
What is syndactyly
Syndactyly is one of the most common hereditary limb malformations depicting the fusion of certain fingers and/or toes 1. The involvement of feet is more frequent than the involvement of hands, and males are affected twice as frequently as females 2. Syndactyly appears in the medical literature under several synonyms. For example, adherent fingers 3, fingers coated with common skin 4, coherence of fingers 5, fingers grown together 6, fingers knit together 3, skin fusion, digits in stocking 7, fingers stuck together 8, symphalanginae, symphalangus syndactylous 9, syndactylia, syndactylous ossification 9, webbed toes 10 and zygodactyly 11.
Ambroise Pare (1510–1590) in the sixteenth century described syndactyly as fingers stuck together and polydactyly as superfluous fingers, respectively 8. Thus, it was established quite early that webbed fingers are not infrequent, appear in various forms, usually without the involvement of other organ systems but frequently witnessed with extra digits 3. Minor webbing types could be easily overlooked; however, severe types required surgical corrections.
Syndactyly is one of the most common hereditary limb malformations depicting a prevalence of 3–10 in 10 000 births, though higher estimates ranging from 10–40/10 000 have been reported 12. Syndactyly may occur as an isolated entity or a component of more than 300 syndromic anomalies. Syndactylies exhibit great inter- and intra-familial clinical variability. Even within a subject, phenotype can be unilateral or bilateral and symmetrical or asymmetrical. At least nine non-syndromic syndactylies with additional sub-types have been characterized. Most of the syndactyly types are inherited as autosomal dominant but two autosomal recessive and an X-linked recessive entity have also been described 1. Whereas the underlying genes/mutations for types II-1, III, IV, V, and VII have been worked out, the cause and molecular basis of the other syndactyly types remain unknown. Despite considerable progress in the understanding of syndactyly at clinical and molecular levels, fundamental questions regarding the disturbed developmental mechanisms leading to fused digits, remain to be answered 1.
Clinically syndactyly is one of the most heterogeneous developmental deformities known in the medical literature. A number of combinations are possible in which the adjacent fingers and/or toes remain connected by a web. It may be unilateral or bilateral, and symmetrical or asymmetrical. Furthermore, inter- and intra-familial phenotypic variability is quite common. The condition is so variable that the same individual may exhibit asymmetrical phenotypes in the upper and lower, and right and left limbs. Syndactyly can be identified as partial or complete, cutaneous or bony, and involving only the phalanges or further extending up to metacarpal/metatarsal or carpal/tarsal levels, sometimes even proximating the distal end of forearm/foreleg. On the minimal extreme, a milder phenotype may only be recognized by the alterations in interphalangeal creases and peculiarities in dermatoglyphics 13.
Syndactyly may segregate as an isolated clinical phenotype. There are at least nine well-characterized syndactylous entities with subdivisions, the majority of which have non-syndromic nature. Most of these entities segregate in Mendelian dominant fashion. However, two autosomal recessive and an X-linked recessive type have also been described. Generally, autosomal dominant phenotypes are rather less severe and demonstrate widely variable expressivity and incomplete penetrances. On the other hand, autosomal recessive syndactylies are clinically more severe with rather consistent phenotypes.
Syndactyly causes
Syndactyly is a failure in the separation of developing digits during organogenesis. Syndactyly may occur as an isolated entity or a component of more than 300 hereditary syndromic malformations 1 or nonsyndromic, existing as 1 of 9 nonsyndromic forms 14. Beyond the well-characterized syndactylies that have genetic etiology there are syndactyly types that occur with congenital ring constrictions, that is, acrosyndactyly/pseudosyndactyly 2.
New syndromes, genes and causative loci are being found as research continues into congenital hand defects and it appears that each new finding gives as many further avenues for further investigation as answers in this field.
Man et al 15 conducted a study which reports a probable association with these conditions and maternal smoking and there are suggestions that syndactyly occurrence is associated with lower nutritional and economic status, including increased meat and egg intake whilst pregnant, although more research is required before suggesting these are causative factors 16.
Syndactyly genetics
The genes involved in syndactyly and polydactyly tend to affect specific bodily regions, including the zone of polarizing activity (ZPA): an area that controls limb structure and positional identity 17. The zone of polarizing activity (ZPA) disappears by day 44 of embryonic development, after which time the phalanges form.10 Aside from the ZPA, which is on the posterior embryo and produces fibroblast growth factor 8 (FGF8), the apical ectodermal ridge, which is on the dorsal-ventral margin and produces FGF8, also aids in limb development.
The HOX genes, hedgehog pathways [Sonic Hedgehog (SHH) and Indian hedgehog (IHH)], FGFs, bone morphogenetic proteins (BMPs), and cartilage-derived morphogenetic proteins are also involved 11.
Thirty-nine HOX genes exist in humans; the HOXA and HOXD clusters have been associated with syndactyly and polydactyly 18. Genes within the HOXD cluster and locus chr2q31 seem to be involved in syndactyly 19: the HOXD13 gene, for example, is linked to syndactyly type V (SD5) and a brachydactyly-syndactyly syndrome 20.
The Sonic Hedgehog (SHH) signaling pathway plays key roles in limb development 21. Sonic Hedgehog (SHH) is affected by—or affects—several transcription factors, namely HAND2, GLI3, ALX4, and certain BMP antagonists (formin and gremlin), changes to which have led to syndactyly and polydactyly 10. If disrupted, as if ZPA activity is impacted, then Sonic Hedgehog (SHH) cannot lead to normal limb development; this often results in syndactyly 22.
The IHH signaling pathway may influence later development, considering its roles in chondrocyte differentiation and ossification 23. Further, IHH is repressed by fibroblast growth factor (FGF) receptors 24, functioning at a different time than WNTs and FGFs, which are involved in the final stages of mesenchymal ossification 25; later, in the last stage of digit formation, there is down-regulation of gremlin and restriction of FGF8 expression 26.
Wingless-type mouse mammary tumor virus integration sit family members 6 and 10B (WNT6, WNT10B) are involved in the developing limb bud and related to the chr2q35 region [a locus implicated in syndactyly type I (SD1)] 27. BMPs and their antagonist noggin (NOG), when blocked, also lead to syndactyly 28. GLI3 may also contribute 29: it is involved in a range of syndromes, and a GLI3 missense mutation led to syndactyly and polydactyly 30. N-Myc and zinc-finger transcription factors have caused soft-tissue syndactyly in mice 31.
Syndactylies and polydactylies also vary in their patterns of inheritance. Most syndactyly types follow autosomal dominant inheritance 32, but SD7 and SD9 are autosomal recessive 33, and SD5 is X-linked recessive 34. Generally, autosomal dominant phenotypes are less severe with variable expressivity and incomplete penetrance.
Syndactyly classification
The ‘current classification scheme’ of syndactyly is an adaptation and extension of Temtamy-McKusick system by incorporating into it the clinical, genetic, and molecular developments in this field. The syndactyly types identified according to the current classification are described below.
Table 1. Current classification of well-characterized syndactyly types
| ID | Type/description | Fingers webbing | Toes webbing | Inheritance | Locus/gene |
| I-a | ZD1; Zygodactyly; Weidenreich type | Normal | 2/3 Toes only | AD | 3p21.31 |
| I-b | SD1; Lueken type | 3/4 Fingers, cutaneous/bony | 2/3 Toes, cutaneous | AD | 2q34-q36 |
| I-c | Montagu type | 3/4 Fingers only, cutaneous/bony | Normal | AD | |
| I-d | Castilla type | Normal | 4/5 Toes only, cutaneous | AD? | |
| II-a | SPD1; Vordingborg type | SPD, mesoaxial (3/4 fingers) | SPD, postaxial (4/5 toes) | AD | 2q31; HOXD13 |
| II-b | SPD2; Debeer type | SPD is central and postaxial | Postaxial syndactyly | AD | 22q13.3; FBLN1 |
| II-c | SPD3; Malik type | SPD is central | SPD postaxial | AD | 14q11.2-q13 |
| III | SDTY3; ODDD; Johnston-Kirby type | 4/5 Fingers; fifth finger short | Normal | AD | 6q21-q23; GJA1 |
| IV-a | SDTY4; Haas type | All fingers webbed; pre-/post-axial polydactyly, cup-shaped hand | Normal | AD | 7q36; ZRS (LMBR1) |
| IV-b | Andersen-Hansen type | All fingers webbed; pre-/post-axial polydactyly, cup-shaped hand | Variable webbing of toes with polydactyly | ||
| V | SDTY5; Dowd type | 4/5 Fingers with metacarpals fusion; hypoplastic metacarpals 4/5 | Mesoaxial webbing | AD | 2q31; HOXD13 |
| VI | Mitten type | 2/5 Fingers | 2/5 Toes | AD | |
| VII-a | Cenani-Lenz type; spoon-hand type | Total synostotic syndactyly with metacarpals fusion, spoon-head shape | Total synostotic syndactyly with metatarsals fusion | AR | 11p12-p11.2; LRP4. |
| VII-b | Oligodactyly type | Few deformed digits | Variable syndactyly of toes | AD | 15q13.3; GREM1-FMN1 |
| VIII-a | Orel-Holmes type | 4/5 Metacarpal fusion | Normal | X-R | |
| VIII-b | Lerch type | 4/5 Metacarpal fusion | Normal | AD | |
| IX | MSSD; Malik-Percin type | Mesoaxial synostotic syndactyly with phlanageal reduction | Preaxial webbing; distal phalangeal hypoplasia | AR | 17p13.3 |
Abbreviations: SPD = synpolydactyly; AD = autosomal dominant; AR = autosomal recessive
[Source 1 ]Figure 1. Syndactyly types – schematic diagrams of syndactyly types (I-a–III).
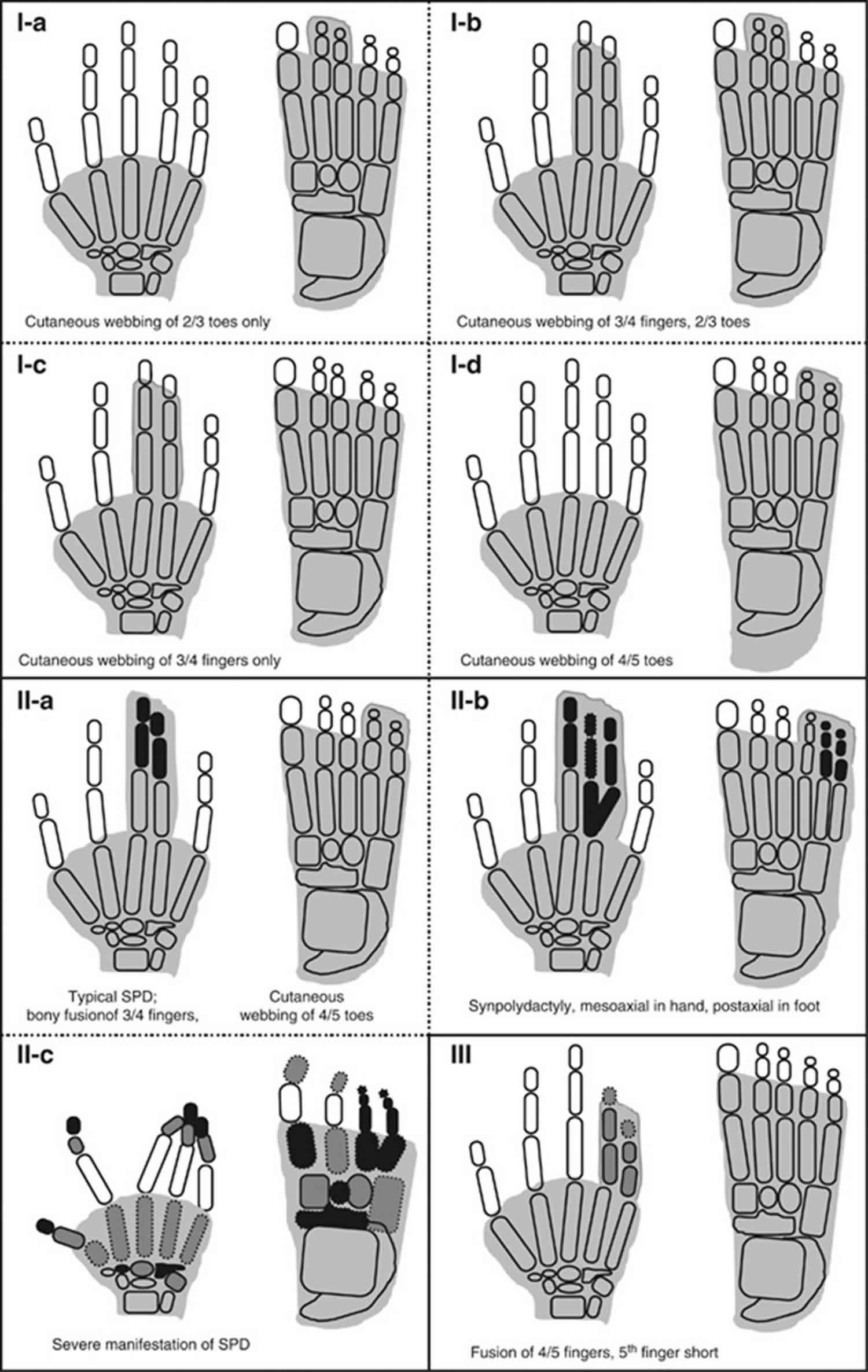
Footnote: Shaded digits depict cutaneous fusion only, while bony synostosis is represented by black digital elements within the shaded area. The grey digital elements show hypoplastic phalanges or clinodactyly/brachydactyly. The digital elements with amorphous borders symbolize dysplastic bones.
[Source 1 ]Figure 2. Syndactyly types – schematic diagrams of syndactyly types (IV-a–IX).
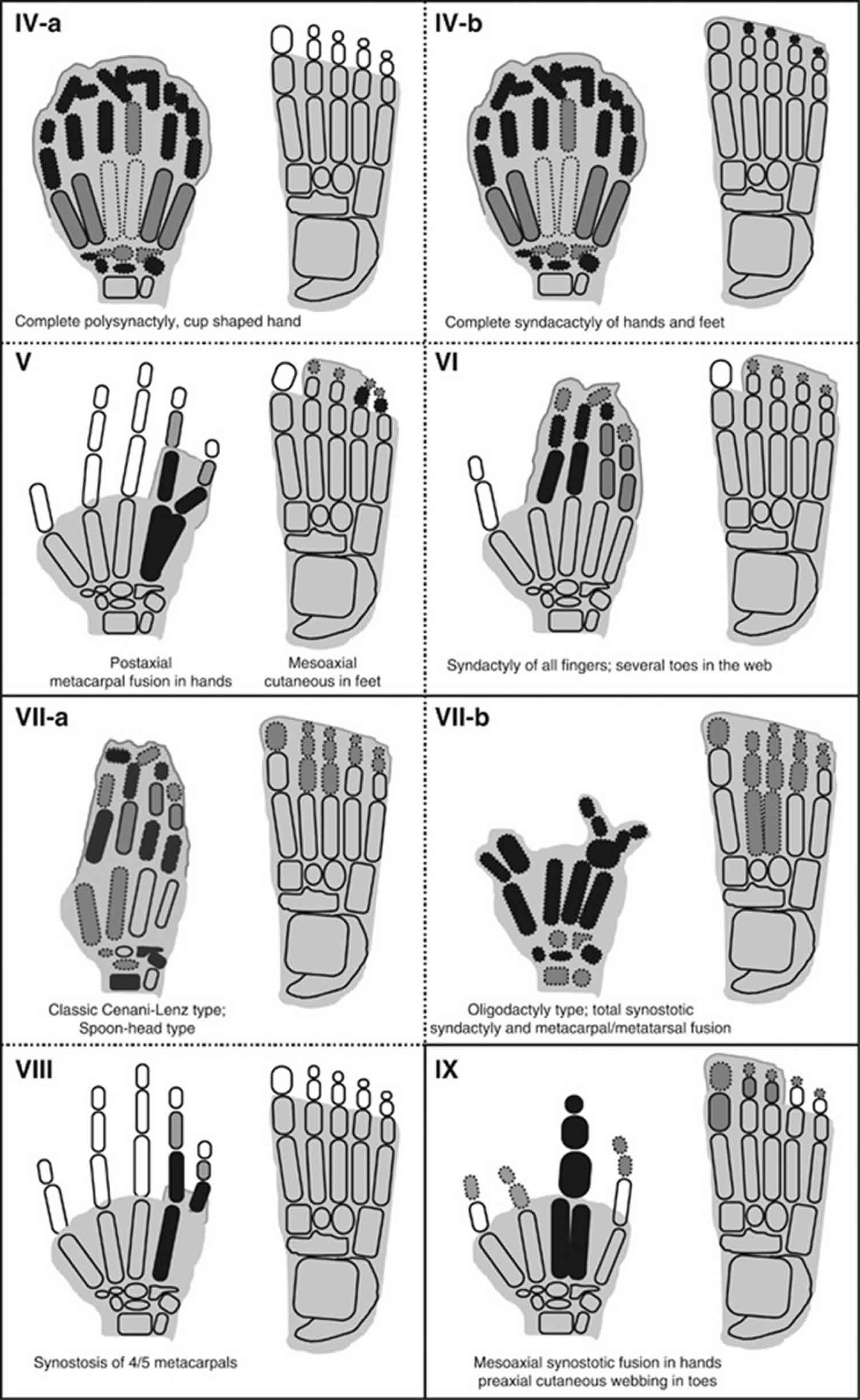
Syndactyly type I
Of all the known non-syndromic syndactylies, type I syndactyly is one of the most common types. It demonstrates mesoaxial webbing: fusion of third and fourth fingers, and/or second and third toes. Several characteristic phenotypic and genetic variants within this type witnessed in distinct families have led to the suggestion of four subtypes for type I syndactyly 35.
Syndactyly type I-a
Syndactyly type I-a (Weidenreich type; zygodactyly; 2/3 toes syndactyly) is an autosomal dominant entity was originally named zygodactyly by Weidenreich 11. Zygodactyly is a minor and least conspicuous type, and often goes unnoticed in the clinical practice. It has an estimated prevalence of 4 in 10 000 men and accounts for 70% of all non-syndromic syndactyly cases 2.
Zygodactyly is characterized by bilateral cutaneous webbing of second and third toes without the involvement of hands (Figure 1). Rarely, other toes are also affected. The phenotype in both feet is usually concordant 36. In its mildest forms, it gives the impression of slight ascent of interdigital web between second and third toes, or it may only be detected by abnormal dermatoglyphics 13. In its extreme form, the web reaches up to phalangeal tips, a rather intimate fusion is witnessed at nails, and the second toe depicts varus inclination. First molecular evidence of zygodactyly being a distinct genetic entity was provided by Malik et al 32 by a mapping study on a large Pakistani family. Cosegregation of zygodactyly was shown with ZD1 locus at chromosome 3p21-p31. However, genetic heterogeneity has been suggested from the linkage data of a German kindred 32.
Syndactyly type I-b
Syndactyly type I-b (Lueken type; 3/4 fingers and 2/3 toes syndactyly) subtype is characterized by bilateral webbing of third and fourth fingers, and second and third toes (Figure 1). Finger’s webbing may exhibit osseous fusion in the form of a bony bridge at the phalangeal tips. In more severe cases, additional fingers from second to fifth and toes from first to fifth may be involved. Reduplication of any fused digit is not a characteristic of this type. In the family reported by Lueken, this dominant phenotype was mapped to the SD1 locus at chromosome 2q34-q35 37.
Syndactyly type I-c
Syndactyly type I-c (Montagu type; 3/4 fingers syndactyly) is a rare autosomal dominant type is characterized by bilateral cutaneous/bony fusion of third and fourth fingers with normal feet (Figure 1) 38. A large Chinese family reported by Hsu 39 had 23 affected subjects demonstrating variable degree of bilateral osseous fusion of third and fourth or third, fourth and fifth fingers. Only one subject also had partial fusion of toes from third to fifth.
Syndactyly type I-d
Syndactyly type I-d (Castilla type; 4/5 toes syndactyly) subtype depicts bilateral cutaneous webbing of fourth and fifth toes (Figure 1) and has been reported to be the second most common type of isolated webbing of toes with a frequency of 0.22 in 10 000 subjects 2. Occasionally, the fifth toe is tucked inside the fibular aspects of the fourth toe, and thus any minor form of webbing could be easily overlooked in the clinical practice. This is particularly the case when the fifth toe is disfigured due to faulty footwear. The inheritance pattern and penetrance estimates for this subtype have not been worked out.
Syndactyly type II
Type II syndactyly/SPD (Vordingborg type; 3/4 fingers and 2/3 toes synpolydactyly), a well-described entity, is clinically and genetically one of the most heterogeneous types. The hallmark features of SPD are cutaneous/bony fusion of third and fourth fingers and second and third toes with partial or complete reduplication of a digital ray within the syndactylous web (Figure 1) 40. It is the only syndactyly type with a mesoaxial superfluous finger. The extreme phenotypic heterogeneity in SPD families has led to the lumping of all clinical variants (∼18 types) into three categories: (A) typical SPD features; (B) minor variants; and (C) unusual phenotypes 40. SPD segregates as an autosomal dominant entity with reduced penetrance. Three SPD loci have been discovered (SPD1-3), however, these entities have not yet been clinically delineated (Table 1) 34. Additionally, detailed clinical and mutation data are available only for SPD1 linked to HOXD13.
Syndactyly type III
Syndactyly type III (Johnston–Kirby type; 4/5 or 3/4/5 fingers fusion) type of syndactyly affects fourth and fifth or third, fourth and fifth fingers (Figure 1). The middle phalanx of the fifth finger is hypoplastic 41. The fourth finger shows valgus deviation in order to accommodate the webbing with little finger particularly when the fusion is complete 13. There is adduction of fused fingers, and the nails of the syndactylyous fingers are fused medially. The fusion may involve a bony bridge at the distal phalanges. The feet are generally unaffected. This type shows an autosomal dominant mode of inheritance with incomplete penetrance.
The fusion of fourth and fifth fingers is, likewise, a feature of ODDD (oculodentodigital dysplasia), which exhibits additional symptoms of eyes, ears, and orofacial region. Schrander–Stumpel et al 42 proposed that oculodentodigital dysplasia and pure syndactyly type III are the respective ends of a clinical spectrum as variable expression of a contiguous gene deletion syndrome. Interestingly, molecular studies revealed that type III syndactyly and/or oculodentodigital dysplasia are caused by mutations in GJA1, a pleiotropic gene encoding connexin 43 43.
Syndactyly type IV
Syndactyly type IV (Haas type; complete syndactyly of all fingers) has a prevalence of 1/300 000 and segregates in an autosomal dominant fashion 2. It manifests itself as complete cutaneous fusion of all fingers accompanied by the presence of extra digital pre- or postaxial ray(s) in the web (Figure 2) 44. The nails may be fused completely or give an impression of separation by a groove only. Flexion of fingers is limited and the union of contiguous fingers gives the hand a cup-shaped appearance. Phalanges may fuse as a conglomerate mass of bones; however, metacarpal synostosis is absent.
In the literature, at least two variants of type IV syndactyly have been reported: (a) typical Haas type without involvement of feet; and (b) complete fusion of all fingers with variable fusion of all five digits in feet (Table 1) 45. Haas type syndactyly has been shown to be allelic to triphalangeal thumb polysyndactyly, which is present at a milder end of phenotype. Both entities are caused by mutations in the ZRS locus at chromosome 7q36 (LMBR1), encompassing a long-range regulator of SHH 46.
Syndactyly type V
The hallmark of syndactyly type V (Dowd type; fusion of 4/5 metacarpals) is the fusion of fourth and fifth metacarpals (Figure 2). Additional symptoms may involve shortening of fused fourth and fifth metacarpals, ulnar deviation of fingers from second to fifth, interdigital cleft between third and fourth fingers, camptodactyly of fifth finger, short distal phalanges, and absent distal interphalangeal creases of the affected fingers 47. In the feet, there is hyperplasia of first ray/metatarsal, and shortening of metatarsals from second to fifth, resulting in varus deviation of metatarsals and valgus deviation of toes/phalanges. Type V syndactyly is inherited as an autosomal dominant entity, and it has been attributed to a missense mutation in homeodomain of HOXD13 in a Chinese family 48.
Syndactyly type VI
Syndactyly type VI (Mitten type; fusion of 2/5 fingers and 2/3 toes). Temtamy and McKusick 13 described a family in which the index subject had fusion of fingers from second to fifth in his right hand, whereas distal and terminal phalanges were amalgamated in a knot-like structure (Figure 2). The feet showed syndactyly involving second and third toes. The maternal second cousin also had the same deformity. Other family members, however, showed only webbing between second and third toes without involvement of fingers. An autosomal dominant mode of inheritance with reduced penetrance and variable expressivity has been suggested for this rare type.
Syndactyly type VII
Syndactyly type VII (Cenani–Lenz syndactyly, CLS; severe bony fusion of all digits and deformed hand) is an autosomal recessive entity manifests severe abnormalities of all digital elements. It is characterized by gross disorganization of all bones of hand to such an extent that no phalangeal element is identifiable (Figure 2).41 The carpals, metacarpals and phalanges show irregular synostosis giving an impression of ‘hands-in-stockings’. The anomaly may involve radius and ulna that are either fused, short, or rudimentary resulting in luxation of the radial head and mesomelic shortening of forearm 13. Lower limbs show changes similar to upper limbs and certain digital rays may be absent. Additionally, there is rare involvement of craniofacial and nephrological features 49.
Harpf et al 31 suggested that there exist two grossly different clinical features in Cenani–Lenz type: (a) spoon-head type, and (b) oligodactyly type (Table 1). They also differentiated between consistent and inconsistent feature for Cenani-Lenz syndactyly. Recently, Li et al 49 have shown that Cenani-Lenz syndactyly, both spoon-head and oligodactyly types, and with or without kidney malformations are caused by mutations in LRP4 (chromosome 11p11.2), which is involved in Wnt/β-Catenin signaling. Interestingly, another molecular study showed that Cenani–Lenz phenotype with renal defects and hearing loss, and an autosomal dominant Cenani–Lenz-like non-syndromic oligodactyly are caused by genomic rearrangements of GREM1-FMN1 locus on chromosome 15q13.3 50.
Syndactyly type VIII
Syndactyly type VIII (Orel-Holmes type; metacarpals 4/5 fusion; X-linked recessive) is characterized by the fusion of fourth and fifth metacarpals with a marked ulnar deviation of the little finger and with no other abnormality (Figure 2). There are shortened fourth and fifth metacarpals with excessive separation between their distal ends, and inability to bring the affected fingers in parallel to other fingers. Fifth metacarpal is hypoplastic and tri-radii c and d are absent 51. This entity shows X-linked recessive inheritance.54, 44 However, there is at least one report depicting autosomal dominant segregation for this phenotype (Table 1) 52.
Syndactyly type IX
Syndactyly type IX (Malik–Percin type; fusion of 3/4 metacarpals with a reduction of mesoaxial finger, and preaxial syndactyly of toes). Percin et al 53 and Malik et al 54 described a consanguineous Turkish and Pakistani family, respectively, in which the affected subjects showed mesoaxial reduction of fingers, osseous synostosis of third and fourth metacarpals culminating into a single digit, malformed thumbs, and hypoplasia and clinodactyly of the fifth finger (Figure 2). In addition, there was preaxial webbing of toes with terminal phalangeal hypoplasia of all toes. Clinically this type is less severe than Cenani–Lenz syndactyly as it is not extending beyond carpals/tarsals, and is not characterized by bizarre arrangement of skeletal elements of hand/foot. This entity segregated in autosomal recessive fashion and was mapped to chromosome 17p13.3 30.
Syndactyly treatment
The current mainstay for the treatment and management of syndactyly is surgery. The indications in general for hand anomalies run true with syndactyly and include 55;
- Functional needs: The degree of function required by the patient; including whether delaying surgery may alter hand function and grip development.
- Aesthetic consideration: The appearance of the deformity and its subsequent effects on psychological and social aspects of the patient’s life.
It must be remembered that surgical intervention is not urgent. Despite this there are age related targets for reconstruction and dependency on the type of anomaly present.
Border digit syndactyly involving the thumb, index and ring fingers is felt to benefit from earlier release, usually between the ages of 6 to 12 months, and should always be released first in multiple digit involvement. The result in delayed surgery can involve deformity of the digit relating to forced flexion, angulation and/or rotation.
Long finger syndactyly usually involves similarly lengthened digits and for this reason is often delayed, and KettleKamp and Flatt 56 found results to be better in those aged greater than 18 months, particularly with long term final appearance of the commissure.
Multiple digit involvement, as stated above, should involve in the first instance release of the border digits, with subsequent digit release after a 6-month waiting period. These subsequent releases involve either radial or ulnar sides of a finger as releasing both sides may endanger the digit viability.
It is generally regarded that operative therapy should be done pre-school age. The main reason for this is that interaction with other children whilst having the anomaly is thought to have potentially detrimental affects on the child’s social, functional and psychological development.
The main concern with surgical therapy is the fact that combined circumference of the two normal digits is approximately 1.4 times the circumference of the fused digits. Pre reconstruction it is felt that massaging the skin where the new web space will be constructed facilitates skin loosening and maximises potential soft tissue needed during the procedure.
Full thickness skin grafts are used for soft-tissue coverage in the majority of cases at present, with altering levels of flap use. The choice of flap, and consequent extra need for full thickness graft coverage, appears to be dependant on the surgeon’s preference, with multiple techniques and variances described in the literature. Several other methods have been used, including split thickness grafts 57 and tissue expanders 58. Both have suffered with less favourable outcomes and increased complications compared to the use of full thickness grafts and have hence failed to establish themselves as alternative techniques. The use of no graft is more common, but this appears only to be of use in mild cases of simple syndactyly.
Follow up should be until skeletal maturity, mainly due to the prevalence of web creep until this age. Complications of all the surgical techniques include web creep, finger deviation (particularly in complex syndactyly) as well as those complications associated with any surgical procedure. The greater the degree of syndactyly, the more need there is for the surgeon to be aware of neurovascular variance and this takes on greater importance in graft and flap survival.
In light of genetic and peri-natal investigations, both these fields have potential for future management input. Ultrasound has allowed earlier diagnosis and this can now be supplemented by genetic review of likely carriers. Constriction bands have been released in-utero allowing a more normal limb development and with minimal scarring 59. However, in utero surgery at present is high risk and currently the benefit of operating does not outweigh the risks and associated complications for non-fatal conditions.
The continuing development in both the genetic basis of syndactyly, and the improvement in tissue bioengineering, bodes that in the future both these avenues will expand management options.
- Malik S. Syndactyly: phenotypes, genetics and current classification. Eur J Hum Genet. 2012;20(8):817–824. doi:10.1038/ejhg.2012.14 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3400728[↩][↩][↩][↩][↩][↩][↩]
- Castilla EE, Paz JE, Orioli-Parreiras IM. Syndactyly: frequency of specific types. Am J Med Genet. 1980;5:357–364.[↩][↩][↩][↩][↩]
- Bell J. On syndactylies and its association with polydactyly The Treasury of Human InheritanceLondon: Cambridge University Press, 1953V(II30–50.[↩][↩][↩]
- Koenner DM. Abnormalities of the hand and feet. J Hered. 1934;25:329–334.[↩]
- Davis JS, German WJ. Syndactylism coherence of the fingers or toes. Arch Surg. 1930;21:32–75.[↩]
- Temtamy SA, McKusick VA. The Genetics of Hand Malformations. New York: Alan R Liss; 1978. Syndactyly; pp. 301–322[↩]
- Schultz AH. Zygodactyly and its inheritance. J Hered. 1922;13:113–117.[↩]
- Lubahn JD. Syndactylyin Fitzgerald RH, Kaufer H, Malkani AL (eds): OrthopaedicsPhiladelphia: Elsevier Health Sciences, 20021895–1896.[↩][↩]
- Straus WL. The nature and inheritance of webbed toes in man. J Morphol Physiol. 1926;41:427–439.[↩][↩]
- Newsholme HP. A pedigree showing bi-parental inheritance of webbed toes. Lancet. 1910;176:1690–1691.[↩][↩]
- Weidenreich F. Die Zygodactylie und ihre Vererbung. Z Abst Vererb. 1923;32:304.[↩][↩][↩]
- Syndactyly: frequency of specific types. Castilla EE, Paz JE, Orioli-Parreiras IM. Am J Med Genet. 1980; 5(4):357-64.[↩]
- Temtamy SA, McKusick VA. The Genetics of Hand Malformations. New York: Alan R Liss; 1978. Syndactyly; pp. 301–322.[↩][↩][↩][↩][↩]
- A simple method for characterising syndactyly in clinical practice. Malik S, Ahmad W, Grzeschik KH, Koch MC. Genet Couns. 2005; 16(3):229-38.[↩]
- Maternal cigarette smoking during pregnancy increases the risk of having a child with a congenital digital anomaly. Man LX, Chang B. Plast Reconstr Surg. 2006 Jan; 117(1):301-8.[↩]
- [A case-control study on genetic and environmental factors regarding polydactyly and syndactyly]. Luo JY, Fu CH, Yao KB, Hu RS, DU QY, Liu ZY. Zhonghua Liu Xing Bing Xue Za Zhi. 2009 Sep; 30(9):903-6.[↩]
- Deciphering skeletal patterning: clues from the limb. Mariani FV, Martin GR. Nature. 2003 May 15; 423(6937):319-25.[↩]
- Spink MS, Lweis GL. The Wellcome Institute for History of Medicine. London; 1973. Albucasus on surgery and instruments.[↩]
- Al-Ghazal SK. Al-Zahrawi (Albucasis) – a light in the dark middle ages in Europe. J Int Soc Hist Islam Med. 2003;1:37–38.[↩]
- Roblot G. La syndactylie conge’nitale. Paris: Maulde, Doumencet Cie; 1906.[↩]
- Kelikian H. Congenital Deformities of the Hand and Forearm. Philadelphia: WB Saunders Company; 1974.[↩]
- Woolf CM, Cone DL. Problem of sex ratio in cases of type I syndactyly. J Med Genet. 1977;14:108–113.[↩]
- Lenz W, Majewski F. Fehlbildungen der Gliedmaßenin Schinz HR (eds): Lehrbuch der Röntgendiagnostik Verlag, Stuttgart: Thieme; 1981935–1032.[↩]
- Swanson AB. A classification for congenital limb malformations. J Hand Surg Am. 1976;1:8–22.[↩]
- Winter RM, Tickle C. Syndactylies and polydactylies: embryological overview and suggested classification. Eur J Hum Genet. 1993;1:96–104.[↩]
- Stoll C, Duboule D, Holmes LB, et al. Classification of limb defects. Am J Med Genet. 1998;77:439–441.[↩]
- Goldstein DJ, Kambouris M, Ward RE. Familial crossed polysyndactyly. Am J Med Genet. 1994;50:215–223.[↩]
- Newsholme HP. A pedigree showing bi-parental inheritance of webbed toes. Lancet. 1910;176:1690–1691[↩]
- Malik S, Arshad M, Amin-Ud-Din M, et al. A novel type of autosomal recessive syndactyly: clinical and molecular studies in a family of Pakistani origin. Am J Med Genet. 2004;126:61–67.[↩]
- Malik S, Percin FE, Ahmad W, et al. Autosomal recessive mesoaxial synostotic syndactyly with phalangeal reduction maps to chromosome 17p13.3. Am J Med Genet. 2005;134:404–408.[↩][↩]
- Harpf C, Pavelka M, Hussl H. A variant of Cenani-Lenz syndactyly (CLS): review of the literature and attempt of classification. Br J Plast Surg. 2005;58:251–257.[↩][↩]
- Malik S, Schott J, Ali SW, et al. Evidence for clinical and genetic heterogeneity of syndactyly type I: the phenotype of second and third toe syndactyly maps to chromosome 3p21.31. Eur J Hum Genet. 2005;13:1268–1274.[↩][↩][↩]
- Hay S. Incidence of selected congenital malformations in Iowa. Am J Epidemiol. 1971;94:572–584.[↩]
- Malik S, Abbasi AA, Ansar M, et al. Genetic heterogeneity of synpolydactyly: a novel locus SPD3 maps to chromosome 14q11.2-q12. Clin Genet. 2006;69:518–524.[↩][↩]
- Malik S, Schott J, Ali SW, et al. Evidence for clinical and genetic heterogeneity of syndactyly type I: the phenotype of second and third toe syndactyly maps to chromosome 3p21.31. Eur J Hum Genet. 2005;13:1268–1274[↩]
- Stiles KA, Hawkins DA. The inheritance of zygodactyly. J Hered. 1946;37:16–18.[↩]
- Bosse K, Betz RC, Lee Y-A, et al. Localization of a gene for syndactyly type 1 to chromosome 2q34-q36. Am J Hum Genet. 2000;67:492–497.[↩]
- Montagu MFA. A pedigree of syndactylism of the middle and ring fingers. Am J Hum Genet. 1953;5:70–72.[↩]
- Hsu CK. Hereditary syndactylia in a Chinese family. Chin Med J. 1965;84:482–485.[↩]
- Malik S, Grzeschik KH. Synpolydactyly: clinical and molecular advances. Clin Genet. 2008;73:113–120.[↩][↩]
- Johnston O, Kirby VV., Jr Syndactyly of the ring and little finger. Am J Hum Genet. 1955;7:80–82.[↩]
- Schrander-Stumpel CT, De Groot-Wijnands JB, De Die-Smulders C, et al. Type III syndactyly and oculodentodigital dysplasia: a clinical spectrum. Genet Couns. 1993;4:271–276.[↩]
- Paznekas WA, Boyadjiev SA, Shapiro RE, et al. Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Am J Hum Genet. 2003;72:408–418.[↩]
- Haas SL. Bilateral complete syndactylism of all fingers. Am J Surg. 1940;50:363–366.[↩]
- Andersen HJ, Hansen AK. Tibial hypo-/aplasia with preaxial syn- and polydactyly. Arch Orthop Trauma Surg. 1990;109:231–233.[↩]
- Wieczorek D, Pawlik B, Li Y, Akarsu NA, et al. A specific mutation in the distant sonic hedgehog (SHH) cis-regulator (ZRS) causes Werner mesomelic syndrome (WMS) while complete ZRS duplications underlie Haas type polysyndactyly and preaxial polydactyly (PPD) with or without triphalangeal thumb. Hum Mutat. 2010;31:81–89.[↩]
- Dowd CN. Cleft hand: a report of a case successfully treated with the use of periosteal flaps. Ann Surg. 1896;24:210.2–21216.[↩]
- Zhao X, Sun M, Zhao J, et al. Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet. 2007;80:361–371.[↩]
- Li Y, Pawlik B, Elcioglu N, et al. LRP4 mutations alter Wnt/beta-catenin signaling and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet. 2010;86:696–706.[↩][↩]
- Dimitrov BI, Voet T, De Smet L, et al. Genomic rearrangements of the GREM1-FMN1 locus cause oligosyndactyly, radio-ulnar synostosis, hearing loss, renal defects syndrome and Cenani–Lenz-like non-syndromic oligosyndactyly. J Med Genet. 2010;47:569–574.[↩]
- Anneren G, Amilon A. X-linked recessive fusion of metacarpals IV∣ and V and hypoplastic metacarpal V. Am J Med Genet. 1994;52:248–250.[↩]
- Lerch H. Erbliche Synostosen der Ossa metacarpalia IV und V. Z Orthop. 1948;78:13–16.[↩]
- Percin EF, Percin S, Egilmez H, et al. Mesoaxial complete syndactyly and synostosis with hypoplastic thumbs: an unusual combination or homozygous expression of syndactyly type I. J Med Genet. 1998;35:868–874.[↩]
- Malik S, Arshad M, Amin-Ud-Din M, et al. A novel type of autosomal recessive syndactyly: clinical and molecular studies in a family of Pakistani origin. Am J Med Genet. 2004;126:61–67[↩]
- Jordan D, Hindocha S, Dhital M, Saleh M, Khan W. The epidemiology, genetics and future management of syndactyly. Open Orthop J. 2012;6:14–27. doi:10.2174/1874325001206010014 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3308320[↩]
- Kettelkamp DB, Flatt AE. An evaluation of syndactylia repair. Surg Gynecol Obstet. 1961;113:471.[↩]
- Toledo LC, Ger E. Evaluation of the operative treatment of syndactyly. J Hand Surg Am. 1979;4(6 ):556–64.[↩]
- d’Arcangelo M, Maffulli N. Tissue expanders in syndactyly a brief review. Acta Chir Plast. 1996;38(1 ):11–3.[↩]
- Crombleholme TM, Dirkes K, Whitney TM, et al. Amniotic band syndrome in fetal lambs I Fetoscopic release and morphometric outcome. J Pediat Sur. 1995;30:974.[↩]





{kind=link}