
Skeletal dysplasia
Skeletal dysplasias also known as osteochondrodysplasias, are a complex heterogeneous group of more than 450 inherited disorders of bone and cartilage, but can also have significant effects on muscle, tendons and ligaments 1 with extraordinary clinical and molecular heterogeneity 2. Skeletal dysplasias are characterized by abnormalities of cartilage and bone growth, resulting in abnormal shape and size of the skeleton and disproportion of the long bones, spine, and head 3. Skeletal dysplasias differ in natural histories, prognoses, inheritance patterns, and etiopathogenetic mechanisms. Typified by short stature (defined as height that is three or more standard deviations below the mean height for age), skeletal dysplasias can be accompanied by involvement of other systems, including the neurologic, respiratory, and cardiac systems.
Fetal skeletal dysplasia affects approximately 2.4–4.5 of 10,000 births 4. Although fetal skeletal dysplasia is associated with few chromosomal abnormalities, skeletal dysplasia is mostly associated with mutations in genes that regulate bone formation 5.
Skeletal dysplasias frequently present in the newborn period with disproportion, radiographic abnormalities, and occasionally other organ system abnormalities 6. For improved clinical care it is important to determine a precise diagnosis to aid in management, familial recurrence and identify those disorders highly associated with mortality. Long-term management of these disorders is predicated on an understanding of the associated skeletal system abnormalities and these children are best served by a team approach to health care surveillance.
Skeletal dysplasia classification changes as scientists learn about their molecular bases. The molecular basis for a large majority of these disorders in now known. Commonly seen skeletal dysplasias include achondroplasia, osteogenesis imperfecta, thanatophoric dysplasia, campomelic dysplasia, and hypochondroplasia.
Over the past decades, substantial advances have been made in understanding the underlying genetic abnormalities responsible for most skeletal dysplasias 7. The 2015 revision of Nosology and Classification of Genetic Skeletal Disorders recognizes 436 genetic skeletal diseases, stemming from mutations in 364 genes, with the disorders organized into 42 groups 8. Classifications based on the underlying molecular genetic cause are regularly updated 9.
Skeletal dysplasias can be inherited as autosomal dominant, autosomal recessive, X-linked, or Y-linked. Examples include the following:
- Autosomal dominant – Fibroblast growth factor 3 ( FGFR3) disorders (achondroplasia, thanatophoric dysplasia, hypochondroplasia) and type II collagen disorders (achondrogenesis II, spondyloepiphyseal dysplasia congenita, Kniest dysplasia)
- Autosomal recessive – Cartilage-hair hypoplasia, Ellis-van Creveld syndrome, hypophosphatasia, osteopetrosis
- X-linked dominant – Chondrodysplasia punctata
- X-linked recessive – Conradi-Hunermann chondrodysplasia punctata
A brief overview of the most common skeletal dysplasias is included below.
Skeletal dysplasia key points
- Children with skeletal dysplasia are ideally care for by a multidisciplinary approach that includes obstetricians (prior to delivery), pediatricians, neonatologists, medical geneticists, endocrinologists, neurosurgeons, otolaryngologists and orthopaedic surgeons.
- Approximately 5% of child with congenital birth defects have skeletal dysplasias
- Lethality usually results from small chest, pulmonary hypoplasia and respiratory compromise
- Diagnosis is made based on clinical and radiographic findings, including molecular diagnosis when possible
- Nonlethal skeletal dysplasias are associated with short stature but overall affected individuals are cognitively normal and have a good quality of life
Skeletal development
The skeletal forms under two distinct processes; endochondral and membraneous ossification 6. Endochondral ossification is responsible for the formation of most of the mammalian appendicular skeleton and it involves a sequence of carefully orchestrated developmental processes. These include embryonic limb bud initiation and its outgrowth from lateral plate mesoderm, specification of mesenchymal cells for the future limb elements, mesenchymal condensations triggering cartilage differentiation, ossification of developing bones, and finally, their proper growth and maturation in the postnatal period 10. Membraneous ossification is the developmental event in which condensing mesenchymal cells progress almost directly to bone cells. The bones of the skull, lateral clavicle and pubis form via mesenchymal ossification. Postnatally, growth continues through the cartilage growth plate in which resting chondrocytes proliferate, undergo hypertrophy, then apoptosis, becoming the growing scaffold of bone 11. Multiple molecular mechanisms (genes) underlie skeleton formation and perturbations to these highly orchestrated processes can lead to skeletal dysplasias 12.
Skeletal dysplasia genetics
The skeletal dysplasias are inherited in an autosomal recessive, autosomal dominant, X-linked recessive, X-linked dominant, and Y-linked manner 1. Appreciation of the mode of inheritance is important because it imparts information to families regarding future recurrences. Family history, including parental and familial heights and growth patterns, should be obtained from the parents of any affected child to determine if there are similarly affected siblings, or other family members, which can lead to a diagnosis or establish the mode of inheritance. There are several X-linked skeletal disorders that recur in male family members and carrier females are either unaffected or only mildly affected (e.g. X-linked Spondyloepiphyseal Tarda) 13. Table 1 lists some of these more commonly seen disorders seen in the neonatal period with their respective inheritance pattern. The Nosology and Classification of Genetic Skeletal Disorders 1 and On-line Inheritance of Man (https://omim.org/) are sources that include information on these disorders and include data on their patterns of inheritance. There are some uncommonly seen patterns of inheritances in the skeletal dysplasias. These include somatic mosaicism, in which one of the parents is mildly affected and their offspring is more severely affected 14. Evaluation of the parents should be considered if there is any question that one of the parents could have a mild skeletal disorder. Gonadal mosaicism is characterized by familial recurrence of a known dominant disorder resultant from one parent carrying heterozygosity for a mutation in one of the cell lineages that comprise the pool of progenitor germ cells and the parent is clinically unaffected 15. This is a rare occurrence, but influences counseling of all dominant disorders because if a newborn is diagnosed with an autosomal dominant disorder, counseling should include a less than 1% recurrence risk based on gonadal mosaicism.
Table 1. Skeletal dysplasia types and mode of inheritance and genes
| Group/name of disorder | Mode of Inheritance | Gene Symbol |
| FGFR3 chondrodysplasia group | ||
| Thanatophoric dysplasia | AD | FGFR3 |
| Achonodroplasia | AD | FGFR3 |
| Hypochondroplasia | AD | FGFR3 |
| Severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN) | AD | FGFR3 |
| Type II collagen disorders | ||
| Achondrogenesis II | AD | COL2A1 |
| Hypochondrogenesis | AD | COL2A1 |
| Spondyloepiphyseal dysplasia congenita (SEDC) | AD | COL2A1 |
| Kniest dysplasia | AD | COL2A1 |
| Type X1 collagen disorders | ||
| Fibrochondrogenesis | AR | COL11A1 |
| Fibrochondrogenesis | AD | COL11A1, COL11A2 |
| Otospondylomegaepiphyseal dysplasia (OSMED) | AR | COL11A2 |
| Sulfation disorders | ||
| Achondrogenesis IB | AR | SLC26A2 |
| Atelosteogenesis II | AR | SLC26A2 |
| Diastrophic dysplasia | AR | SLC26A2 |
| Chondrodysplasia with congenital joint dslocations | AR | CHST3 |
| Perlecan disorders | ||
| Dyssegmental dysplasia | AR | PLC |
| Dyssegmental dysplasia, Silverman-Handmaker type | AR | PLC |
| Dyssegmental dysplasia, Rolland Desbuquois type | AR | PLC |
| Filamin Disorders and similar disorders | ||
| Otopalatodigital syndrome I and II | XLD | FLNA |
| Osteodysplasty, Melnick-Needles | XLD | FLNA |
| Atelosteogenesis types I and III | AD | FLNB |
| Larsen syndrome | AD | FLNB |
| Spondylo-carpal-tarsal dysplasia | AR | FLNB |
| Serpentine fibula-polycystic kidney syndrome | AD | NOTCH2 |
| TRPV4 disorders | ||
| Metatopic dysplasia | AD | TRPV4 |
| Short-rib dysplasias (with and without polydactyly) | ||
| Chondroectodermal dysplasia (Ellis-van Creveld (EVC) | AR | EVC1, EVC2 |
| Short-rib polydactyly syndrome I, II, III and IV including Asphxiating Thoracic Dystrophy | AR | DYNC2H1 |
| IFT80 | ||
| NEK | ||
| WDR35 | ||
| WDR19 | ||
| WDR34 | ||
| Thoracolaryngeal dysplasia | AD | unknown |
| Metaphyseal dysplasias | ||
| Cartilage-hair hypoplasia | AR | RMRP |
| Metaphyseal dysplasia, Jansen type | AD | PTHR1 |
| Spondylo-epi-(meta)-physeal dysplasia | ||
| SEMD, short limb abnormal calcification type | AR | DDR2 |
| Severe spondylodysplastic dysplasias | ||
| Achondrogenesis 1A | AR | GMAP210 |
| Schneckenbecken dysplasia | AR | SLC35D1 |
| Opsismodysplasia | AR | INPPL1 |
| Acromesomelic disorders | ||
| Acromesomelic dysplasia, type Maroteaux | AR | NPR2 |
| Mesomelic and rhizo-mesomelic dysplasias | ||
| Langer type (homozygoud dyschondrosteosis | pseudo-AR/XLD | SHOX |
| Omodysplasia | AR | GPC6 |
| Robinow syndrome, recessive | AR | ROR2 |
| Robinow syndrome, dominant | AD | WNT5 |
| Bent bone dysplasias | ||
| Campomelic dysplasia | AD | SOX9 |
| Stuve-Wiedemann dysplasia | AR | LIFR |
| Bent bone dysplasia FGFR2 type | AD | FGFR2 |
| Slender bone dysplasias | ||
| Microcephalic osteodysplastic primordial dwarfism (MOPD1) | AR | RNU4ATAC |
| Microcephalic osteodysplastic primordial dwarfism (MOPD2) | AR | PCNT |
| Osteocraniostenosis | FAM111A | |
| Dysplasias with multiple joint dislocations | ||
| Desbuquois dysplasia | AR | CANT1, XYLT1 |
| Pseudodiatrophic dysplasia | AR | unknown |
| Chondrodysplasia punctata group (CDP) | ||
| CDP, X-linked dominant | XLD | EBP |
| Conradi-Hunermann type (CDPX2) | XLR | ARSE |
| brachytelephalangic type (CDPX1) | XLD | NSDHL |
| CHILD syndrome | XLD | EBP |
| Greenberg dysplasia | AR | LBR |
| Rhizomelic CDP type 1 | AR | PEX7 |
| Rhizomelic CDP type 2 | AR | DHPAT |
| Rhizomelic CDP type 3 | AR | AGPS |
| Neonatal osteosclerotic dysplasias | ||
| Bloomstrand dysplasia | AR | PTHR1 |
| Desmosterolosis | AR | DHCR24 |
| Caffey disease (infantile) | AD | COL1A1 |
| Raine dysplasia | AR | FAM20C |
| Increased bone density group | ||
| Osteopetrosis (severe neonatal or infantile forms) | AR | TCIRG1 |
| Osteopetrosis (severe neonatal or infantile forms) | AR | CLCN7 |
| Dysosteosclerosis | AR | SLC29A3 |
| Lenz-Majewski hyperostostic dysplasia | SP | PTDSS1 |
| Osteogenesis imperfecta and decreased bone density group | ||
| Osteogenesis imperfecta, moderate, severe and perinatal lethal | AD | COL1A1, COL1A2 |
| IFITM5 | ||
| Osteogenesis imperfecta, moderate, severe and perinatal lethal | AR | CRTAP |
| P3H1 | ||
| PPBI | ||
| FKBP10 | ||
| HSP47 | ||
| SP7 | ||
| WNT1 | ||
| TMEM33B | ||
| Bruck syndrome | PLOD2 | |
| FKBP10 | ||
| Osteoporosis-pseudoglioma syndrome | AR | LRP5 |
| Cole-Carpenter dysplasia | SP | unknown |
| Abnormal mineralization group | ||
| Hypophosphatasia, perinatal and infantile forms | AR | ALPL |
Abbreviations: AD = autosomal dominant; AR = autosomal recessive; XLD = X-linked dominant inheritance
[Source 6 ]Skeletal dysplasia types
In the 2010 revision of the Nosology and Classification of Genetic Skeletal Disorders, 456 conditions were classified into 40 groups defined by molecular, biochemical and radiographic criteria 16. Among these conditions, 316 conditions were associated with mutations in one or more of 226 different genes, providing a basis for the molecular genetic diagnosis of fetal skeletal dysplasia.
Groups of skeletal dysplasia conditions organized according to their molecular bases 2:
- FGFR3 chondrodysplasia group
- Type 2 collagen group and similar disorders
- Type 11 collagen group
- Sulfation disorders group
- Perlecan group
- Aggrecan group
- Filamin group and related disorders
- TRPV4 group
Groups of skeletal dysplasia conditions organized according to their clinical presentations:
- Short-ribs dysplasias (with or without polydactyly) group
- Short-ribs dysplasias (with or without polydactyly) group
- Multiple epiphyseal dysplasia and pseudoachondroplasia group
- Metaphyseal dysplasias
- Spondylometaphyseal dysplasias
- Spondylo-epi-(meta)-physeal dysplasias
- Severe spondylodysplastic dysplasias
- Acromelic dysplasias (extremities of the limbs)
- Acromesomelic dysplasias (extremities and middle portion of the limbs)
- Mesomelic and rhizo-mesomelic dysplasias (proximal and middle portions of the limbs)
- Bent bones dysplasias
- Slender bone dysplasia group
- Dysplasias with multiple joint dislocations
- Chondrodysplasia punctata group
- Neonatal osteosclerotic dysplasias
- Increased bone density group (without modification of bone shape)
- Increased bone density group with metaphyseal and/or diaphyseal involvement
- Osteogenesis imperfecta and decreased bone density group
- Abnormal mineralization group
- Lysosomal storage diseases with skeletal involvement (dysostosis multiplex group)
- Osteolysis group
- Disorganized development of skeletal components group
- Overgrowth syndromes with skeletal involvement
- Genetic inflammatory/rheumatoid-like osteoarthropathies
- Cleidocranial dysplasia and isolated cranial ossification defects group
- Craniosynostosis syndromes
- Dysostoses with predominant craniofacial involvement
- Dysostoses with predominant vertebral with and without costal involvement
- Patellar dysostoses
- Brachydactylies (with or without extraskeletal manifestations)
- Limb hypoplasia—reduction defects group
- Polydactyly-Syndactyly-Triphalangism group
- Defects in joint formation and synostoses
FGFR3 chondrodysplasia group
FGFR3 chondrodysplasia group of disorders all result from heterozygosity for activation mutations in the gene that encodes Fibroblast Growth Factor Receptor 3 (FGFR3) 17. FGFR3 chondrodysplasia group of disorders includes thanatophoric dysplasia, achondroplasia, hypochondroplasia and the very rare severe achondroplasia with developmental delay and acanthosis nigricans (SADDAN). Thanatophoric dysplasia is almost uniformly lethal without aggressive management. Radiographic findings include macrocephaly with narrow skull based, long narrow trunk with handlebar clavicles, flat vertebral bodies, small flared iliac bones with very narrow sacrosciatic notches and generalized micromelia.
Thanatophoric dysplasia
Thanatophoric dysplasia named because it often results in early death, is characterized by micromelia (abnormally small and imperfectly developed extremities) with bowed femurs, short ribs, narrow thorax, macrocephaly, distinctive facial features, brachydactyly, hypotonia. Radiographically, there is rhizomelic shortening of the long bones with irregular metaphyses, platyspondyly, small foramen magnum with brain stem compression, bowed femurs (thanatophoric dysplasia type I) and cloverleaf skull (always in thanatophoric dysplasia type II; sometimes in thanatophoric dysplasia type I). CNS abnormalities include temporal lobe malformations, hydrocephaly, brain stem hypoplasia and neuronal migration abnormalities.
Achondroplasia
This is the most common nonlethal skeletal dysplasia, occurring in 1:26,000-1:28,000 live births and affecting 250,000 individuals worldwide 18.
Achondroplasia is characterized by small stature with rhizomelia (shortened limbs – arms and legs) and redundant skin folds, limitation of elbow extension and genu varum, short fingers with trident configuration of the hands. Craniocervical junction compression is a major complication which may occur and requires surveillance for early detection and management. There is also thoracolumbar kyphosis, lumbar lordosis, and a large head with frontal bossing with midface hypoplasia. The radiographic findings include short tubular bones with metaphyseal flaring, narrowing of the interpediculate distance of the lumbar spine, rounded ilia and horizontal acetabula, narrow sacrosciatic notch and proximal femoral radiolucency. In hypochondroplasia, there are similar but milder clinical and radiological findings, the head is large but there is no midface hypoplasia.
The molecular mechanism for achondroplasia is a G380R mutation in the FGFR3 transmembrane domain. A gain-of-function mutation, it is present in 99% of affected individuals. Inheritance is autosomal dominant, with 80% of cases involving de novo mutations.
Major characteristics of achondroplasia include the following 18:
- Disproportionate short stature with rhizomelic (proximal) shortening of the arms and legs and large head with frontal bossing
- Trident hand configuration
- Average final height: 130 cm for men and 125 cm for women
- Normal intelligence and lifespan
Major complications include the following:
- Craniocervical junction compression
- Middle ear infections
- Obstructive apnea
- Spinal stenosis
Hypochondroplasia
Hypochondroplasia is a autosomal dominant disorder characterized by short-limbed dwarfism, lumbar lordosis, short and broad bones, and caudad narrowing of the interpediculate distance of the lumbar spine. It shows some resemblance to achondroplasia, but is much milder and can be distinguished on clinical and radiographic grounds 19.
Camptodactyly, tall stature, and hearing loss syndrome (CATSHL)
Camptodactyly-tall stature-scoliosis-hearing loss syndrome is characterized by camptodactyly 20, tall stature, scoliosis, and hearing loss (CATSHL). It has been described in around 30 individuals from seven generations of the same family. The CATSHL syndrome is caused by a missense mutation in the FGFR3 gene, leading to a partial loss of function of the encoded protein, which is a negative regulator of bone growth.
Type 2 collagenopathies group
Type 2 collagenopathies are a heterogenous group of disorders that all show some relationship to each other clinically and radiographically 21. Type 2 collagenopathies typically result from heterozygosity for mutations in the type II collagen gene, COL2A1. Achondrogenesis II and hychondrogenesis are lethal disorders that present with large skulls, very small and short ribs, almost lack of mineralization of most vertebral bodies. The pelvis has small iliac wings with absent ischia, pubic bones and sacral elements. The extremities show severe rhizomelia and mesomelia with relative sparing of the hands.
Stickler syndrome
Stickler syndrome is characterized by ocular findings of myopia, cataract, and retinal detachment, sensorineural and conductive hearing loss, flat mala and cleft palate (alone or as part of the Robin sequence), mild spondyloepiphyseal dysplasia and early-onset arthritis 22.
Spondyloepiphyseal dysplasia congenita
Spondyloepiphyseal dysplasia congenita presents with disproportionate short stature (short trunk), abnormal epiphyses, and flattened vertebral bodies. Spondyloepiphyseal dysplasia is usually nonlethal and clinical presentation includes severely short trunked, short-limbed infant with normal appearing hands, feet and skull. There may be micrognathia and a Pierre Robin sequence and attention should be paid to the upper cervical spine, which can be hypomineralized. Radiographic findings in the newborn period include short thorax with short ribs, pear shaped or oval vertebral bodies, absent pubic ossification, short long bones and unossified talus and calcaneus (which are typically ossified by about 24 weeks of gestation) 23. Pediatricians should be aware that these children can have some respiratory issues based on small rib cage, feeding difficulties due to micrognathia and flat mid face and are at risk for recurrent ear infections. Families should be counseled that serial evaluation of the cervical spine by a team of specialists who can management odontoid hypoplasia in childhood as part of long-term care for these children.
Osteogenesis imperfecta
Osteogenesis imperfecta also known as brittle bone disease, is a heterogenous group of heritable connective tissue disorders, is another common skeletal dysplasia, with a prevalence of 1:15,000-1:20,000 births. Severe forms may present in the newborn period with fractures sustained in-utero. Osteogenesis imperfecta has been classified in numerous forms, mild, moderate, severe and perinatal forms. These classifications are used by clinicians to manage the complications and predict some of the natural history associated with the disorder, and are somewhat arbitrary 24. In the last decade there has been enormous progress in unwinding the molecular genetics associated with osteogenesis imperfecta 25. About 90% of the cases result from mutations in the genes that encode type I collagen, COL1A1 and COL1A2. Most of the remaining cases result from gene mutations that are recessively inherited 25. These recessively inherited cases usually present with severe forms of osteogenesis imperfecta, some of them perinatal lethal 26. They are extremely rare, and unless there is a family history consistent with autosomal recessive inheritance (previously affected sibling or consanguinity), most osteogenesis imperfecta cases result from dominantly or sporadically inherited mutations.
Newborns with osteogenesis imperfecta present with relative macrocephaly, flat facies, short limbs, narrow thorax, deformed and fractured extremities, with normal appearing hands and feet. Radiographs show poor ossification of the skull, fractured ribs, under-mineralized bone with fractures and in very severe cases, femurs that are accordion in appearance, diffuse osteopenia including the vertebrae, flattened acetabulae and relatively normal appearing hands and feet. Similarly to the aforementioned skeletal dysplasias, there is a broad phenotypic spectrum in osteogenesis imperfecta. The perinatal lethal form usually presents with a very small chest and deformed extremities. Care should be taken if a newborn with osteogenesis imperfecta and in-utero occurring fractured ribs requires ventilator assistance. There is probable underlying pulmonary insufficiency due to a small chest and ventilation may cause ongoing fractures in a probable lethal condition; thus the degree and period of active intervention should be discussed with the family.
For those newborns who are stable, but present with fractures, orthopaedic surgeons should evaluate the patient. In many instances, the treatment modality depends on the site of the fracture and degree of angulation at the site. Many of these fractures are managed by immobilization using strapping (e.g. femoral fractures in the subtrochanteric region are managed by strapping of the thigh to abdomen) while fractures of the shaft of bones may be managed by casting 27. In nonlethal but severe cases, consultation should be obtained from medical genetics or pediatric endocrinology for consideration of treatment with intravenous bisphosphonates while in the nursery. Studies have shown that early treatment with bisphosphonates decreases the number of subsequent fractures 28. Individuals with osteogenesis imperfecta also have fragile skin and blood vessels because type I collagen is an important component of connective tissue. Care in handling of these children should be discussed with staff.
Inheritance in osteogenesis imperfecta is as follows:
- Types I-IV (Sillence classification, 85% of cases): Autosomal dominant
- Types V-XII: Autosomal recessive, except for type V (autosomal dominant)
Major characteristics of the most common forms of osteogenesis imperfecta are as follows.
All types
All forms are characterized by bone fragility and susceptibility to fracture from minimal trauma.
Osteogenesis imperfecta type I – mild form with diagnosis in early childhood
This is characterized by the following:
- Sclera may be blue
- Dentinogenesis imperfecta (subtype IB)
- Normal stature reached
- Hearing loss in 50% of patients
Osteogenesis imperfecta type II – perinatal lethal form
The condition is characterized as follows:
- Patients may survive the neonatal period
- Later mortality can occur secondary to pneumonia and respiratory insufficiency
Osteogenesis imperfecta type III – progressive deforming form
This form is characterized as follows:
- Moderate deformity at birth
- Development of chest wall deformities
- Most patients are wheelchair dependent
- Very short stature
- Variable sclera
- Dentinogenesis imperfecta and hearing loss are common.
Osteogenesis imperfecta type IV – moderately severe form
Characteristics include the following:
- Mild to moderate bone deformity
- Variable short stature
- Hearing loss occurs in some families
- Variable sclera
Major complications
- These include bone fractures, bone deformity, and growth deficiency.
Skeletal dysplasia diagnosis
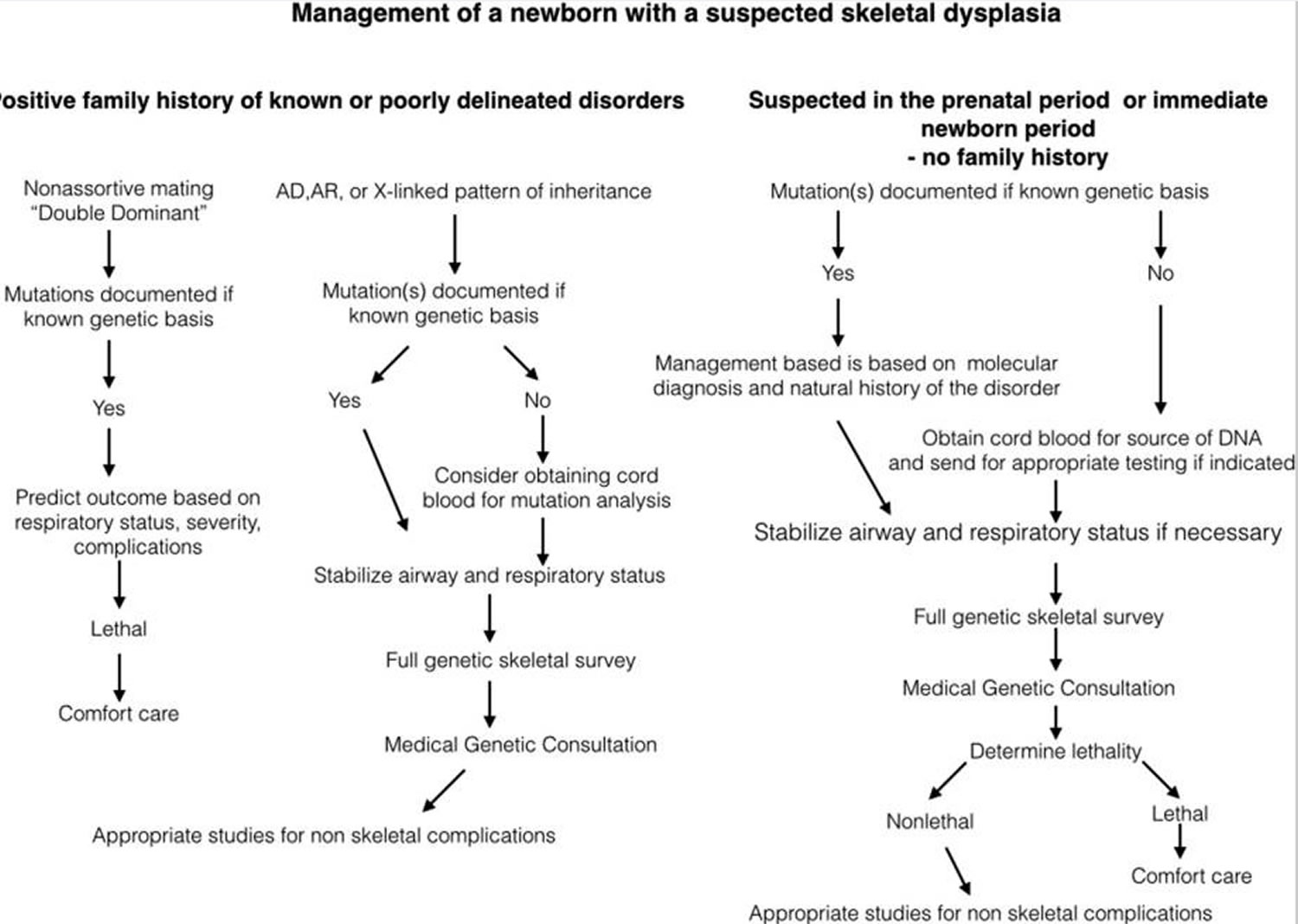
Diagnosis of skeletal dysplasias requires a multidisciplinary approach and includes radiologic and genetic evaluations. This approach is required for accurate diagnosis and the determination of the best treatment options, as well as for accurate counseling on outcomes and risk of recurrence. The approach to the initial evaluation of newborn with a suspected skeletal dysplasia is outlined in Figure 1 below.
If the diagnosis has been determined in the prenatal period or based on family history, then laboratory testing or radiographs should be obtained to confirm the clinical or molecular diagnosis. If the skeletal dysplasia is undiagnosed or unexpected then a similar approach should be taken. Once the newborn is stable, a thorough physical exam should be undertaken. Key measurements include head circumference (HC), birth weight (BW), birth length (BL), chest circumference if it appears small, and palm and middle finger lengths. Carefully delineation of any dysmorphic facial features should be performed and include evaluation of the fontanels, nasal bridge, midface, philtrum, mandible, palate and ears. Frequently, the neck will appear short and if the chest is small the nipples may appear widely spaced. In many disorders the hands will appear small, and the fingers will appear to have wide spaces between them because they are short. Attention should be paid to the proportion of the upper and mid sections of the arm. In most newborns they appear subjectively one to one, and in skeletal disorders, there is frequently subjective disproportion. Rhizomelia is common in achondroplasia and spondyloepiphyseal dysplasia congenita, two of the commonly seen nonlethal disorders. Very significant mesomelia relative to the rhizomelic segment suggests a group of specific disorders, the mesomelic dysplasias. Frequently, when there is rhizomelia and mesomelia, there are increased skin creases and skin folds due to the abnormal underlying bone length. A significantly curved long bone may appear substantially much shorter externally than by radiographs.
After clinical evaluation, it is critical to obtain conventional radiographic examination of the dysplastic skeleton. The skeletal survey should include the skull (anteroposterior [AP], lateral views), chest (AP), spine (AP and lateral views, including dedicated lateral view of the cervical spine), pelvis (AP), tubular bones (AP), and/or hands and feet (AP) 29. Radiographic evaluation should start with overall assessment of epiphyseal ossification to determine if they are delayed or irregular for age, then there should be consideration for an epiphyseal dysplasia. If the metaphyses are widened, flared or irregular then the diagnosis of a metaphyseal chondrodysplasia should be entertained. If diaphyseal abnormalities are present, such as widening and/or cortical thickening, or marrow space expansion then a diaphyseal dysplasia is implied. Combination of the aforementioned abnormalities helps categorize the disorder, e.g. epimetaphyseal disorder. If the vertebral bodies are affected then there is a spondylo-component present, further categorizing the disorder. Once the extent of radiographic abnormalities are determined and placed with a category, e.g. spondylometaphyseal dysplasia, radiographic textbooks can aid in refining the diagnosis.
Organ system abnormalities beyond the skeleton are occasionally seen and can be clues to diagnosis. Congenital heart defects are commonly seen in the skeletal ciliopathies (chondroectodermal dysplasia, asphyxiating thoracic dysplasia, short rib polydactyly syndromes), abnormal formed genitalia in the skeletal ciliopathies, campomelic dysplasia, omodysplasia, Robinow dysplasia, Antley-Bixler and severe disorders of cholesterol metabolism 30. Immune deficiency and Hirschprung disease are seen in metaphyseal chondrodysplasia, McKusick type (Cartilage hair hypoplasia) 31.
In addition, genetic testing plays a critical role in the diagnosis and management of skeletal dysplasias. Molecular diagnostic techniques have led to the identification of the underlying gene disorders in about two thirds of known skeletal dysplasias.
Prenatal diagnosis
Rapid advances of in both imaging modalities and molecular diagnostics has improved doctors abilities to recognize osteochondrodysplasias in the prenatal period 32. For those families with a parent affected by an autosomal dominant disorder, either molecular diagnostics via invasive techniques or by ultrasound imaging, can aid in predicting whether the fetus will be similarly affected (Figure 1). For at-risk families based on a previously affected child with an autosomal recessive disorder, the same above-mentioned approach can be utilized. However, affected neonates with skeletal dysplasias are often the first affected children in their respective families.
Many pregnant women are offered an array of noninvasive tests to determine if their fetuses are at risk for genetic disorders 33. These molecular screening panels are for autosomal recessive and X-linked disorders including skeletal dysplasias such as diastrophic dysplasia; however it is important to note that many genes and mutations that can cause skeletal dysplasias are presently not included in these panels. Usually if one parent screens positive for a disorder, then the other parent is screened, determining a baseline risk for a disorder. First trimester ultrasound analysis, usually used to identify aneuploidy, is also affective in identifying severe, usually lethal skeletal dysplasias including osteogenesis imperfecta, thanatophoric dysplasia, and the short-rib polydactyly syndromes, as some examples 34. If a neonate with a skeletal disorder was noted to have abnormal ultrasound findings in the first trimester including a small crown-rump length for gestational age and increased nuchal fold thickness, the likelihood is that the fetus has a severe, probable lethal skeletal dysplasia.
Many prenatal skeletal dysplasias are diagnosed in the late second trimester when many pregnant women are screened by ultrasound for congenital anomalies 35. The advantages to early detection (with and without a precise diagnosis) allows for preparation prior to delivery. This includes discussion of active resuscitation, proper assembly of consultants, collection of appropriate material for molecular diagnosis from cord blood, and smoother transition for the fetus from the prenatal to neonatal period. This is particularly important if the neonate is predicted to have a severe, but non-lethal skeletal disorder.
Experience has informed us that many skeletal disorders are not diagnosed in the late second trimester, but either in the third trimester or at birth. These disorders tend to be milder, with a less profound effect on the skeletal, including the thorax, thus severe respiratory compromise is not usually encountered. Achondroplasia, Spondyloepiphyseal Dysplasia Congenita and nonlethal forms of Osteogenesis Imperfecta are frequency first encountered in the newborn period. Many parents are distraught if the suggestion of a skeletal dysplasia is made at birth, and question if “something” was missed at second trimester ultrasound evaluation. It is helpful to explain that for the milder skeletal disorders, long bone measurements are frequently on the normal growth curves at 20 weeks, and fall off occurs in the third trimester since in most of these disorders the defect is in endochondral ossification (not condensation or other earlier-occurring processes) which is a process that is most active in the third trimester 36.
Beyond ultrasound and enhanced carrier screening panels, noninvasive prenatal testing from maternal blood for Mendelian disorders may soon be a clinical reality. Screening for cell free fetal DNA in maternal blood during the first trimester of gestation is employed for the detection of aneuploidy, including disorders of copy number variations 37. Reports of de novo FGFR3 mutations (thanatophoric dysplasia) from the fetal cell free DNA fraction in maternal blood illustrates that this technology has the future potential to diagnose de novo autosomal dominant skeletal disorders 38.
Figure 1. Scheme of management in newborns with a skeletal dysplasia
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Skeletal dysplasia management
Treatment is supportive. Medical care for individuals with skeletal dysplasia should be directed at preventing neurologic and orthopedic complications due to spinal cord compression, joint instability, and long bone deformity.
Surgical intervention depends on the signs and symptoms of skeletal dysplasia. For example, progressive kyphosis, which may lead to spinal cord compression and spastic paraparesis, is best treated by anterior and posterior fusion. Lumbar lordosis with spinal stenosis responds to extensive lumbar laminectomy. Surgical decompression is required to relieve edema of the cervicomedullary cord secondary to bony compression.
Defining Lethality
If a newborn has been diagnosed with a known lethal disorder in the fetal period, then treating physicians should offer comfort care for the newborn, but not aggressive management. Defining lethality in the prenatal period can be accomplished by two means; molecular diagnosis of a known lethal disorder, precise diagnosis by ultrasound or chest size abnormalities seen by ultrasound that correlate with lethality. If the chest to abdominal circumference ratio is less than 0.7, the heart to chest circumference is >50 percent or the abdomen to femur length ratio is < 0.16, these objective measures are highly correlated with lethality. However, these measurements can be gestational age dependent and may not apply to the infant 32 with significant prematurity and a skeletal dysplasia; the presenting disorder may be a known lethal disorder, but the aforementioned criteria may not quite have been reached, or conversely a severely affected child may survive for a prolonged period of time. When lethality is suspected based only on prenatal ultrasound measurements, it is important to counsel parents that while a lethal condition is suspected that only at birth will a true assessment of lethality be possible.
Postnatally, similar criteria can apply to management of a newborn. If clinical and radiographic findings and/or molecular findings indicate a known lethal diagnosis, then supportive care is indicated. Some of the more difficult clinical management scenarios result from the clinical spectrum of disease that is associated individual gene defects. This is particularly true for the disorders listed in the categories in Table 1. For example, in type II collagen disorders, hypochonodrogenesis diagnosed by radiographic criteria is often associated with lethality due to a small chest and respiratory compromise, yet some of these infants survive the neonatal period and then are categorized as severe spondyloepiphyseal dysplasia congenita 21. Thus for each group of skeletal dysplasias, clinical judgment must be used to determine the severity of the skeletal disorder and whether aggressive management is indicated. If highly aggressive ventilation is necessary because of severe respiratory insufficiency particularly due to a small chest, respiratory failure and lethality commonly occurs 39. For all newborns that die of complications from skeletal disorders, radiographs and a source of DNA should be obtained, and families should be offered autopsy. Collection of postmortem material aids in final diagnosis, which helps to provide parents emotional closure and determine recurrence risk for families through clinical molecular diagnosis or research participation.
If the newborn is known to have a uniformly lethal disorder, then supportive care is indicated. Similarly, if the diagnosis is known prior to delivery then management is predicated on the knowledge regarding the natural history of the disorder. Newborns with recognized skeletal disorders present in the newborn period with disproportion. Dependent on the skeletal disorder, there are some common findings; this includes relative macrocephaly, narrow chest appearance relative to the abdomen, rhizomelia (short upper portion extremity), mesomelia (short mid-portion extremity), and frequently brachydactyly (short hands, including phalanges). Frequently the face is normal but there are numerous disorders associated with a flat nasal bridge, frontal bossing and midface hypoplasia and include many of the lethal skeletal disorders, achondroplasia, campomelic dysplasia, chondrodysplasia punctate (all forms), type II collagen disorders, Larsen syndrome, and the mucopolysaccharidoses (most forms). Disorders with micrognathia include some of the following disorders: type II collagen disorders, acrofacial dysostoses, Robinow syndrome and again, many of the lethal skeletal dysplasias. Attention must be paid to the Robin-Pierre Robin sequence (small mandible and posterior cleft palate) associated with these disorders. Few of these newborns are delivered by EXIT procedures because of the mandibular abnormality and safety of the airway 40, but some of these children require post delivery intubation and subsequent tracheostomies until definitive surgery for jaw retraction or time, awaiting facial growth. If the newborn has Pierre-Robin sequence, timing of the repair of the cleft palate should be deferred to the craniofacial surgery team or plastic surgeons 41.
Care should be taken regarding the trachea in the skeletal dysplasias. The trachea is composed of fifteen to twenty incomplete C-shaped tracheal rings that contain cartilage that reinforces the front and sides of the trachea to protect and maintain the airway 42. Pediatricians, neonatologists and anesthesiologists should be informed that many skeletal disorders have been associated with tracheal anatomic changes complicating respiratory status, intubation and any manipulations. The abnormalities include tracheal agenesis, congenital stenosis, premature cartilage calcifications, short trachea and tracheomalacia, as well as disorders associated with tracheoesphageal fistulas 43. Caretakers should be aware of this when caring for newborns with skeletal abnormalities, and can affect prognosis in many of the nonlethal skeletal disorders.
Long-term follow-up
Children with skeletal dysplasia are best managed by a multidisciplinary approach that includes obstetricians (prior to delivery), pediatricians, neonatologists, medical geneticists, endocrinologists, neurosurgeons, otolaryngologists and orthopaedic surgeons. Sub-speciality care should be directed toward the individual presenting issue. Families should be prepared for ongoing visits to supervise health management. For most children with nonlethal skeletal dysplasias, their quality of life is good. Families should be encourage to seek out support from organizations dedicated to the well being of individuals with forms of dwarfism that include Little People of America (https://www.lpaonline.org/) and the Osteogenesis Imperfecta Foundation (http://www.oif.org/site/PageServer). Pediatricians should be aware that these children with normal immune systems should receive standard doses of vaccines on routine schedules, as well as recognizing the potential for increased ear infections in disorders associated with midface hypoplasia, hearing loss due to increased infection and abnormalities of the stapes, as well as hydrocephalous in disorders associated with foramen magnum stenosis (achondroplasia, hypochondroplasia, diastrophic dysplasia and metatropic dysplasia).
- Warman ML, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2010 revision. American journal of medical genetics. Part A. 2011 May;155A(5):943–968.[↩][↩][↩]
- Campeau P, Schlesinger AE. Skeletal Dysplasias. [Updated 2017 Jan 30]. In: Feingold KR, Anawalt B, Boyce A, et al., editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279130[↩][↩]
- Skeletal dysplasia. https://emedicine.medscape.com/article/943343-overview[↩]
- Liu Y, Wang L, Yang YK, et al. Prenatal diagnosis of fetal skeletal dysplasia using targeted next-generation sequencing: an analysis of 30 cases. Diagn Pathol. 2019;14(1):76. Published 2019 Jul 13. doi:10.1186/s13000-019-0853-x https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6626426[↩]
- Zhou X, Chandler N, Deng L, Zhou J, Yuan M, Sun L. Prenatal diagnosis of skeletal dysplasias using a targeted skeletal gene panel. Prenat Diagn. 2018;38:692–699. doi: 10.1002/pd.5298[↩]
- Krakow D. Skeletal dysplasias. Clin Perinatol. 2015;42(2):301–viii. doi:10.1016/j.clp.2015.03.003 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4456691[↩][↩][↩]
- Ikegawa S. Genetic analysis of skeletal dysplasia: recent advances and perspectives in the post-genome-sequence era. J Hum Genet. 2006. 51(7):581-6.[↩]
- Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A. 2015 Dec. 167A (12):2869-92.[↩]
- Chen C, Jiang Y, Xu C, et al. Skeleton Genetics: a comprehensive database for genes and mutations related to genetic skeletal disorders. Database (Oxford). 2016. 2016[↩]
- Provot S, Schipani E. Molecular mechanisms of endochondral bone development. Biochemical and biophysical research communications. 2005;328(3):658–665.[↩]
- Ballock RT, O’Keefe RJ. The biology of the growth plate. The Journal of Bone & Joint Surgery. 2003;85(4):715–726.[↩]
- Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423(6937):332–336.[↩]
- Gedeon ÁK, Colley A, Jamieson R, et al. Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nature genetics. 1999;22(4):400–404.[↩]
- Edwards MJ, Wenstrup RJ, Byers PH, Cohn DH. Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a mutation in the COL1A2 gene of type I collagen. The mosiac parent exhibits phenotypic features of a mild form of the disease. Human mutation. 1992;1(1):47–54.[↩]
- Cohn D, Starman B, Blumberg B, Byers P. Recurrence of lethal osteogenesis imperfecta due to parental mosaicism for a dominant mutation in a human type I collagen gene (COL1A1). American journal of human genetics. 1990;46(3):591.[↩]
- Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, Mortier G, Mundlos S, Nishimura G, Rimoin DL, Robertson S, Savarirayan R, Sillence D, Spranger J, Unger S, Zabel B, Superti-Furga A. Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011;155A:943–968. doi: 10.1002/ajmg.a.33909[↩]
- Vajo Z, Francomano CA, Wilkin DJ. The Molecular and Genetic Basis of Fibroblast Growth Factor Receptor 3 Disorders: The Achondroplasia Family of Skeletal Dysplasias, Muenke Craniosynostosis, and Crouzon Syndrome with Acanthosis Nigricans 1. Endocrine reviews. 2000;21(1):23–39.[↩]
- Pauli RM, Legare JM. Achondroplasia. 1998 Oct 12 [Updated 2018 May 10]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1152[↩][↩]
- Walker BA, Murdoch JL, McKusick VA, Langer LO, Beals RK. Hypochondroplasia. Am J Dis Child. 1971;122(2):95–104. doi:10.1001/archpedi.1971.02110020029001[↩]
- a medical condition that causes one or more fingers to be permanently bent[↩]
- Kannu P, Bateman J, Savarirayan R. Clinical phenotypes associated with type II collagen mutations. Journal of paediatrics and child health. 2012;48(2):E38–E43.[↩][↩]
- J. Spranger, A. Winterpacht, B. Zabel, The type II collagenopathies: a spectrum of chondrodysplasias. European journal of pediatrics 153, 56 (Feb, 1994).[↩]
- Unger S. A genetic approach to the diagnosis of skeletal dysplasia. Clinical orthopaedics and related research. 2002;401:32–38.[↩]
- Sillence D. Osteogenesis imperfecta: an expanding panorama of variants. Clinical Orthopaedics and Related Research. 1981;159:11–25.[↩]
- Marini JC, Blissett AR. New genes in bone development: what’s new in osteogenesis imperfecta. The Journal of Clinical Endocrinology & Metabolism. 2013;98(8):3095–3103.[↩][↩]
- Baldridge D, Schwarze U, Morello R, et al. CRTAP and LEPRE1 mutations in recessive osteogenesis imperfecta. Human mutation. 2008;29(12):1435–1442.[↩]
- Kancherla R, Sankineani SR, Naranje S, et al. Birth-related femoral fracture in newborns: risk factors and management. Journal of children’s orthopaedics. 2012;6(3):177–180.[↩]
- Cheung MS, Glorieux FH. Osteogenesis imperfecta: update on presentation and management. Reviews in Endocrine and Metabolic Disorders. 2008;9(2):153–160.[↩]
- Parnell SE, Phillips GS. Neonatal skeletal dysplasias. Pediatr Radiol. 2012 Jan. 42 Suppl 1:S150-7.[↩]
- Baujat G, Le Merrer M. Ellis-van Creveld syndrome. Orphanet J Rare Dis. 2007;2(6):27.[↩]
- Mäkitie O, Kaitila I. Cartilage-hair hypoplasia—clinical manifestations in 108 Finnish patients. European journal of pediatrics. 1993;152(3):211–217.[↩]
- Krakow D, Lachman RS, Rimoin DL. Guidelines for the prenatal diagnosis of fetal skeletal dysplasias. Genetics in medicine : official journal of the American College of Medical Genetics. 2009 Feb;11(2):127–133.[↩][↩]
- Levenson D. New test could make carrier screening more accessible. American Journal of Medical Genetics Part A. 2010;152(4):fm vii–fm viii.[↩]
- Parilla BV, Leeth EA, Kambich MP, Chilis P, MacGregor SN. Antenatal detection of skeletal dysplasias. Journal of Ultrasound in Medicine. 2003;22(3):255–258.[↩]
- Schramm T, Gloning K, Minderer S, et al. Prenatal sonographic diagnosis of skeletal dysplasias. Ultrasound in Obstetrics & Gynecology. 2009;34(2):160–170.[↩]
- Krakow D, Williams J, Poehl M, Rimoin D, Platt L. Use of three – dimensional ultrasound imaging in the diagnosis of prenatal – onset skeletal dysplasias. Ultrasound in obstetrics & gynecology. 2003;21(5):467–472.[↩]
- Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proceedings of the National Academy of Sciences. 2008;105(42):16266–16271.[↩]
- Saito H, Sekizawa A, Morimoto T, Suzuki M, Yanaihara T. Prenatal DNA diagnosis of a single-gene disorder from maternal plasma. The Lancet. 2000;356(9236):1170.[↩]
- Harding CO, Green CG, Perloff WH, Pauli RM. Respiratory complications in children with spondyloepiphyseal dysplasia congenita. Pediatric pulmonology. 1990;9(1):49–54.[↩]
- Costello BJ, Edwards SP, Clemens M. Fetal diagnosis and treatment of craniomaxillofacial anomalies. Journal of Oral and Maxillofacial Surgery. 2008;66(10):1985–1995.[↩]
- Schaefer RB, Gosain AK. Airway management in patients with isolated Pierre Robin sequence during the first year of life. Journal of Craniofacial Surgery. 2003;14(4):462–467.[↩]
- Minnich DJ, Mathisen DJ. Anatomy of the trachea, carina, and bronchi. Thoracic surgery clinics. 2007;17(4):571–585.[↩]
- Wells AL, Wells TR, Landing BH, Cruz B, Galvis DA. Short trachea, a hazard in tracheal intubation of neonates and infants: syndromal associations. Anesthesiology. 1989;71(3):367–373.[↩]


{kind=link}