gene in each cell. Affected people inherit one mutated copy of the gene from each parent, who is referred to as a carrier. Carriers of an autosomal recessive condition typically are unaffected. When 2 carriers of an autosomal recessive condition have children, each child has a:
- 25% chance to be affected
- 50% chance to be an unaffected carrier like each parent
- 25% chance to be unaffected and not a carrier.
Figure 4. Retinitis pigmentosa autosomal recessive inheritance
Retinitis pigmentosa X-linked inheritance
X-linked inheritance means that the responsible gene is located on the X chromosome. Males have one X chromosome (and one Y chromosome), while females have two X chromosomes. Males who have a mutation on their X chromosome will be affected, while female carriers of the mutation may be affected or unaffected, because they have another X chromosome with a normal copy of the gene.
- All the daughters of an affected male will inherit the mutation; none of his sons will inherit the mutation.
- The sons of a female with a mutation have a 50% chance to inherit the mutation and be affected; the daughters have a 50% chance to inherit the mutation (and be affected or unaffected).
Figure 5. Retinitis pigmentosa X-linked inheritance (mother is the carrier of retinitis pigmentosa)
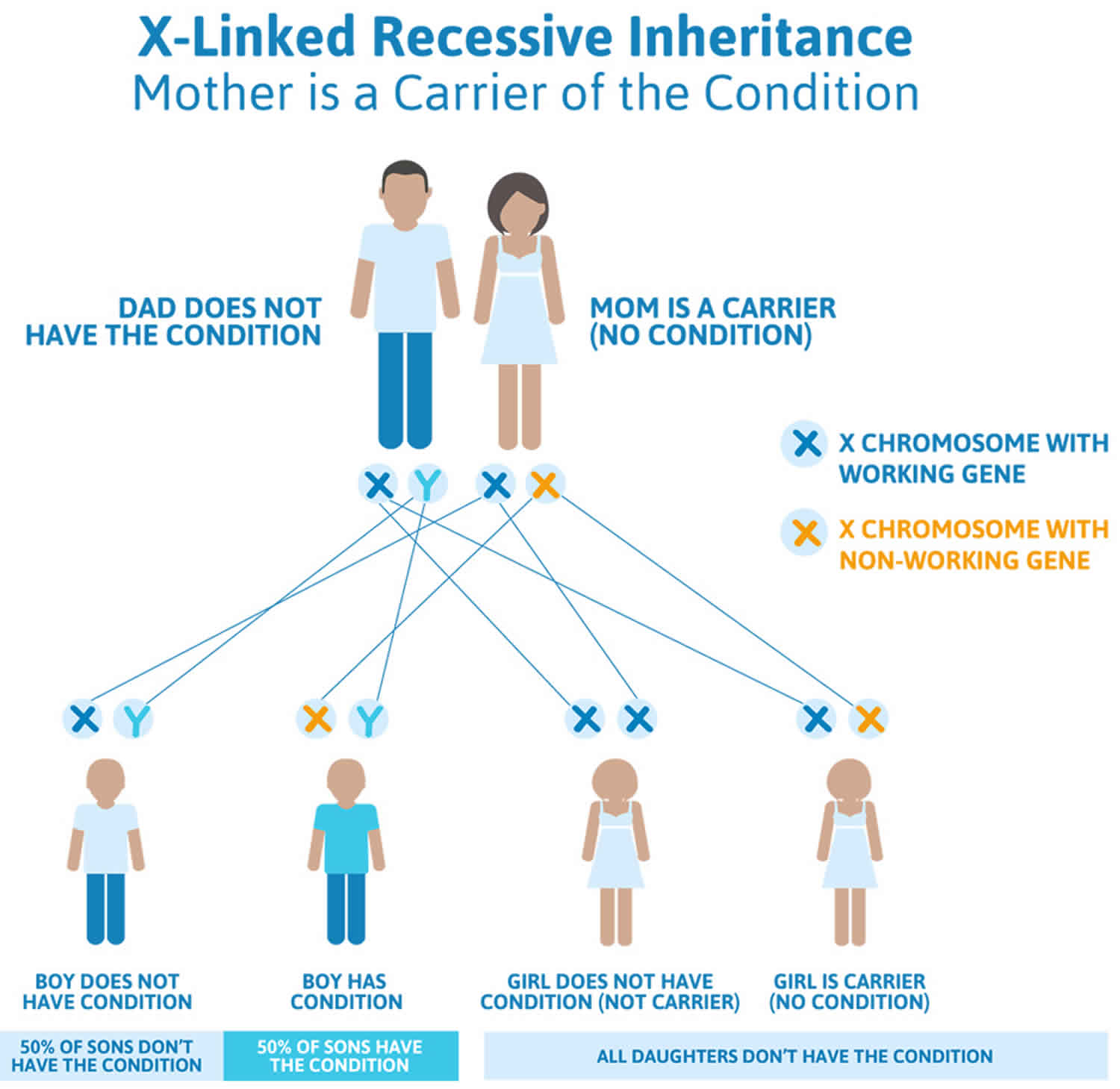
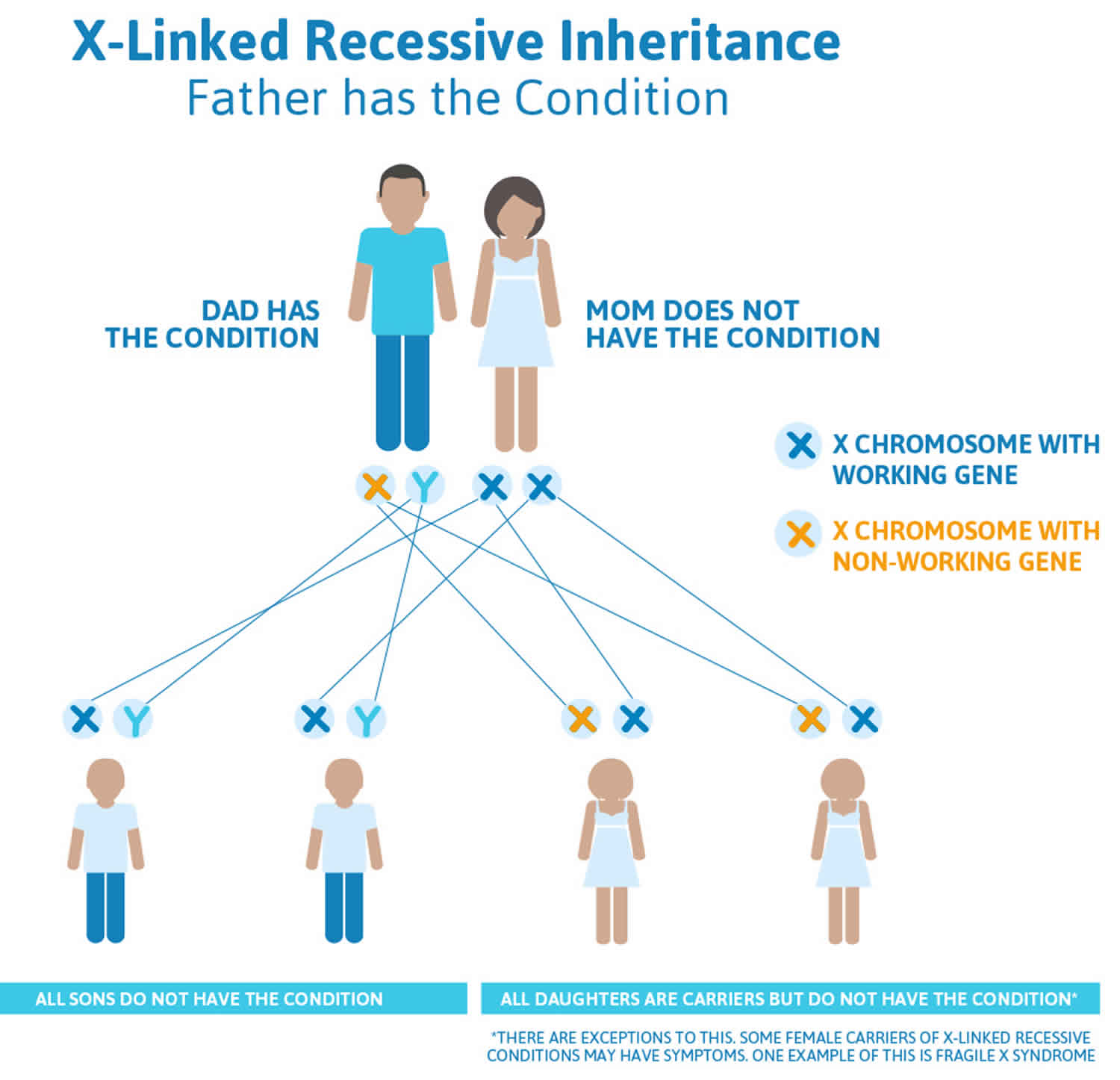
Figure 6. Retinitis pigmentosa X-linked inheritance (father has retinitis pigmentosa)
What are the chances for me and my children to develop retinitis pigmentosa if my father is affected?
The chances for children and grandchildren of an affected person to develop retinitis pigmentosa (retinitis pigmentosa) depend on the mode of inheritance in the family (autosomal dominant, autosomal recessive, or X-linked – see the description above).
Each child of a person with autosomal dominant retinitis pigmentosa has a 50% chance of inheriting the mutation 3. If a child does not inherit the mutation, it cannot be passed on to future generations (i.e. the grandchildren will be unaffected). If a child does inherit the mutation, the chance for each grandchild to be affected would likewise be 50%.
Each child of a person with autosomal recessive retinitis pigmentosa will be an unaffected carrier of autosomal recessive retinitis pigmentosa. A carrier of autosomal recessive retinitis pigmentosa is at risk to have affected children only if his/her partner is also a carrier.
The risk to children and grandchildren of a male with X-linked retinitis pigmentosa depends on the sex of the children. All the daughters of an affected male will inherit the mutation and be carriers; none of his sons will inherit the mutation (they will be unaffected) 3.
People with personal questions about the genetic cause and inheritance of this condition are encouraged to speak with a genetic counselor or other genetics professional. A genetics professional can help by:
- thoroughly evaluating the family history
- addressing questions and concerns
- assessing recurrence risks
- facilitating genetic testing if desired
- discussing reproductive options
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counsellors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Is it possible to avoid having a child with retinitis pigmentosa?
With advanced planning and appropriate testing, it may be possible to find out if a fetus or child will be affected, and/or to avoid having a child with retinitis pigmentosa. The optimal time for determining genetic risk and discussing the availability of prenatal testing is before pregnancy. If the disease-causing mutation(s) in a family have been identified, prenatal testing or preimplantation genetic diagnosis for a pregnancy at increased risk may be an option 3.
Prenatal genetic testing is performed during a pregnancy to determine if a fetus has inherited the disease-causing mutation(s) in a family. Genetic testing may be performed on a sample obtained by chorionic villus sampling (at about 10 to 12 weeks gestation), or by amniocentesis (usually performed at about 15 to 18 weeks gestation). If the condition is confirmed in the fetus by either method, planning for an affected child and/or pregnancy management options may be discussed with a health care provider.
As an alternative to prenatal diagnosis during the pregnancy, preimplantation genetic diagnosis (PGD) before a pregnancy may be an option. Preimplantation genetic diagnosis is done after in vitro fertilization (IVF) to diagnose a genetic condition in an embryo before it is introduced into the uterus. When having preimplantation genetic diagnosis, only embryos known to be unaffected are introduced into the uterus for a possible pregnancy.
Additional options for couples with a substantial risk to have an affected child may include adoption, gamete (egg or sperm) donation, and embryo donation.
People interested in learning more about their reproductive options should speak with a genetic counselor or other genetics professional. Requests for prenatal testing for diseases such as retinitis pigmentosa are not common. Medical professionals and individuals within families may have different opinions about the use of prenatal testing, particularly if it is being considered for the purpose of pregnancy termination (rather than early diagnosis). While most centers offering prenatal testing consider decisions to belong to the parents, discussion of these issues with a qualified professional is appropriate 3.
How does one know what type of retinitis pigmentosa they have?
Frequently (around 40-50% of the time), a person with retinitis pigmentosa has no family history of the condition. This may be because the retinitis pigmentosa mutation was a new event in that person, other family members have retinitis pigmentosa but have not been diagnosed, or the retinitis pigmentosa mutation has been in the family for a long time, but by chance, no other family members have been affected. For autosomal recessive retinitis pigmentosa, carriers may have been present in the mother’s and father’s side of the family for several generations, but a child won’t develop retinitis pigmentosa unless both parents are carriers and both pass on a mutation to their child 14.
There are many different gene mutations that lead to a diagnosis of retinitis pigmentosa, retinitis pigmentosa mutations can be inherited in different ways, and people with even the same genetic mutation can present with very different symptoms 6. As a result discovering the underlying mechanism for specific cases of retinitis pigmentosa can be challenging. As you seek to learn more about the cause of your retinitis pigmentosa, we recommend that you work with a genetic professional. A genetic professional will review your personal and family health history and can discuss with you your genetic testing options.
Retinitis pigmentosa prognosis
Symptoms of retinitis pigmentosa vary greatly, even among affected family members. Retinitis pigmentosa disease severity and long term outlook can be difficult to predict, even when assessed by a healthcare provider experienced in treating these conditions. Results from a electroretinogram (a record of the electrical signals in the retina produced when you see things) can be a useful aid to doctors as they estimate the long-term visual outlook for individual patients 15.
People with retinitis pigmentosa usually do not go completely blind, but in the later stages of the disease, may be considered “legally blind” 14. Legal blindness is defined as having best corrected vision equal to or worse than 20/200 or less than 20 degrees on a visual field in both eyes 14. Many people with retinitis pigmentosa meet criteria for legal blindness by age 40 15. Studies in retinitis pigmentosa patients have estimated the visual field to diminish at a rate of around 5 to 12 percent each year on average 15. Visual acuity declines more slowly. Age of symptom onset (e.g., childhood vs adulthood) is not a reliable predictor of disease severity 15. Knowing the specific retinitis pigmentosa causing gene mutation and type of retinitis pigmentosa occasionally provides additional information regarding the condition’s severity.
Some studies suggest that the rate of progression, age of onset, and eventual visual loss are related to the mode of inheritance. Autosomal dominant retinitis pigmentosa has the best prognosis, with the majority of patients under 30 years having visual acuity of 20/30 or better 2. X-linked is the most severe form with appreciable impairment of central visual acuity to 20/200 or less by the fifth decade of life. Autosomal recessive and sporadic cases were intermediate in severity 16. In terms of visual field loss, a study of 104 patients with autosomal dominant retinitis pigmentosa shows 93% of patients under age 20, 89% of those from 20-40, and 60% over the age of 40 had a central visual field radius of 10 degrees or greater with the IV4e test object 17.
Retinitis pigmentosa symptoms
Patients typically presents with night vision problems (unable to see in the dark or slow to adjusting to dark), progressive peripheral vision restriction, and tunnel vision at later stage of retinitis pigmentosa 2. It is rare for patients to lose all vision in both eyes 2. In a large study involving close to 1,000 patients with retinitis pigmentosa and Usher Syndrome at age 45 or older, one fourth of the patients had a visual acuity of 20/200 or worse in both eyes, and more than half had a visual acuity of 20/40 or better in at least one eye 2. Only 0.5% of patients were completely blind in both eyes 18. In one study, about 50% of retinitis pigmentosa patients reported having headaches, and 35% of retinitis pigmentosa patients reported light flashes 19.
Retinitis pigmentosa symptoms:
- Night blindness (nyctalopia) — Hallmark; most commonly the earliest symptom in retinitis pigmentosa 6
- Visual loss, usually peripheral; in advanced cases, central visual loss
- Photopsia (seeing flashes of light)
Symptoms of retinitis pigmentosa are most often recognized in children, adolescents and young adults, with progression of the disease continuing throughout the individual’s life. The pattern and degree of visual loss are variable.
As retinitis pigmentosa progresses and more rod cells breakdown, patients lose their peripheral vision (tunnel vision). Individuals with retinitis pigmentosa often experience a ring of vision loss in their periphery, but retain clear central vision. Others report the sensation of tunnel vision, as though they see the world through a straw. Many patients with retinitis pigmentosa retain a small degree of central vision throughout their life.
Other forms of retinitis pigmentosa, sometimes called cone-rod dystrophy, first affect central vision. Patients first experience a loss of central vision that cannot be corrected with glasses or contact lenses. With the loss of cone cells also comes disturbances in color perception. As the disease progresses, rod cells degenerate causing night blindness and peripheral vision.
The symptoms described above may not necessarily mean that you have retinitis pigmentosa. However, if you experience one or more of these symptoms, contact your eye doctor for a complete exam.
Retinitis pigmentosa diagnosis
Because retinitis pigmentosa is a collection of many inherited diseases, significant variability exists in the physical findings. Ocular examination involves assessment of visual acuity and pupillary reaction, as well as anterior segment, retinal, and funduscopic evaluation 6.
Medical History
Patients with retinitis pigmentosa characteristically develop night blindness and difficulty with mid-peripheral visual field in adolescence. Timing of onset can vary among pedigrees. As their condition progresses, they lose mid-peripheral followed by far-peripheral visual field, but often maintain central vision until the very end stage of retinitis pigmentosa 13.
Physical examination and signs
The classic clinical triad of retinitis pigmentosa is arteriolar attenuation, retinal pigmentary changes (could be either hypopigmentation and/or hyperpigmentation in form of bone-spicule and pigment clumpings), and waxy disc pallor 2. The characteristic pigmentary changes occur in the mid-peripheral fundus, which is predominantly populated by rods. There is often a high degree of symmetry in the fundus findings between the two eyes. Other common signs include vitreous cells, depigmentation and atrophy of the RPE, posterior subcapsular cataracts, cystic macular lesions, and refractive errors including myopia and astigmatism 20.
Systemic examination for retinitis pigmentosa can be helpful to rule out syndromic retinitis pigmentosa, which are conditions that have pigmentary retinopathy and mimic retinitis pigmentosa, such as the following:
- Syndromes associated with retinitis pigmentosa and hearing loss: Usher syndrome 1, Waardenburg syndrome, Alport syndrome, Refsum disease
- Kearns-Sayre syndrome: External ophthalmoplegia, lid ptosis, heart block, and pigmentary retinopathy
- Abetalipoproteinemia: Fat malabsorption, fat-soluble vitamin deficiencies, spinocerebellar degeneration, and pigmentary retinal degeneration
- Mucopolysaccharidoses (e.g., Hurler syndrome, Scheie syndrome, Sanfilippo syndrome): Can be affected with pigmentary retinopathy
- Bardet-Biedl syndrome: Polydactyly, truncal obesity, kidney dysfunction, short stature, and pigmentary retinopathy
- Neuronal ceroid lipofuscinosis: Dementia, seizures, and pigmentary retinopathy; infantile form is known as Jansky-Bielschowsky disease, juvenile form is Vogt-Spielmeyer-Batten disease, and adult form is Kufs syndrome
In general, the diagnosis of retinitis pigmentosa is established when the following findings are present 21.
- Bilateral involvement (can be asymmetric);
- Impairment of night vision and loss of peripheral vision;
- Rod dysfunction evidenced by elevated rod final threshold on dark adaptation and/or rod responses on ERG testing that are either reduced in b-wave amplitude and prolonged in implicit time or are essentially non-detectable(extinguished ERG);
- Progressive loss in photoreceptor function.
Testing
The following laboratory tests are useful in excluding masquerading diseases or in detecting conditions that are associated with retinitis pigmentosa:
- Infectious studies for syphilis (VDRL, FTA-ABS), toxoplasmosis (when suspected; serum IgG)
- Inherited/syndromic disease studies for Refsum disease (serum phytanic acid in the presence of other neurologic abnormalities), gyrate atrophy (ornithine levels), Kearns-Sayre syndrome (ECG to help rule out heart block), and abetalipoproteinemia (lipid profile with possible protein electrophoresis)
- Neoplasm related studies for antiretinal antibodies (particularly antirecoverin antibodies), especially in cancer-associated retinopathy (CAR) or in severe retinitis pigmentosa
Other studies that may be helpful include the following:
- Full-Field Electroretinogram (ERG): Most critical diagnostic test for retinitis pigmentosa. ERG measures the electrical potential generated by rods and cones after a light stimulus and is essential in the diagnosis of retinitis pigmentosa. The most important parameters being measured include a- and b-wave amplitudes and implicit times. In early stages of the disease, there is reduction in a- and b-wave amplitudes but implicit time can be prolonged or normal. Patients with advanced stages have non-detectable ERG.
- Electro-oculogram (EOG): Electrooculogram (EOG) is a measurement of standing potential between the cornea and the retina and is a measurement of function of the RPE and photoreceptors. It is usually abnormal in retinitis pigmentosa. However, ERG is considered a more sensitive test for detection of photoreceptor function and consequently EOG is not routinely done. Not helpful in diagnosing retinitis pigmentosa, but central macular changes, normal ERG findings, and abnormal EOG findings suggest Best vitelliform macular dystrophy (Best disease)
- Formal visual field testing: Most useful measure for ongoing follow-up care of patients with retinitis pigmentosa; Goldmann (kinetic) perimetry is recommended. Kinetic perimetry with Goldmann perimeter characteristically shows a ring scotoma in the mid-periphery of the visual field. They usually start as a group of isolated scotomas around 20 degrees from fixation, and gradually coalesce to form a partial followed by a complete ring. The outer edge of the ring expands relatively quickly to the periphery, while the inner edge constricts slowly toward fixation. Patients often have good central vision from a small central island (“tunnel vision”) until their 50’s or 60’s 22. Visual field testing is useful in monitoring the progression of disease and document the status of legal blindness.
- Color testing: Commonly, mild blue-yellow axis color defects, although most patients with retinitis pigmentosa do not clinically complain of major difficulty with color perception
- Dark adaptation study: Visual threshold is the minimum intensity of light that will stimulate the rods or cones to elicit a subjective response. Dark adaptometry measures the absolute threshold of rods at given time intervals as the retina adapts to the dark. In retinitis pigmentosa, there is increased absolute rod threshold and dark adaptation is usually prolonged. This test maybe useful in detecting early cases of retinitis pigmentosa 23.
- Genetic subtyping: Definitive test for diagnosis to identify the particular defect.
- Optical coherence tomography (OCT): OCT is a quick, inexpensive, and widely available tool to detect cystic macular lesions, epiretinal membrane, and vitreomaular traction syndrome observed in some retinitis pigmentosa patients with decreased central vision. One study also showed mild inner retinal layer thinning and severe outer retinal layer thinning using spectral domain OCT 24.
- Fluorescein angiography (FA): FA may have a role in documenting early deterioration of the retinal pigment epithelium and especially in female carriers of X-linked retinitis pigmentosa. It has a role in patients with cystic macular lesions and exudative vasculopathy.
Imaging tests
Fluorescein angiography is rarely useful in diagnosing retinitis pigmentosa; however, the presence of cystoid macular edema can be confirmed by this test. Likewise, although optical coherence tomography (OCT) is not useful in helping to establish a diagnosis of retinitis pigmentosa, this imaging study can be helpful to document the extent and/or presence of cystoid macular edema.
Procedures
Biopsy for histologic examination in patients with retinitis pigmentosa is not clinically helpful, owing to the general good health of these patients and the chronic nature of the disease. Generally, specimens are obtained only on chronically atrophic retinas.
Retinitis pigmentosa treatment
Available treatments aim to slow the progression of the disease and primarily include light avoidance and the use of low-vision aids. Some practitioners also consider vitamin A as a possible treatment option. However, taking too much vitamin A can be toxic and the effects of vitamin A on the disease appear to be relatively weak 7. Studies have explored potential treatment with docosahexaenoic acid (DHA), an omega-3 fatty acid naturally found in fish. While DHA (docosahexaenoic acid) is known to play a structural role in retinal cells, more research is needed to determine whether supplements should be recommended 8.
Current research is focused on the development of new treatments including gene therapy, retinal transplantation, and the use of a retinal prosthesis. Stem cell transplantation would involve the injection and integration of stem cells into the retina, in hopes these cells will replace dead cells and provide the missing enzymes and chemicals needed for sight. Gene therapy could potentially be used when the disease-causing mutation is known and would aim to restore production of the missing or abnormal protein. Studies with retinal prosthetics have tested devices that transform light into electrical signals that can be sent directly to the inner retina and brain, avoiding the diseased part of the outer retina. Though challenges remain, preliminary research into these technologies has been promising 5, 8.
FDA-Approved Treatments
The medication(s) listed below have been approved by the Food and Drug Administration (FDA) as orphan products for treatment of this condition.
- Voretigene neparvovec-rzyl (Brand name: Luxturna) – Manufactured by Spark Therapeutics, Inc. FDA-approved indication: An adeno-associated virus vector-based gene therapy indicated for the treatment of patients with confirmed biallelic RPE65 mutation-associated retinal dystrophy. Patients must have viable retinal cells determined by a treating physician.
Medications
Medications used in the management of retinitis pigmentosa include the following 6:
- Fat-soluble vitamins (e.g., vitamin A, vitamin E, ascorbic acid)
- Calcium-channel blockers (e.g., diltiazem)
- Carbonic anhydrase inhibitors (eg, acetazolamide, methazolamide)
The following are medications with potential adverse effects in retinitis pigmentosa 6:
- Isotretinoin (Accutane)
- Sildenafil (Viagra)
- High-dose vitamin E
Many treatments have been explored without proven benefit for the isolated forms of retinitis pigmentosa 13. These include various vitamins and minerals, vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy, injections of a hydrolysate of yeast RNA, ultrasound, transfer factor, dimethyl sulfoxide, ozone, muscle transplants, and subretinal injections of fetal retinal cells 2. None of the above treatments were conducted in randomized, controlled clinical trials. It is important to note that anecdotal treatment with subjective improvement of visual function should be interpreted with caution due to fluctuation in visual acuity and visual fields in this disease. Electroretinogram (ERG) is a better objective measure of remaining retinal function. Any potential therapy will likely require several years of follow-up to assess efficacy due to the nature of slow progression of this disease.
Controversies exist regarding the use of high dose vitamin A, docosahexaenoic acid (DHA), and lutein to slow the progression of retinitis pigmentosa. Berson et al. 25, 26, 27 conducted three large randomized, controlled, double-masked trials. In the first study, 601 adult patients were randomized to one of four treatment groups: vitamin A, 15,000 IU/day plus vitamin E 3 IU/day; vitamin A 75 IU/day plus vitamin E, 3 IU/day; vitamin A, 15,000 IU/day plus vitamin E, 400 IU/day; and vitamin A, 75 IU/day plus vitamin E, 400 IU/day. The main outcome variable was the 30-Hz cone flicker ERG. In summary, patients who are on the higher dose of vitamin A had the slowest annual rate of decline in remaining ERG amplitude (8.3% of decline per year) while those on high dose vitamin E had the fastest (11.8%). The results were more significant in the cohort with higher amplitudes to start with (i.e., > 0.68 μV).
In the second study, patients who were given vitamin A palmitate 15,000 IU/day were randomized to either DHA capsules (1200 mg/day) or control fatty acid capsules. The main outcome variable was the total point score of the 30-2 Humphrey visual field. Overall, DHA supplementation by capsules did not slow the course of retinitis pigmentosa over a 4-year interval. However, for those who are taking vitamin A for the first time, a subgroup analysis concluded DHA supplement slowed the rate of visual field loss and log ERG amplitude loss in years 1 and 2, but not in years 3 and 4 after the start of treatment.
In the third study, they evaluated the supplemental effects of lutein 12 mg/day combined with high dose vitamin A and high dietary intake of DHA on the rate of retinitis pigmentosa visual field loss. The investigators reported no difference between groups in the rate of decline in the total point score for the HFA 30-2 program (primary outcome measure, p=0.66), nor loss of HFA 30-2 plus 60-4 total point score, logERG amplitude, and logMAR visual acuity (secondary outcomes). However, they did report a significant effect of treatment on the rate of decline for the HFA 60-4 total point score.
Based on these studies, the authors concluded that patients with retinitis pigmentosa would benefit from taking 12 mg of lutein per day in addition to 15,000 IU/d of vitamin A and weekly meals of oily fish, of which DHA is a major component. However, there were some debates regarding these recommendations 28. For example, members of the Data and Safety Monitoring Committee from the first study reported that much of the originally reported significant difference was a consequence of pooling the data and could be attributed to early and consistently large differences between the vitamin E group and all of the other groups 29. In the 2nd and 3rd study, conclusions were drawn based on secondary outcomes and subgroup analyses, rather than primary outcome 28. Therefore, the use of high dose vitamin A and other supplements must be weighed against their potential side effects (see complications).
The precise mechanism by which vitamin A supplementation provides its benefit is not known. It has been speculated that vitamin A rescues remaining cones, thereby explaining how one supplement may help a group of patients with different rod-specific gene defects. Vitamin E may lead to an adverse effect on the course of retinitis pigmentosa by inhibiting the absorption or transport of vitamin A. DHA is thought to facilitate the release of vitamin A from its carrier protein (interphotoreceptor retinoid binding protein) in the subretinal space.
Other treatment considerations
Patients who develop cystic macular lesions (about 30%) may benefit from oral acetazolamide 30, topical dorzolamide drops 31, and intravitreal steroids in some cases. Anti-VEGF intravitreal injection has also been shown to be effective in a small case series 32. The long-term efficacy of topical dorzolamide in improving the macular cystic lesions in patients with retinitis pigmentosa and Usher syndrome has been been demonstrated in a retrospective series with a mean follow-up of 39 months 33.
Although light deprivation has not been shown to be of benefit in altering the course of retinal degeneration 34, it is generally advisable for patients to use ultraviolet and short-wavelength (blue) blocking sunglasses for outdoor activities. Audiology consults should be considered for patients with possible or known diagnosis of Usher syndrome. Low vision services are designed to benefit those whose ability to function is compromised by visual impairment. A low vision examination may be useful to help optimize the use of remaining visual function. Genetic counseling can provide patients and families with information on the inheritance and implications of their genetic disorders and can help them make informed medical and personal decisions.
Medical follow up
Annual ocular examinations usually are sufficient to measure visual acuity and Goldmann visual field. If medical treatment is initiated, more frequent visits and laboratory blood work may be indicated. For example, patients with red blood cell (RBC) docosahexaenoic acid (DHA) level of at least 4% of total RBC fatty acids has been reported to have, on average, a slower rate of decline of visual field sensitivity than those with lower levels 35. Vitamin A levels and liver function tests should also be done annually if treatment has been initiated (see Complications).
Complications
In general, toxicity from vitamin A treatment is rare. As a safety measure, patients should have a pretreatment assessment of fasting serum vitamin A levels and liver function and annually thereafter. Because of the potential for birth defects, women who are pregnant or planning to conceive are advised not to take high doses of vitamin A (15,000 IU/day). In older adults, long-term vitamin A supplementation has been associated with a decrease in bone density and up to a 1% increased risk of hip fractures 36. Therefore, postmenopausal women and men over the age of 49 who are taking vitamin A should consult with their primary care physician regarding their bone health. Patients with renal failure or renal transplant should not take vitamin A due to excessive renal re-absorption. Finally, vitamin A should not be given to patients on chronic doxycycline because the combination can lead to increased intracranial pressure.
The 5 year study of the ARGUS II Implant supports the long term safety and benefit of the implant for those blind from retinitis pigmentosa 37. A collaborative has also recently published their recommendations to optimize patient outcomes 38. Most common complications are conjunctival erosion and hypotony. It is rare that the implant would require removal.
Surgery
There is now an FDA approved Humanitarian Device, called the ARGUS II implant, which may help patients with end-stage retinitis pigmentosa. It is approved for use in patients with bare light to no light perception. It consists of 3 parts: a video recorder, a transmitter and the implant itself. The implant is an epiretinal electrode chip coated in silicone that stimulates the retina electrically. It is connected to a silicone strip that carries the electrodes from the receiver. This strip encircles the eyeball and is surgically sewn onto the sclera. The wireless receiver receives electrical signals from a video recorder which is mounted to glasses on the patient’s face. The video unit converts the video images into electrical impulses which are transmitted to the receiver. The retinal stimulation results in the patient seeing lines or dots of light that indicate edges or objects in the patient’s field of vision. The patient does not see in color and the resolution does not allow for “seeing faces or small details.” Prior research on the ARGUS II showed that patients are better able to find doors, walk along a path and identify the location and movement of objects with the device turned “on” than without the device 39.
In patients with another form of retinitis pigmentosa, Leber’s variant, gene therapy for RPE 65 is being performed 2. This technique requires the injection of the gene into the eye, specifically into the space under the center of the retina (macular subretinal space). Replacement of the gene in younger patients (versus adults) has allowed patients to gain vision. This treatment is produced by Spark Therapeutics.
If the patient develops a cataract, it is generally advisable to defer surgical removal until the patient can no longer read with the better eye. In one study of 30 patients with retinitis pigmentosa, 83% improved by 2 lines on the Snellen visual acuity chart with cataract surgery 40.
Investigational procedures with potential in managing retinitis pigmentosa include the following:
- Surgical placement of growth factors
- Transplantation of retinal or retinal pigment epithelial (retinitis pigmentosaE) tissue
- Placement of retinal prosthesis or phototransducing chip
- Subretinal gene therapy
Future directions
Retinitis pigmentosa gene therapy
Although there is currently no cure for retinitis pigmentosa, well-characterized animal models and a developed understanding of the genetic basis of the disease allow gene therapy to be a potentially viable therapeutic strategy 41. For example, in the Rds mouse model which carries a mutation in Prph2, the rhodopsin promoted delivery of Prph2 rescues both ultrastructure and function in differentiated photoreceptors 42. In the Rd mouse, which carries a cyclic GMP phosphodiesterase (PDE) mutation, expression of this gene prolongs photoreceptor survival and induces a twofold increase in light sensitivity 43. In general, better improvements are generally seen in younger mice compared to adult mice 44. In humans, retinal gene therapy mediated by adeno-associated virus (AAV) based gene transfer was shown to be safe and effective in improving photoreceptor function in some patients with an inherited retinal blinding disorder associated with mutations in the retinitis pigmentosaE65 gene known as Leber Congenital Amaurosis 45. While this is very exciting for autosomal recessive diseases with well defined genetic mutations, one big challenge for retinitis pigmentosa is the heterogeneous nature of genetic mutations 46. For example, in autosomal dominant forms of retinitis pigmentosa, where one mutant allele causes disease, researchers must develop strategies to knock down expression of the mutated allele, while adding the normal allele by gene transfer. For those patients suffering from retinitis pigmentosa with unknown mutations, an AAV based transfer of bacterial forms of rhodopsin in the central retina might be an option to reactivate residual cones in the future.
Ciliary neurotrophic factor
Ciliary neurotrophic factor has been shown to slow retinal degeneration in a number of animal models. In human, an encapsulated form of retinitis pigmentosaE cells producing ciliary neurotrophic factor (Neurotech) was implanted into the eye of three patients with Usher syndrome and retinitis pigmentosa 47. Sham surgery was performed in the opposite eye as control. After 24 months of follow-up, although there is significant difference in structure (thicker retina and higher cone density in treated group), there is no significant change in visual acuity, visual field sensitivity, or ERG response 48. Recently, the ciliary neurotrophic factor trial results showed no long term benefit (60-96 months), with regards to efficacy for visual acuity, visual field sensitivity, or OCT measures of retinal structure with the implant 49.
Retinal prothesis
A retinal prosthesis or phototransducing chip can be surgically placed on the retinal surface and the healthy ganglion cell layer of the retina can be stimulated 50. Subjective improvements were noted in a few patients. Post approval, a paper has studied the outcomes in the real word 51.
Are there clinical trials investigating the use of gene therapy for treatment of retinitis pigmentosa?
ClinicalTrials.gov (https://www.clinicaltrials.gov/) currently lists several studies that either use gene therapy or are aimed at the development of this technology for the treatment of retinitis pigmentosa. Since gene therapy is still relatively new, the studies are fairly small and are focused on very specific genes. To access the studies currently recruiting participants, click here (https://clinicaltrials.gov/ct2/results?term=retinitis+pigmentosa+AND+gene+therapy&recr=Open). After you click on a study, review its “eligibility” criteria to determine its appropriateness. Use the study’s contact information to learn more. Check this site often for regular updates.
You can contact the Patient Recruitment and Public Liaison Office at the National Institutes of Health (https://clinicalcenter.nih.gov/) if you have questions.
Are there clinical trials involving retinal cell transplant or retinal prosthesis that are enrolling people with retinitis pigmentosa?
Yes. You can learn more about ongoing trials through ClinicalTrials.gov (https://www.clinicaltrials.gov/). Currently there are several clinical trials identified as enrolling individuals with retinitis pigmentosa. One trial, titled “Safety Study in Retinal Transplantation for Retinitis Pigmentosa” (https://clinicaltrials.gov/ct2/show/NCT00345917) involves retinal cell transplant, another trial, titled “Safety and Efficacy of Subretinal Implants for Partial Restoration of Vision in Blind Patients” (https://clinicaltrials.gov/ct2/show/NCT01024803) involves inserting a retinal prosthesis. Click on the study titles above and review the ‘eligibility’ criteria to determine their appropriateness. Use the study’s contact information to learn more.
Click here (https://clinicaltrials.gov/ct2/results?term=retinitis+pigmentosa+AND+gene+therapy&recr=Open) to view other retinitis pigmentosa trials, including trials that are completed as well as ones that are ongoing but not accepting new participants at this time. We suggest checking ClinicalTrials.gov (https://www.clinicaltrials.gov/) often for regular updates
You can also contact the Patient Recruitment and Public Liaison Office at the National Institutes of Health (https://clinicalcenter.nih.gov/).
Is retinal cell transplant and gene therapy the same thing?
No. Gene therapy introduces a healthy copy of a defective gene into the patient’s cells, while the aim of retinal cell transplant is to transplant neural retinal tissue and retinal pigment epithelium into the eyes of people with retinitis pigmentosa.
The National Human Genome Research Institute provides the following description regarding current aims for retinitis pigmentosa research on their Web site at the following link: (https://www.genome.gov/13514348)
“Research is also being conducted in areas such as gene therapy research, transplant research, and retinal prosthesis. Since retinitis pigmentosa is usually the result of a defective gene, gene therapy has become a widely explored area for future research. The goal of such research would be to discover ways healthy genes can be inserted into the retina. Attempts at transplanting healthy retinal cells into sick retinas are being made experimentally and have not yet been considered as clinically safe and successful. Retinal prosthesis is also an important area of exploration because the prosthesis, a man-made device intended to replace a damaged body part, can be designed to take over the function of the lost photoreceptors by electrically stimulating the remaining healthy cells of the retina. Through electrical stimulation, the activated ganglion cells can provide a visual signal to the brain. The visual scene captured by a camera is transmitted via electromagnetic radiation to a small decoder chip located on the retinal surface. Data and power are then sent to a set of electrodes connected to the decoder. Electrical current passing from individual electrodes stimulate cells in the appropriate areas of the retina corresponding to the features in the visual scene.”
References- Saihan Z, Webster AR, Luxon L, Bitner-Glindzicz M. Update on Usher syndrome. Curr Opin Neurol. 2009 Feb. 22(1):19-27.
- Retinitis pigmentosa. http://eyewiki.org/Retinitis_Pigmentosa
- Fahim AT, Daiger SP, Weleber RG. Nonsyndromic Retinitis Pigmentosa Overview. 2000 Aug 4 [Updated 2017 Jan 19]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1417
- Retinitis pigmentosa. https://ghr.nlm.nih.gov/condition/retinitis-pigmentosa
- Zarbin M. Cell-Based Therapy for Degenerative Retinal Disease. Trends in Molecular Medicine. February 2016; 22(2):115-34. http://www.ncbi.nlm.nih.gov/pubmed/26791247
- Retinitis Pigmentosa. https://emedicine.medscape.com/article/1227488-overview#showall
- Learning About Retinitis Pigmentosa. https://www.genome.gov/13514348/
- Retinitis pigmentosa: Treatment. https://www.uptodate.com/contents/retinitis-pigmentosa-treatment
- Mizuno K, Nishida S: Electron microscopic studies of human retinitis pigmentosa. Part I. Two cases of advanced retinitis pigmentosa. Am J Ophthalmol. 1967;63(4):791-803.
- Bunt-Milam AH, Kalina RE, Pagon RA. Clinical-ultrastructural study of a retinal dystrophy. Invest Ophthalmol Vis Sci. 1983;24(4):458-469.
- Ben-Arie-Weintrob Y, Berson EL, Dryja TP. Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalmic Genet. 2005;26(2):91-100
- Pagon RA. Retinitis Pigmentosa. Surv Ophthalmol. 1988;33(3):137-177.
- Berson EL. Retinitis Pigmentosa and Allied Diseases. In: Albert D, Miller J, Azar D, Blodi B, eds. Albert and Jakobiec, 3rd edn. Philadelphia, PA: Elsevier; 2008:Ch. 177.
- Retinitis Pigmentosa (RP). https://www.umkelloggeye.org/conditions-treatments/retinitis-pigmentosa-rp
- Givre S, Garg S. Retinitis pigmentosa: Clinical presentation and diagnosis. In: Basow, DS . UpToDate. Waltham, MA: UpToDate; 2012
- Fishman GA, Farber MD, Derlacki DJ. X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol. 1988;106(3):369-375.
- Lyness AL, Ernst W, Quinlan MP, et al. A clinical, psychophysical, and electroretinographic survey of patients with autosomal dominant retinitis pigmentosa. Br J Ophthalmol. 1985;69(5):326-339.
- Grover S, Fishman GA, Anderson RJ, et al. Visual acuity impairment in patients with retinitis pigmentosa at age 45 years or older. Ophthalmology. 1999;106(9):1780-1785.
- Heckenlively JR, Yoser SL, Friedman LH et al. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 1988; 105(5):504-511.
- Kanski J. Retinitis Pigmentosa. In: Kanski J, Sehmi K, Bolton A (eds.) Clinical Ophthalmology: A Systemic Approach, 6th edn. Philadelphia, PA: Elsevier; 2008:Ch. 18.
- Marmor MF, Aguirre G, Arden G, et al. Retinitis pigmentosa, a symposium on terminology and methods of examination. Ophthalmology. 1983;90(2):126-131.
- Grover S, Fishman GA, Brown J Jr. Patterns of visual field progression in patients with retinitis pigmentosa. Ophthalmology. 1998;105(6):1069-1075.
- Alexander KR, Fishman GA. Prolonged rod dark adaptation in retinitis pigmentosa. Br J Ophthalmol. 1984;68(8):561-569.
- Lim JI, Tan O, Fawzi AA, et al: A pilot study of Fourier-domain optical coherence tomography of retinal dystrophy patients. Am J Ophthalmol. 2008;146(3):417-426.
- Berson EL, Rosner B, Sandberg MA, et al. Clinical trial of lutein in patients with retinitis pigmentosa receiving vitamin A. Arch Ophthalmol. 2010;128(4):403-411.
- Berson EL, Rosner B, Sandberg MA, et al: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch Ophthalmol. 2004; 122(9):1306-1314.
- Berson EL, Rosner B, Sandberg MA, et al: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol. 1993; 111(6):761-772.
- Massof RW, Fishman GA. How strong is the evidence that nutritional supplements slow the progression of retinitis pigmentosa? Arch Ophthalmol. 2010;128(4):493-495.
- Norton EWD. A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa [letter to the editor]. Arch Ophthalmol. 1993;111(11):1460.
- Cox SN, Hay E, Bird AC: Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol. 1988; 106(9):1190-1195.
- Grover S, Apushkin MA, Fishman GA: Topical Dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am J Ophthalmol. 2006; 141(5):850-858.
- Yuzbasioglu E, Artunay O, Rasier R, et al. Intravitreal bevacizumab (Avastin) injection in retinitis pigmentosa. Curr Eye Res. 2009;34(3):231-237.
- Genead MA, Fishman GA. Efficacy of sustained topical dorzolamide therapy for cystic macular lesions in patients with retinitis pigmentosa and Usher syndrome. Arch Ophthalmol. 2010;128(9):1146-1150.
- Berson EL: Light deprivation and retinitis pigmentosa. Vision Res. 1980;20(12):1179-1184.
- Berson EL, Rosner B, Sandberg MA, et al: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch Ophthalmol. 2004; 122(9):1306-1314
- Michaelsson K, Lithell H, Vessby B, Melhus H: Serum retinol levels and the risk of fracture. N Engl J Med. 2003;348(4):287-294
- deCruz et al. Five-Year Safety and Performance Results from the Argus II Retinal Prosthesis. Ophthalmology. 2016 Oct;123(10):2248-54
- Ghodasra DH, et al. Worldwide Argus II implantation: recommendations to optimize patient outcomes. BMC Ophthalmol. 2016 May 6;16:52. doi: 10.1186/s12886-016-0225-1.
- Luo YH, Zhong JJ, da Cruz L. The use of Argus® II retinal prosthesis by blind subjects to achieve localisation and prehension of objects in 3-dimensional space. Graefes Arch Clin Exp Ophthalmol. 2014 Dec 31.
- Bastek JV, Heckenlively JR, Straatsma BR. Cataract surgery in retinitis pigmentosa patients. Ophthalmology 1982;89(8):880-884.
- Liu MM, Tuo J, Chan C-C. Gene therapy for ocular diseases. Br J Ophthalmol. 2011;95(5):604-612
- Ali RR, Sarra GM, Stephens C, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25(3):306-310.
- Jomary C, Vincent KA, Grist J, et al. Rescue of photoreceptor function by AAV mediated gene transfer in a mouse model of inherited retinal degeneration. Gene Ther. 1997;4(7):683-690.
- Sarra GM, Stephens C, De AM, et al. Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina. Hum Mol Genet. 2001;10(21):2353-2361.
- Maguire AM, Simonelli F, Pierce EA, et al. Safety and Efficacy of gene transfer for Leber congenital amaurosis. N Engl J Med. 2008;358(21):2240-2248
- Stieger K, Lorenz B. Gene therapy for vision loss — recent developments. Discov Med. 2010;10(54):425-433.
- Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA. 2006;103(10):3896-3901.
- Talcott KE, Ratnam K, Sundquist SM, et al. Longitudinal study of cone photoreceptors during retinal degeneration and in response to ciliary neurotrophic factor treatment. Invest Ophthalmol Vis Sci. 2011;52(5):2219-2226.
- Birch DG, Bennett LD, Duncan JL, Weleber RG, Pennesi ME. Long-term Follow-up of Patients With Retinitis Pigmentosa Receiving Intraocular Ciliary Neurotrophic Factor Implants. Am J Ophthalmol. 2016 Oct;170:10-14.
- Humayun MS, Weiland JD, Fujii GY, et al. Visual perception in a blind subject with a chronic microelectronic retinal prosthesis. Vision Res. 2003;43(24):2573-2581.
- Ghodasra DH, Chen A, Arevalo JF, Birch DG, Branham K, Coley B, Dagnelie G, Juan ED; Devenyi RG, Dorn JD, Fisher A, Geruschat DR, Gregori NZ, Greenberg RJ, Hahn P, Ho AC, Howson A, Huang S, Iezzi R, Khan N, Lam BL, Lim JI, Locke KG, Markowitz M, Ripley AM, Rankin M, Schimitzek H, Tripp F, Weiland JD, Yan J, Zacks DN, Jayasundera KD, Jayasundera KT. Worldwide Argus II Implantation: Recommendations to Optimize Patient Outcomes. BMC Ophthalmology 2016;16(1):52.




{kind=link}