Systemic mastocytosis
Systemic mastocytosis is often called systemic mast cell disease, is a blood disorder called mastocytosis, that results in an excessive number of mast cells accumulating in your body such as in internal tissues and organs such as the liver, spleen, bone marrow, and small intestines 1, 2, 3, 4, 5, 6, 7, 8, 9. Systemic mastocytosis can affect many different body systems. Mast cells normally help protect you from disease and aid in wound healing by releasing substances such as histamine and leukotrienes. But if you have systemic mastocytosis, excess mast cells generally build up in your skin, bone marrow, gastrointestinal tract and bones. When triggered, these mast cells release substances that can overwhelm your body and result in signs and symptoms such as facial flushing, itching, a rapid heartbeat, abdominal cramps, feeling lightheaded or even loss of consciousness. Common triggers include alcohol, temperature changes, spicy foods and certain medications.
Systemic mastocytosis is estimated to occur in 1 per 10,000 to 20,000 individuals worldwide. Individuals with systemic mastocytosis can develop signs and symptoms at any age, but it usually appears after adolescence.
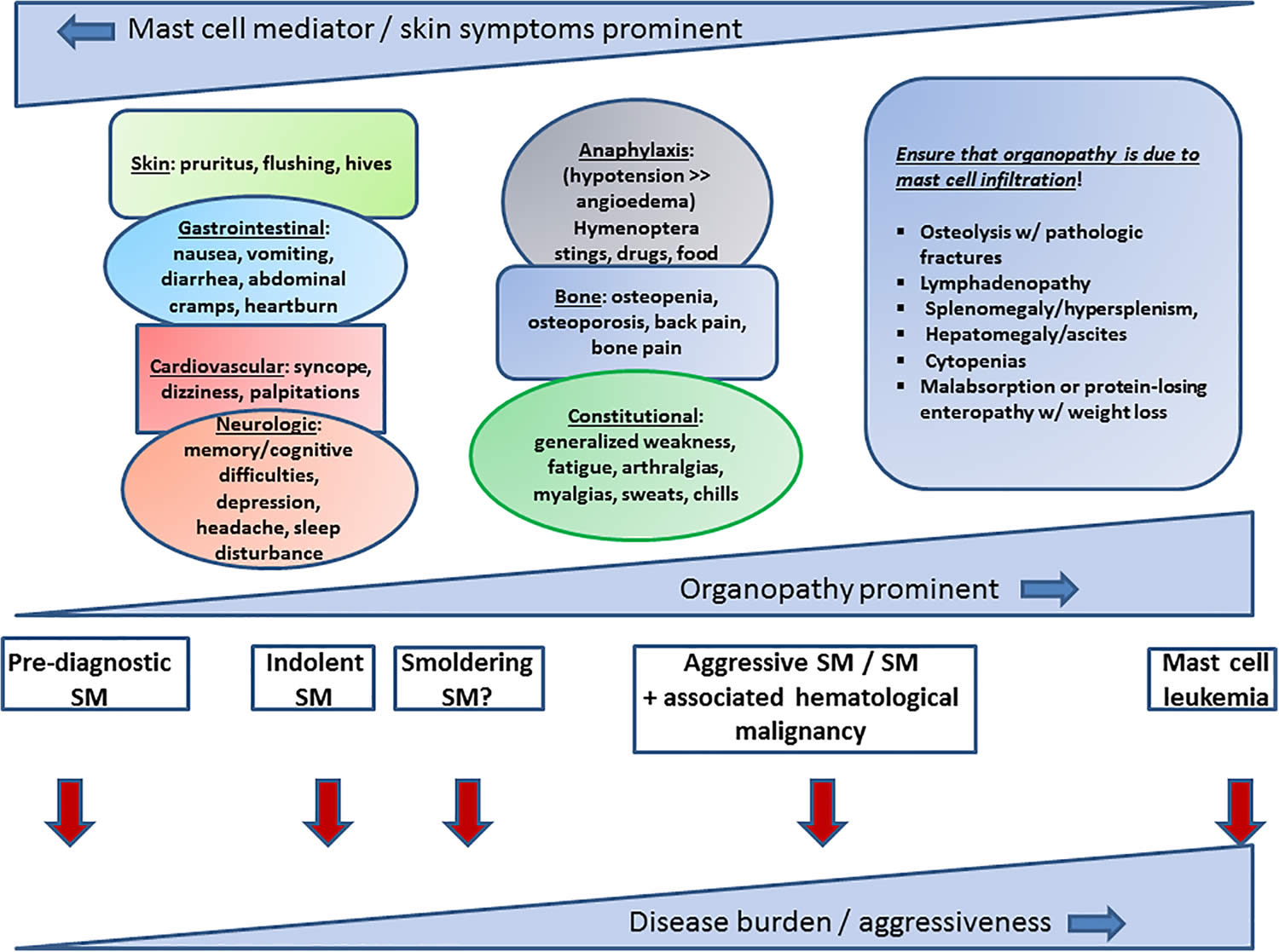
Clinically, there are six subtypes of systemic mastocytosis based on the 2022 World Health Organization (WHO) classification, which are differentiated by their severity and the signs and symptoms 6. Non-advanced systemic mastocytosis subtypes are by far the most frequent and include bone marrow mastocytosis (BMM), indolent systemic mastocytosis (ISM), and smoldering systemic mastocytosis (SSM). Advanced systemic mastocytosis comprises aggressive systemic mastocytosis (ASM), systemic mastocytosis with an associated hematologic neoplasm (SM-AHN), and mast cell leukemia (MCL); advanced systemic mastocytosis subtypes are associated with organ damage and poor overall survival 5. In a recent study, Arock et al. 10 proposed a diagnostic algorithm to differentially diagnose systemic mastocytosis based on clinical, histopathologic criteria, and molecular markers.
- Indolent systemic mastocytosis (ISM) – the most common form of systemic mastocytosis with a slowly progressive (worsening over time) clinical course.
- Bone marrow mastocytosis (BMM) – Indolent systemic mastocytosis with bone marrow involvement but no skin lesions, no B-finding(s), no dense systemic mastocytosis infiltrates in an extramedullary organ, where the serum tryptase is often normal or near normal (< 125 ng/mL), no signs/criteria of mast cell leukemia (MCL), and an associated hematologic neoplasm 11, 12, 13, 14, 15, 16, 17. Bone marrow mastocytosis (BMM) is associated with osteoporosis and severe anaphylaxis 11, 12, 13, 14, 15, 16, 17. Patients with bone marrow mastocytosis (BMM) and low disease burden (defined as absence of B-findings and a tryptase level < 125 ng/mL) have a superior prognosis to patients with typical indolent systemic mastocytosis (ISM) or smoldering systemic mastocytosis (SSM) 17; therefore, the importance to accurately diagnose this entity. Of note, in the absence of skin lesions, if B-findings are present and/or serum tryptase is over 125 ng/mL, the disease would be considered indolent systemic mastocytosis (ISM) and not bone marrow mastocytosis (BMM) 7. For these reasons, bone marrow mastocytosis (BMM) was recognized as a separate systemic mastocytosis (ISM) subtype in the recent consensus proposal 7 and the 2022 WHO classification 6. In bone marrow mastocytosis (BMM), the KIT D816V mutation appears to be restricted to mast cell compartments, and allele burden and mast cell infiltrate in the bone marrow are low, occasionally with a well-differentiated morphology, which may explain the indolent course of bone marrow mastocytosis (BMM) 16. Nevertheless, bone marrow mastocytosis (BMM) is strongly associated with potentially life-threatening anaphylactic reactions; thus, recognizing bone marrow mastocytosis (BMM) early is of paramount importance 14.
- Smoldering systemic mastocytosis (SSM) – WHO (World Health Organization) criteria for systemic mastocytosis met, typically with skin lesions, with 2 or more B findings, but no C findings (see below). There is a greater possibility that the disease will progress to a more aggressive variant.
- Systemic mastocytosis with an associated hematologic non-mast cell lineage disorder (systemic mastocytosis with an associated hematologic neoplasm [SM-AHN]) – a form of systemic mastocytosis associated with other blood disorders (such as myeloproliferative or myelodysplastic conditions). These patients fit the criteria for systemic mastocytosis and they fit the WHO criteria for myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), myelodysplastic syndrome or myeloproliferative neoplasm overlap disorder, or acute myeloid leukemia (AML), with or without skin lesions 18. Patients are treated for both the systemic mastocytosis component and for the associated hematologic neoplasm.
- Aggressive systemic mastocytosis (ASM) – a severe form of systemic mastocytosis usually characterized by organ impairment or organ failure (usually liver, gut, bone or bone marrow) due to aggressive mast cell infiltration
- Mast cell leukemia (MCL) – Mast cell leukemia is a very rare and aggressive form of systemic mastocytosis that is associated with greater than 10% immature mast cells in the peripheral blood or greater than 20% immature mast cells in bone marrow cultures. These mast cells are immature and atypical with round nuclei rather than spindle-shaped as seen in systemic mastocytosis. Mast cell leukemia is rare; however, it is associated with the worst prognosis among all mastocytosis varieties. In a considerable number of patients with mast cell leukemia (MCL), the leukemic spread into peripheral blood is less overt or even absent 19. In this setting, when mast cells comprise less than 10% of all circulating blood leukocytes, the disease is termed “aleukemic” mast cell leukemia 19. Mast cell leukemia (MCL) is further subdivided into primary mast cell leukemia (absence of prior systemic mastocytosis) and secondary mast cell leukemia (progression following a previous established systemic mastocytosis). Furthermore, mast cell leukemia (MCL) can be subdivided into acute mast cell leukemia and chronic mast cell leukemia when C-findings are present and absent, respectively 7, 19. Patients with chronic mast cell leukemia may respond to KIT-targeting drugs and have a better prognosis in comparison to acute mast cell leukemia 19. Nevertheless, over time, many patients may progress to acute mast cell leukemia 20.
- Mast cell sarcoma (MCS) – A mast cell sarcoma is a single tumor composed of abnormal mast cells invading the tissue. This condition is very rare and often is not associated with additional skin involvement. More aggressive forms of mastocytosis, mast cell leukemias and mast cell sarcomas are very rarely encountered.
“B” findings
- High mast cell burden shown on bone marrow biopsy: >30% infiltration of cellularity by mast cells (focal, dense aggregates) and serum total tryptase level >200 ng/ml.
- Signs of dysplasia or myeloproliferation, in non-mast cell lineage(s), but insufficient criteria for definitive diagnosis of an associated hematological neoplasm (AHN), with normal or only slightly abnormal blood counts.
- Hepatomegaly without impairment of liver function, palpable splenomegaly without hypersplenism, and/or lymphadenopathy on palpation or imaging.
“C” findings
- Bone marrow dysfunction caused by neoplastic mast cell infiltration, manifested by ≥1 cytopenia(s) (ANC <1.0 × 109/L, Hgb <10 g/dl, and/or platelet count <100 × 109/L).
- Palpable hepatomegaly with impairment of liver function, ascites and/or portal hypertension.
- Skeletal involvement with large osteolytic lesions with/without pathological fractures
- Pathological fractures caused by osteoporosis do not qualify as a “C” finding.
- Palpable splenomegaly with hypersplenism.
- Malabsorption with weight loss due to gastrointestinal mast cell infiltrates.
Once a person is diagnosed, the category of systemic mastocytosis must be determined, as treatment and prognosis differ for each 21.
The most common and mildest forms of systemic mastocytosis— indolent systemic mastocytosis (ISM) — progresses slowly. Individuals with these types tend to have only the general signs and symptoms of systemic mastocytosis described above. Individuals with smoldering systemic mastocytosis (SSM) may have more organs affected and more severe features than those with indolent mastocytosis. The indolent type is the most common type of systemic mastocytosis.
The second most common form is systemic mastocytosis associated with a second blood disorder. Another type, aggressive systemic mastocytosis, develops rapidly and is often associated with organ damage. Mast cell leukemia and mast cell sarcoma are extremely rare forms of systemic mastocytosis.
The severe types include aggressive systemic mastocytosis, systemic mastocytosis with an associated hematologic neoplasm, and mast cell leukemia. These types are associated with a reduced life span, which varies among the types and affected individuals. In addition to the general signs and symptoms of systemic mastocytosis, these types typically involve impaired function of an organ, such as the liver, spleen, or lymph nodes. The organ dysfunction can result in an abnormal buildup of fluid in the abdominal cavity (ascites). Aggressive systemic mastocytosis is associated with a loss of bone tissue (osteoporosis and osteopenia) and multiple bone fractures. Systemic mastocytosis with an associated hematologic neoplasm and mast cell leukemia both involve blood cell disorders or blood cell cancer (leukemia). Mast cell leukemia is the rarest and most severe type of systemic mastocytosis.
Systemic mastocytosis is generally not inherited but arises from a somatic mutation in the body’s cells that occurs after conception.
The median age at diagnosis of systemic mastocytosis in adults is 55 years. Patients with indolent systemic mastocytosis were younger and symptomatic for a longer duration of time as compared with patients with aggressive systemic mastocytosis or systemic mastocytosis with an associated hematologic non-mast cell lineage disorder (with other hematological disorders) 22.
The therapeutic approach in indolent systemic mastocytosis (ISM) patients is primarilly directed at anaphylaxis and prevention/symptom control/therapy for osteoporosis; however, patients with advanced systemic mastocytosis often need mast cells cytoreductive therapy to improve disease-related organ dysfunction 23. Patients with advanced systemic mastocytosis who were treated with small molecule kinase inhibitors targeting KIT D816V, such as midostaurin 24, 25 and avapritinib 26, 27, demonstrated high response rates. These medications have changed the therapeutic landscape of systemic mastocytosis. Midostaurin is a multi-kinase inhibitor that the FDA approved in 2017 for advanced systemic mastocytosis. Midostaurin targets mutant KIT D816V besides wild-type KIT and other kinases. Avapritinib, which received regulatory approval in June 2021 for advanced systemic mastocytosis, selectively and potently targets KIT D816V with a tenfold higher potency compared to midostaurin 26, 27, 28, 29, 30. Avapritinib elicited high overall response rates (75%) with median response of 2 months and drastically reduced the mast cell burden (≥50%) and KIT D816V VAF in the EXPLORER 31 and PATHFINDER 32 clinical trials, which supported avapritinib’s regulatory approval for advanced systemic mastocytosis. A recent systematic literature review of the reports on the trials for avapritinib (EXPLORER and PATHFINDER) and midostaurin (D2201 and A2213 trials), after adjusting for differences in important features of patients with advanced systemic mastocytosis, demonstrated the superior efficacy of avapritinib versus midostaurin with respect to overall response rates, disease burden, and survival 33. Bezuclastinib, a highly selective inhibitor of KIT D816V that exhibits minimal penetrance of the blood–brain barrier is currently being studied in phase 2 clinical trials for indolent systemic mastocytosis (ISM), smoldering systemic mastocytosis (SSM), and advanced systemic mastocytosis 34.
Treatment of aggressive systemic mastocytosis with imatinib mesylate (multi-kinase inhibitor) was approved in 2006. Imatinib’s therapeutic role is limited to systemic mastocytosis patients with wild-type KIT 35, 36 and imatinib-sensitive transmembrane (F522C) 37 and juxtamembrane (V560G) KIT mutations 36, 38 as seen in cases of well-differentiated systemic mastocytosis (WDSM) 39, 40; the kinase domain with KIT D816V mutation shows intrinsic resistance to imatinib as certain juxtamembrane mutations do (V559I) 41.
Management of systemic mastocytosis with an associated hematologic neoplasm (SM-AHN) predominantly targets the associated hematologic neoplasm (AHN) component, especially if an aggressive disease such as acute myeloid leukemia (AML) is present 29. Allogeneic stem cell transplant may be suggested in patients with relapsed or refractory advanced systemic mastocytosis 23.
Figure 1. Systemic mastocytosis subtypes
Footnote: SM = systemic mastocytosis
[Source 9 ]Systemic mastocytosis causes
Systemic mastocytosis occurs when white blood cells called mast cells, which are produced in bone marrow, abnormally accumulate in one or more tissues. In most cases of systemic mastocytosis, the accumulated mast cells have a mutation in a gene called KIT. The KIT gene provides instructions for making a protein that plays an important role in development and activity of mast cells. The KIT protein stimulates chemical signaling pathways that are involved in the growth and division (proliferation) of many types of cells, including mast cells. In systemic mastocytosis, KIT gene mutations are somatic, which means they are acquired during a person’s lifetime. These mutations result in a KIT protein that is always turned on (activated). As a result, signaling pathways are overactive, leading to increased production and accumulation of mast cells.
In systemic mastocytosis, mast cells most often accumulate in the bone marrow, which is where new blood cells are made. Mast cells can also gather in other tissues such as the gastrointestinal tract, lymph nodes, spleen, or liver. In severe cases, excessive accumulation of mast cells can interfere with normal organ functioning. Mast cells normally trigger inflammation during an allergic reaction. When mast cells are activated by an environmental trigger, they release proteins (called mediators) that signal an immune response. In systemic mastocytosis, excess mast cells mean more mediator proteins are being released in the tissues where the cells accumulate, leading to an increased immune response. In affected individuals, triggers that can activate mast cells include changes in temperature, friction and minor trauma, surgery, insect stings, vaccines, anxiety, and stress. Certain medications can also be triggers, including aspirin, opioids, or non-steroidal anti-inflammatory drugs (NSAIDs).
Mutations in additional genes seem to modify the severity of systemic mastocytosis, often resulting in a more aggressive disease and shorter survival. These genes primarily play roles in controlling the proliferation of cells or regulating the activity of other genes that are important in development.
Systemic mastocytosis symptoms
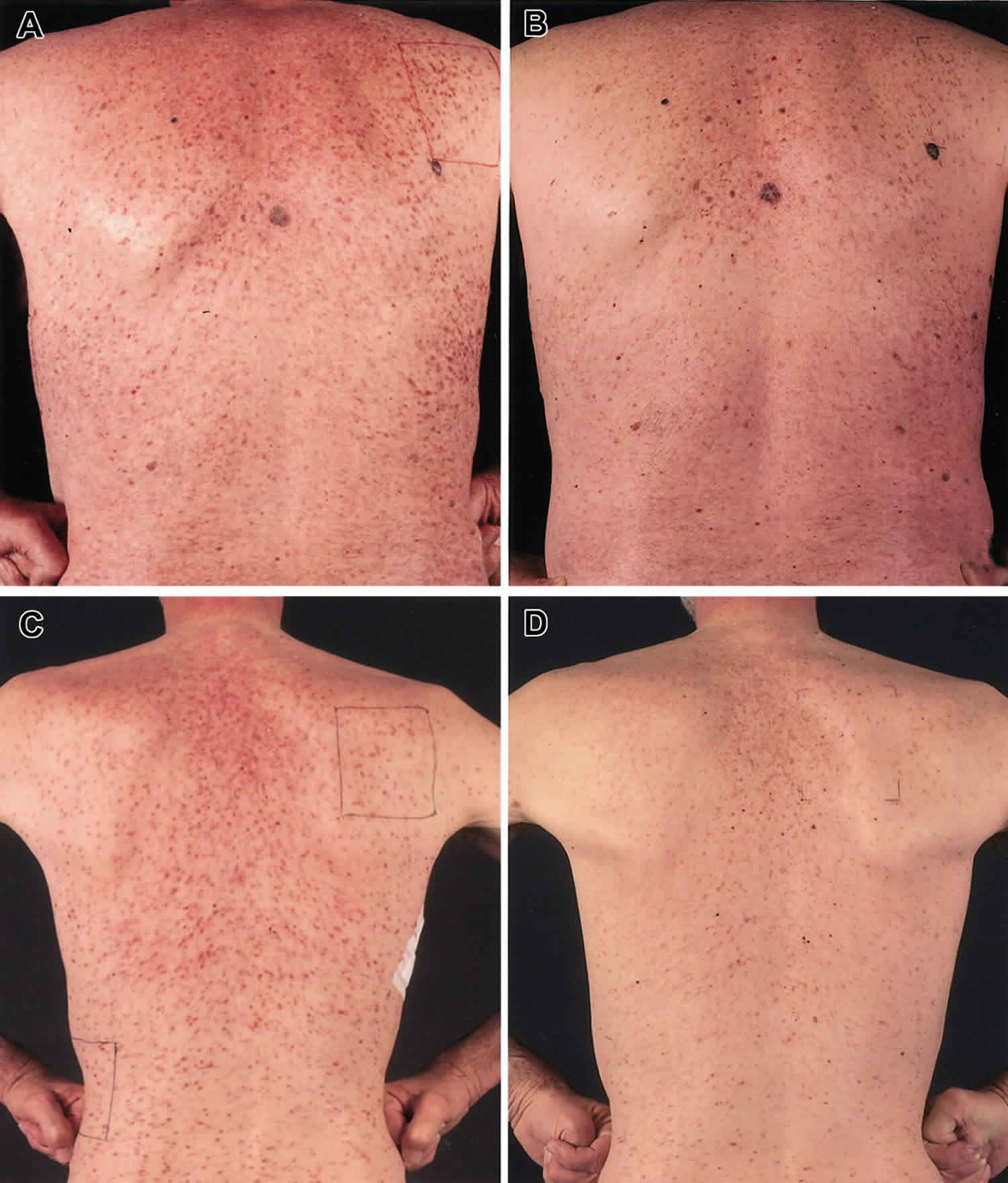
Systemic mastocytosis signs and symptoms often include extreme tiredness (fatigue), skin redness and warmth (flushing), nausea, abdominal pain, bloating, diarrhea, the backflow of stomach acids into the esophagus (gastroesophageal reflux), nasal congestion, shortness of breath, low blood pressure (hypotension), lightheadedness, and headache. Some affected individuals have attention or memory problems, anxiety, or depression. Many individuals with systemic mastocytosis develop a skin condition called urticaria pigmentosa, which is characterized by raised patches of brownish skin that sting or itch with contact or changes in temperature. Nearly half of individuals with systemic mastocytosis will experience severe allergic reactions (anaphylaxis).
The severity of the symptoms associated with mastocytosis may vary from mild to life-threatening. In general, symptoms occurring in mastocytosis are mainly due to the release of chemicals from the mast cells and thus produce symptoms associated with an allergic reaction. Flushing and gastric acid hypersecretion due to mast cell-associated histamine release are common symptoms. Heartburn, stomach aches, abdominal discomfort and diarrhea may occur. The liver, spleen and lymph nodes may become enlarged in some patients; therefore regular follow-up is necessary. Bones affected by mastocytosis may become softened (osteoporosis) and deteriorate, although some new bone growth may occur with thickening of the outer portions or spongy inner areas of the bones. In aggressive systemic mastocytosis, a decrease in blood cells (cytopenia), break-down of bones (osteolysis), swelling of the lymph nodes (lymphadenopathy), swelling of the liver (hepatomegaly), impaired liver function, ascites or portal hypertension, and malabsorption, may also occur.
Massive chemical release from the mast cells (degranulation) may lead to life-threatening episodes of anaphylaxis (anaphylactic shock). The most common triggers include, but are not limited to, certain foods, insect stings, physical stress (heat, cold, mechanical irritation of the skin), emotional stress, alcohol, and medications, including aspirin and non-steroidal anti-inflammatory drugs (NSAIDS), narcotics, muscle relaxants, radiocontrast material, among others. These are similar in nature to severe allergic reactions and may involve decreased blood pressure (hypotension), increased heart rate and loss of consciousness. Recent studies have found that up to 10% of patients with severe allergic reactions to bee stings may have mastocytosis. Additional non-specific symptoms that can be seen with mastocytosis include pain, nausea, headache, and/or malaise. Patients with an associated hematologic disorder may have symptoms of that disorder such as fatigue and weight loss.
Manifestations of systemic mastocytosis may include the following:
- Anemia and coagulopathy
- Abdominal pain is the most common GI symptom, followed, by diarrhea, nausea, and vomiting
- Symptoms and signs of gastroesophageal reflux disease (GERD)
- Pruritus and flushing
- Anaphylactoid reaction (eg, to Hymenoptera stings, general anesthetics, intravenous contrast media, other drugs, foods) 42
Findings on physical examination may include the following:
- Signs of anemia (eg, pallor)
- Hepatomegaly (27%)
- Splenomegaly (37%)
- Lymphadenopathy (21%)
- Urticaria (41%)
- Osteolysis and pathological fractures (rare)
Systemic mastocytosis is known to be associated with a number of other hematologic diseases, including the following:
- Hypereosinophilic syndrome
- Castleman disease
- Monoclonal gammopathy
- Hairy cell leukemia
- Non-Hodgkin lymphoma
- Polycythemia vera
- Primary thrombocythemia
Systemic mastocytosis diagnosis
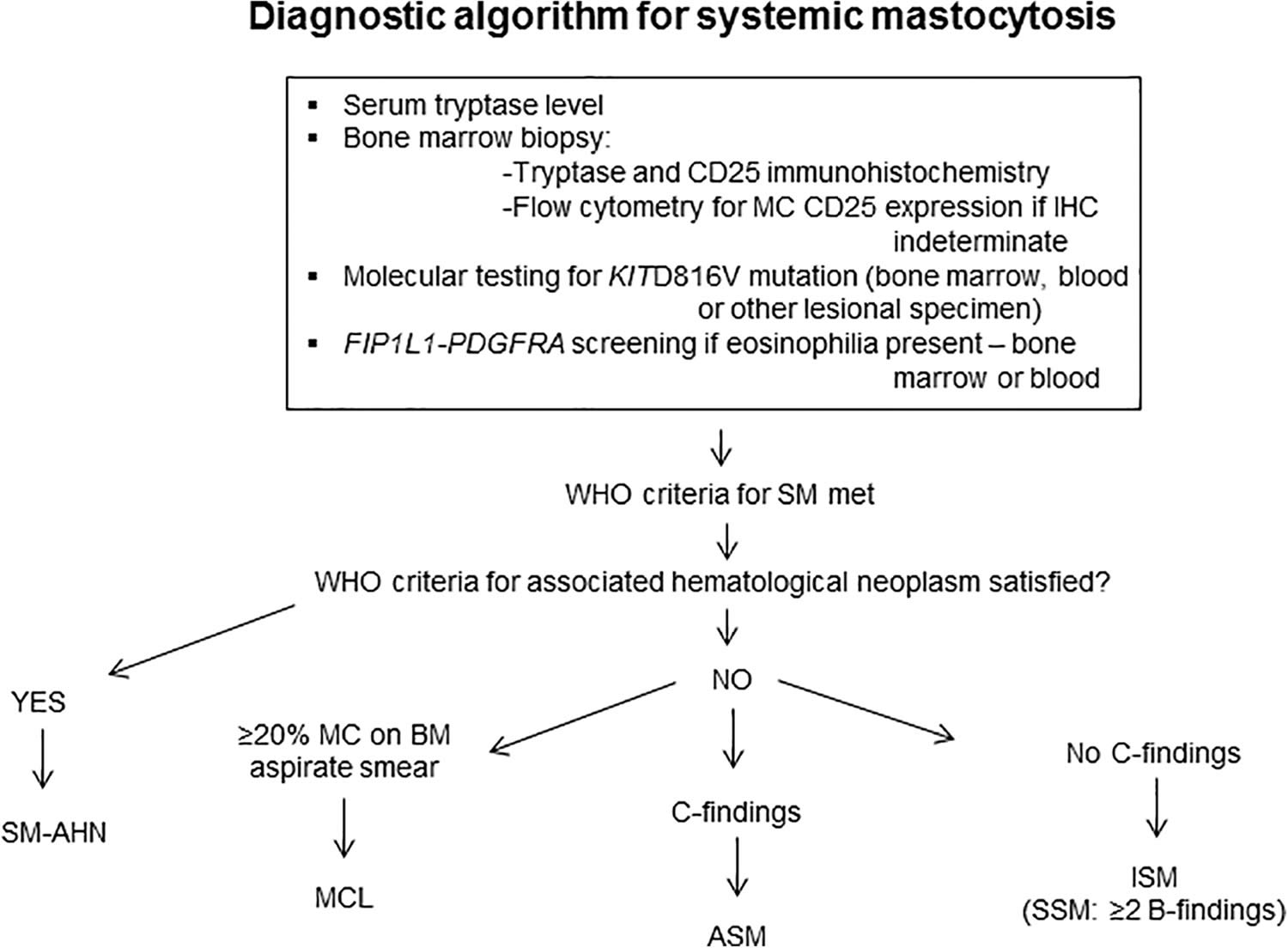
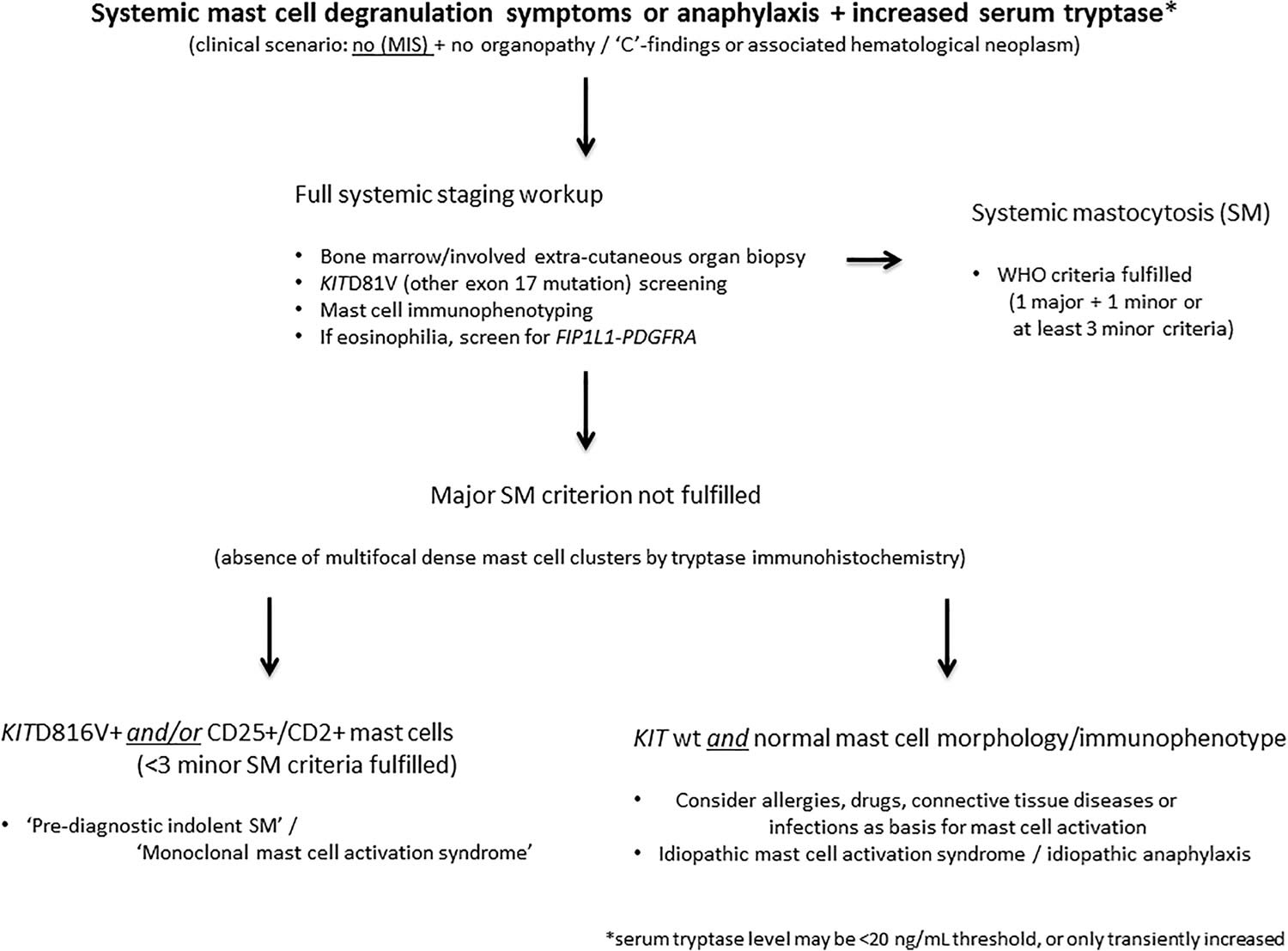
Your doctor may diagnose systemic mastocytosis through blood or urine tests or imaging tests like X-rays, ultrasounds, and CT scans. You may need a test to collect a sample of bone marrow to study in a lab. These tests look for high levels of mast cells or the substances they release.
Figure 2. Systemic mastocytosis diagnostic algorithm
Laboratory studies
The peripheral blood picture can show anemia, thrombocytopenia, and leukocytosis. The most common abnormality found in the peripheral blood is anemia (45%). In some patients with systemic mastocytosis, the following abnormalities can be observed in peripheral blood:
- Eosinophilia
- Basophilia
- Thrombocytosis
- Monocytosis
The combination of anemia, thrombocytopenia, hypoalbuminemia, and excess bone marrow blasts (>5%) portends a poor prognosis 43.
Total serum tryptase levels of 20 ng/mL or higher in a baseline serum sample that is associated with a ratio of total–to–beta-tryptase ratio greater than 20:1 is suggestive of systemic mastocytosis 44. More than 50% of patients reveal a high tryptase level using the cut-off value of 11.5 ng/mL. However, a normal serum tryptase level does not exclude the diagnosis of systemic mastocytosis 45.
Measurements of urinary N-methyl imidazole are useful in some patients with systemic mastocytosis.
Lueke et al 46 developed an assay for measurement of urinary leukotriene E4 (LTE4) and confirmed that median urine LTE4 concentrations were significantly higher in patients with systemic mastocytosis (median 97 pg per mg creatinine versus 50 pg/mg cr.; P< 0.01), with 48% sensitivity and 84% specificity for the disorder. These authors incorporated LTE4 into a panel of urinary biomarkers to provide a screening tool for systemic mastocytosis. Additional biomarker results and accuracy were as follows:
- N-methyl histamine (NMH): >200 ng/mL – Clinical sensitivity 71%
- 11ß-prostaglandin F2α (BPG): >1000 ng/mL – Clinical sensitivity 53%
Imaging studies
Some imaging studies may be necessary in patients with systemic mastocytosis in order to identify the extent and stage of the disease, as follows:
- Patients with abdominal pain may require GI radiography, ultrasonography, or liver-spleen computed tomography (CT) scanning
- Skeletal surveys and bone CT scanning may be necessary in patients with suspected bone involvement.
Other Tests
Cytogenetic data indicate that about 20% of patients with systemic mastocytosis have an abnormal karyotype. These include trisomy 8; monosomy 7; del (13q); del(5q); trisomy 10, 6, and 19; del(20q); and trisomy X. Cytogenetic abnormalities are more often seen with aggressive systemic mastocytosis (ASM) and systemic mastocytosis with associated hematological non–mast cell disorder (SM-AHNMD) than with indolent systemic mastocytosis 43.
Molecular testing for KIT D816V mutation is universally positive, whereas JAK2 V617F is rarely positive (4%).
The mast cell clone is CD117 positive and CD25 and/or CD2 positive.
Bone marrow mast cells reveal 54% CD2 positivity and 93% CD25 positivity on flow cytometry, whereas on immunohistochemistry CD2 positivity is 17% and CD25 positivity is 100% 43.
Expression of CD25 on mast cells is seen in systemic mastocytosis but is not noted in reactive states of mast cell hyperplasia 47.
Diagnostic procedures
The following procedures may be indicated in patients with systemic mastocytosis:
- When gastrointestinal symptoms are present, perform gastrointestinal procedures (eg, barium studies, endoscopy) to help confirm the diagnosis
- Patients with hepatomegaly can show evidence of mast cell infiltration on liver biopsy specimens
- Skin biopsy may be warranted in patients with skin manifestations
- Bone marrow aspiration and biopsy are essential
Bone Marrow Biopsy
Bone marrow aspiration and biopsy is essential for the diagnosis of systemic mastocytosis 48. Histologic findings provide both major and minor diagnostic criteria.
Diagnostic criteria
The major diagnostic criterion for systemic mastocytosis is the presence of dense infiltrates of mast cells in bone marrow or other extracutaneous tissues. Mast cells should be seen in aggregates of 15 or more.
The diagnosis of systemic mastocytosis can be made when the major criterion and one minor criterion or at least three minor criteria are present 6, 7, 8, 9:
Major diagnostic criteria for systemic mastocytosis are as follows 6, 7, 8, 9:
- Multifocal, dense infiltrates of mast cells (≥ 15 mast cells in aggregates) detected in sections of bone marrow and/or other extracutaneous organs.
Major criteria may be absent in early disease. In this situation, the minor criteria are used to make the pathological diagnosis.
Three of the following four minor criteria are required to make the diagnosis 6, 7, 8, 9:
- A) In biopsy sections of bone marrow or other extracutaneous organs, >25% of all mast cells are atypical cells (type 1 or type 2) on bone marrow smears or are spindle-shaped in dense and diffuse mast cell infiltrates in bone marrow or other extracutaneous organs. a
- B) Activating KIT point mutations at codon 816 or in other critical regions of KIT b in the bone marrow or other extracutaneous organs.
- C) Mast cells in bone marrow, blood, or other extracutaneous organs aberrantly express one or more of the following antigens: CD2, CD25, CD30. c
- D) Baseline serum tryptase concentration > 20 ng/mL in the absence of a myeloid associated hematologic neoplasm. d In the case of a known hereditary alpha-tryptasemia, the tryptase level should be adjusted. e
Notes:
- a In tissue sections, an abnormal mast cell morphology counts in both a dense infiltrate and a diffuse mast cell infiltrate. In the bone marrow smear, an atypical morphology of mast cells does not count as a systemic mastocytosis criterion when mast cells are located in or adjacent to bone marrow particles. Morphologic criteria of atypical mast cells were referenced in the consensus proposal 7.
- b Any type of KIT mutation is considered a minor systemic mastocytosis criterion when published solid evidence regarding its transforming behavior is available [an overview of potentially activating KIT mutations was provided in the supplementary material that can be found here 7].
- c Expression has to be confirmed by either flow cytometry or immunohistochemistry or by both techniques. Mast cell CD25 is the more sensitive marker, by both flow cytometry and immunohistochemistry.
- d Myeloid neoplasms can lead to increased serum tryptase levels; thus, this criterion does not count in cases of systemic mastocytosis-associated hematologic neoplasm.
- e A possible method of adjustment has been proposed for known hereditary alpha-tryptasemia 7; the basal tryptase level is divided by 1 plus the extra copy numbers of the alpha tryptase gene. For example, when the tryptase level is 30 and 1 extra copy of the alpha tryptase gene is found, the HαT-corrected tryptase level is 15 (30/2 = 15), and therefore, it is not a minor SM criterion in this case.
“B” findings
- High mast cell burden shown on bone marrow biopsy: >30% infiltration of cellularity by mast cells (focal, dense aggregates) and serum total tryptase level >200 ng/ml.
- Signs of dysplasia or myeloproliferation, in non-mast cell lineage(s), but insufficient criteria for definitive diagnosis of an associated hematological neoplasm (AHN), with normal or only slightly abnormal blood counts.
- Hepatomegaly without impairment of liver function, palpable splenomegaly without hypersplenism, and/or lymphadenopathy on palpation or imaging.
“C” findings
- Bone marrow dysfunction caused by neoplastic mast cell infiltration, manifested by ≥1 cytopenia(s) (ANC <1.0 × 109/L, Hgb <10 g/dl, and/or platelet count <100 × 109/L).
- Palpable hepatomegaly with impairment of liver function, ascites and/or portal hypertension.
- Skeletal involvement with large osteolytic lesions with/without pathological fractures
- Pathological fractures caused by osteoporosis do not qualify as a “C” finding.
- Palpable splenomegaly with hypersplenism.
- Malabsorption with weight loss due to gastrointestinal mast cell infiltrates.
Systemic mastocytosis treatment
Therapy for systemic mastocytosis is primarily symptomatic; no therapy is curative. Treatment for systemic mastocytosis may include medications like antihistamines, aspirin, and drugs that work against the substances released by mast cells in your body. If you have a severe allergic reaction, you may need an injection of epinephrine. Aggressive forms of systemic mastocytosis may need powerful chemotherapy drugs to destroy mast cells.
Treatment modalities include the management of the following:
- Anaphylaxis and related symptoms
- Pruritus and flushing
- Intestinal malabsorption
Agents for symptomatic relief include the following:
- Epinephrine is used in acute anaphylaxis
- H1 and H2 receptor blockers are used to control anaphylactic symptoms
- Corticosteroids have been used to control malabsorption, ascites, and bone pain and to prevent anaphylaxis
- Cromolyn is helpful for decreasing bone pain and headaches and for improving skin symptoms
- Patients with osteopenia that does not respond to therapy may receive a trial of interferon alfa-2b
- First-generation histamine H1 antagonists (eg, diphenhydramine, hydroxyzine) have been used to treat pruritus and flushing
- Histamine H2 antagonists and proton pump inhibitors have been used to treat gastric hypersecretion and peptic ulcer disease
- Aspirin can be used when H1 and H2 receptor blockers do not prevent vascular collapse
- Mast cell stabilizers (eg, ketotifen) have been used to treat pruritus and whealing
- Leukotriene antagonists (eg, zafirlukast, montelukast) have been used
- Cromolyn is helpful for decreasing bone pain and headaches and for improving skin symptoms
- Psoralen ultraviolet A therapy may provide transient relief of pruritus and may cause fading of skin lesions
- Anticholinergics have been used in the treatment of diarrhea
- Disodium cromolyn has been used in the treatment of abdominal cramping and diarrhea
Chemotherapy has not been particularly successful in the management of systemic mastocytosis, but the following regimens have been tried 49:
- Interferon-alfa may be beneficial, especially in patients with aggressive systemic mastocytosis
- 2-Chlorodeoxyadenosine (cladribine [Leustatin])
- Thalidomide in advanced disease
- Imatinib mesylate (Gleevec) in patients who do not have mutations of the codon 816 on the c-kit gene and carry the wild-type kit, or who carry the FIP1L1-PDGFRA rearrangement 50
- Midostaurin (Rydapt) is approved by the FDA for aggressive systemic mastocytosis (ASM), systemic mastocytosis with associated hematological neoplasm (SM-AHN), or mast cell leukemia (MCL), collectively referred to as advanced systemic mastocytosis 51
Systemic mastocytosis prognosis
The long-term outlook (prognosis) for people with systemic mastocytosis varies. Young children and those who present with primarily cutaneous (skin) and flushing symptoms tend to have little or no progression of the disease over a considerable length of time. Older patients and those with extensive, systemic disease involving other organ systems have a poorer prognosis. The median duration of survival is in these cases is not known but appears to be a few years 52.
Features that may be associated with a poorer prognosis may include 52:
- elevated lactate dehydrogenase levels
- anemia
- thrombocytopenia (low platelet count)
- hypoalbuminemia (low albumin)
- excess bone marrow blasts (>5%)
- high alkaline phosphatase
- hepatosplenomegaly (enlarged liver and/or spleen)
- ascites
Systemic mastocytosis life expectancy
Systemic mastocytosis is a progressive neoplastic disorder that has no known curative therapy. Individuals with the milder forms of systemic mastocytosis (indolent systemic mastocytosis) generally have a normal or near normal life expectancy, while those with the more severe forms typically survive months or a few years after diagnosis. Median survival ranges from 198 months in patients with indolent systemic mastocytosis to 41 months in aggressive systemic mastocytosis and 2 months in mast cell leukemia 52. Median survival with aggressive systemic mastocytosis is 41 months and that with systemic mastocytosis associated hematological non– mast cell disorder is 24 months 52. Mast cell leukemia (MCL) has the poorest prognosis with a median survival of 2 months 52.
Early evolution into acute leukemia may occur in as many as 32% of patients with aggressive mastocytosis 53. Leukemic transformation is rare with indolent systemic mastocytosis 43.
References- Velloso EDRP, Padulla GA, de Cerqueira AMM, de Sousa AM, Sandes AF, Traina F, Seguro FS, Nogueira FL, Pereira GF, Boechat JL, Pagnano KBB, Marchi LL, Ensina LF, Giavina-Bianchi M, Aun MV, Agondi RC, Santos FPS, Giavina-Bianchi P. Diagnosis and treatment of systemic mastocytosis in Brazil: Recommendations of a multidisciplinary expert panel. Hematol Transfus Cell Ther. 2022 Oct-Dec;44(4):582-594. doi: 10.1016/j.htct.2022.04.006
- González-López O, Muñoz-González JI, Orfao A, Álvarez-Twose I, García-Montero AC. Comprehensive Analysis of Acquired Genetic Variants and Their Prognostic Impact in Systemic Mastocytosis. Cancers (Basel). 2022 May 18;14(10):2487. doi: 10.3390/cancers14102487
- Zanotti R, Tanasi I, Crosera L, Bonifacio M, Schena D, Orsolini G, Mastropaolo F, Tebaldi M, Olivieri E, Bonadonna P. Systemic Mastocytosis: Multidisciplinary Approach. Mediterr J Hematol Infect Dis. 2021 Nov 1;13(1):e2021068. doi: 10.4084/MJHID.2021.068
- Taylor F, Akin C, Lamoureux RE, Padilla B, Green T, Boral AL, Mazar I, Mar B, Shields AL, Siebenhaar F. Development of symptom-focused outcome measures for advanced and indolent systemic mastocytosis: the AdvSM-SAF and ISM-SAF©. Orphanet J Rare Dis. 2021 Oct 9;16(1):414. doi: 10.1186/s13023-021-02035-5
- El Hussein S, Chifotides HT, Khoury JD, Verstovsek S, Thakral B. Systemic Mastocytosis and Other Entities Involving Mast Cells: A Practical Review and Update. Cancers (Basel). 2022 Jul 17;14(14):3474. doi: 10.3390/cancers14143474
- Khoury J.D., Solary E., Abla O., Akkari Y., Alaggio R., Apperley J.F., Hochhaus A. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. 2022;36:1703–1719. doi: 10.1038/s41375-022-01613-1
- Valent P., Akin C., Hartmann K., Alvarez-Twose I., Brockow K., Hermine O., Niedoszytko M., Schwaab J., Lyons J.J., Carter M.C., et al. Updated Diagnostic Criteria and Classification of Mast Cell Disorders: A Consensus Proposal. HemaSphere. 2021;5:e646. doi: 10.1097/HS9.0000000000000646
- Valent P, Akin C, Metcalfe DD. Mastocytosis: 2016 updated WHO classification and novel emerging treatment concepts. Blood. 2017 Mar 16;129(11):1420-1427. doi: 10.1182/blood-2016-09-731893
- Pardanani, A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am J Hematol. 2021; 96: 508– 525. https://doi.org/10.1002/ajh.26118
- Arock M., Hoermann G., Sotlar K., Hermine O., Sperr W.R., Hartmann K., Brockow K., Akin C., Triggiani M., Broesby-Olsen S., et al. Clinical impact and proposed application of molecular markers, genetic variants, and cytogenetic analysis in mast cell neoplasms: Status 2022. J. Allergy Clin. Immunol. 2022;149:1855–1865. doi: 10.1016/j.jaci.2022.04.004
- Horny HP, Metcalfe DD, Akin C, et al. In: S Swerdlow, E Campo, NL Harris, E Jaffe, S Pileri, H Stein, J Thiele. eds. Mastocytosis, in WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues. Lyon: International Agency for Research and Cancer (IARC); 2017: 62- 69.
- Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage). Blood. 2013; 20: 2013- 3094. https://doi.org/10.1182/blood-2013-01-453183
- Pardanani A, Lim KH, Lasho TL, Finke CM, McClure RF, Li CY, Tefferi A. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010 Jan 7;115(1):150-1. doi: 10.1182/blood-2009-10-249979
- Zanotti R., Tanasi I., Bernardelli A., Orsolini G., Bonadonna P. Bone Marrow Mastocytosis: A Diagnostic Challenge. J. Clin. Med. 2021;10:1420. doi: 10.3390/jcm10071420
- Arber D.A., Orazi A., Hasserjian R., Thiele J., Borowitz M.J., Le Beau M.M., Bloomfield C.D., Cazzola M., Vardiman J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544
- Zanotti R., Bonadonna P., Bonifacio M., Artuso A., Schena D., Rossini M., Perbellini O., Colarossi S., Chilosi M., Pizzolo G. Isolated bone marrow mastocytosis: An underestimated subvariant of indolent systemic mastocytosis. Haematologica. 2011;96:482–484. doi: 10.3324/haematol.2010.034553
- Zanotti R., Bonifacio M., Lucchini G., Sperr W.R., Scaffidi L., van Anrooij B., Oude Elberink H.N., Rossignol J., Hermine O., Gorska A., et al. Refined diagnostic criteria for bone marrow mastocytosis: A proposal of the European competence network on mastocytosis. Leukemia. 2022;36:516–524. doi: 10.1038/s41375-021-01406-y
- Gotlib J, Pardanani A, Akin C, Reiter A, George T, Hermine O, et al. International Working Group-Myeloproliferative Neoplasms Research and Treatment (IWG-MRT) & European Competence Network on Mastocytosis (ECNM) consensus response criteria in advanced systemic mastocytosis. Blood. 2013 Mar 28;121(13):2393-401. http://www.ncbi.nlm.nih.gov/pubmed/23325841
- Valent P., Sotlar K., Sperr W.R., Reiter A., Arock M., Horny H.P. Chronic mast cell leukemia: A novel leukemia-variant with distinct morphological and clinical features. Leuk. Res. 2015;39:1–5. doi: 10.1016/j.leukres.2014.09.010
- Valent P., Blatt K., Eisenwort G., Herrmann H., Cerny-Reiterer S., Thalhammer R., Mullauer L., Hoermann G., Sadovnik I., Schwarzinger I., et al. FLAG-induced remission in a patient with acute mast cell leukemia (MCL) exhibiting t(7;10)(q22;q26) and KIT D816H. Leuk. Res. Rep. 2014;3:8–13. doi: 10.1016/j.lrr.2013.11.001
- Mariana C Castells and Cem Akin. Systemic mastocytosis: Determining the category of disease. UpToDate. Waltham, MA: UpToDate; August, 2016
- Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009 Jun 4. 113(23):5727-36.
- Pardanani A. Systemic mastocytosis in adults: 2021 Update on diagnosis, risk stratification and management. Am. J. Hematol. 2021;96:508–525. doi: 10.1002/ajh.26118
- Gotlib J., Kluin-Nelemans H.C., George T.I., Akin C., Sotlar K., Hermine O., Awan F.T., Hexner E., Mauro M.J., Sternberg D.W., et al. Efficacy and safety of midostaurin in advanced systemic mastocytosis. N. Engl. J. Med. 2016;374:2530–2541. doi: 10.1056/NEJMoa1513098
- DeAngelo D.J., George T.I., Linder A., Langford C., Perkins C., Ma J., Westervelt P., Merker J.D., Berube C., Coutre S., et al. Efficacy and safety of midostaurin in patients with advanced systemic mastocytosis: 10-year median follow-up of a phase II trial. Leukemia. 2018;32:470–478. doi: 10.1038/leu.2017.234
- Evans E.K., Gardino A.K., Kim J.L., Hodous B.L., Shutes A., Davis A., Zhu X.J., Schmidt-Kittler O., Wilson D., Wilson K., et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017;9:eaao1690. doi: 10.1126/scitranslmed.aao1690
- Bose P, Verstovsek S: Avapritinib for Systemic Mastocytosis. Expert. Rev. Hematol. 2021;14:687–696. doi: 10.1080/17474086.2021.1959315
- Shomali W., Gotlib J. The new tool “KIT” in advanced systemic mastocytosis. Hematol. Am. Soc. Hematol. Educ. Program. 2018;2018:127–136. doi: 10.1182/asheducation-2018.1.127
- Reiter A., George T.I., Gotlib J. New developments in diagnosis, prognostication, and treatment of advanced systemic mastocytosis. Blood. 2020;135:1365–1376. doi: 10.1182/blood.2019000932
- Baird J.H., Gotlib J. Clinical validation of KIT inhibition in advanced systemic mastocytosis. Curr. Hematol. Malig. Rep. 2018;13:407–416. doi: 10.1007/s11899-018-0469-3
- DeAngelo D.J., Radia D.H., George T.I., Robinson W.A., Quiery A.T., Drummond M.W., Bose P., Hexner E.O., Winton E.F., Horny H.P., et al. Safety and efficacy of avapritinib in advanced systemic mastocytosis: The phase 1 EXPLORER trial. Nat. Med. 2021;27:2183–2191. doi: 10.1038/s41591-021-01538-9
- Gotlib J., Reiter A., Radia D.H., Deininger M.W., George T.I., Panse J., Vannucchi A.M., Platzbecker U., Alvarez-Twose I., Mital A., et al. Efficacy and safety of avapritinib in advanced systemic mastocytosis: Interim analysis of the phase 2 PATHFINDER trial. Nat. Med. 2021;27:2192–2199. doi: 10.1038/s41591-021-01539-8
- Pilkington H., Smith S., Roskell N., Iannazzo S. Indirect treatment comparisons of avapritinib versus midostaurin for patients with advanced systemic mastocytosis. Future Oncol. 2022;18:1583–1594. doi: 10.2217/fon-2021-1509
- Siebenhaar F., Gotlib J., Deininger M.W., DeAngelo D.J., Payumo F., Mensing G., George T.I. A 3-part, phase 2 study of Bezuclastinib (CGT9486), an oral, selective, and potent KIT D816V inhibitor, in adult patients with nonadvanced systemic mastocytosis (NonAdvSM) Blood. 2021;138:3642. doi: 10.1182/blood-2021-147072
- Zermati Y., De Sepulveda P., Feger F., Letard S., Kersual J., Casteran N., Gorochov G., Dy M., Ribadeau Dumas A., Dorgham K., et al. Effect of tyrosine kinase inhibitor STI571 on the kinase activity of wild-type and various mutated c-kit receptors found in mast cell neoplasms. Oncogene. 2003;22:660–664. doi: 10.1038/sj.onc.1206120
- Akin C., Brockow K., D’Ambrosio C., Kirshenbaum A.S., Ma Y., Longley B.J., Metcalfe D.D. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp. Hematol. 2003;31:686–692. doi: 10.1016/S0301-472X(03)00112-7
- Akin C., Fumo G., Yavuz A.S., Lipsky P.E., Neckers L., Metcalfe D.D. A novel form of mastocytosis associated with a transmembrane c-kit mutation and response to imatinib. Blood. 2004;103:3222–3225. doi: 10.1182/blood-2003-11-3816
- Ma Y., Zeng S., Metcalfe D.D., Akin C., Dimitrijevic S., Butterfield J.H., McMahon G., Longley B.J. The c-KIT mutation causing human mastocytosis is resistant to STI571 and other KIT kinase inhibitors; kinases with enzymatic site mutations show different inhibitor sensitivity profiles than wild-type kinases and those with regulatory-type mutations. Blood. 2002;99:1741–1744. doi: 10.1182/blood.V99.5.1741
- Alvarez-Twose I., Matito A., Morgado J.M., Sanchez-Munoz L., Jara-Acevedo M., Garcia-Montero A., Mayado A., Caldas C., Teodosio C., Munoz-Gonzalez J.I., et al. Imatinib in systemic mastocytosis: A phase IV clinical trial in patients lacking exon 17 KIT mutations and review of the literature. Oncotarget. 2017;8:68950–68963. doi: 10.18632/oncotarget.10711
- Piris-Villaespesa M., Alvarez-Twose I. Systemic mastocytosis: Following the tyrosine kinase inhibition roadmap. Front. Pharmacol. 2020;11:443. doi: 10.3389/fphar.2020.00443
- Nakagomi N., Hirota S. Juxtamembrane-type c-kit gene mutation found in aggressive systemic mastocytosis induces imatinib-resistant constitutive KIT activation. Lab. Investig. 2007;87:365–371. doi: 10.1038/labinvest.3700524
- Bonadonna P, Zanotti R, Pagani M, Caruso B, Perbellini O, Colarossi S. How much specific is the association between hymenoptera venom allergy and mastocytosis?. Allergy. 2009 Sep. 64(9):1379-82
- Lim KH, Tefferi A, Lasho TL, et al. Systemic mastocytosis in 342 consecutive adults: survival studies and prognostic factors. Blood. 2009 Jun 4. 113(23):5727-36
- Schwartz LB, Irani AM. Serum tryptase and the laboratory diagnosis of systemic mastocytosis. Hematol Oncol Clin North Am. 2000 Jun. 14(3):641-57.
- Jawhar M, Schwaab J, Horny HP, Sotlar K, Naumann N, Fabarius A, et al. Impact of Centralized Evaluation of Bone Marrow Histology in Systemic Mastocytosis. Eur J Clin Invest. 2016 Feb 23
- Lueke AJ, Meeusen JW, Donato LJ, Gray AV, Butterfield JH, Saenger AK. Analytical and clinical validation of an LC-MS/MS method for urine leukotriene E4: A marker of systemic mastocytosis. Clin Biochem. 2016 Feb 18. 264 (1):217-22.
- Horny HP. Mastocytosis: an unusual clonal disorder of bone marrow-derived hematopoietic progenitor cells. Am J Clin Pathol. 2009 Sep. 132(3):438-47
- Jawhar M, Schwaab J, Horny HP, Sotlar K, Naumann N, Fabarius A, et al. Impact of Centralized Evaluation of Bone Marrow Histology in Systemic Mastocytosis. Eur J Clin Invest. 2016 Feb 23.
- Ustun C, Corless CL, Savage N, et al. Chemotherapy and dasatinib induce long-term hematologic and molecular remission in systemic mastocytosis with acute myeloid leukemia with KIT D816V. Leuk Res. 2009 May. 33(5):735-41
- Pardanani A, Ketterling RP, Brockman SR, et al. CHIC2 deletion, a surrogate for FIP1L1-PDGFRA fusion, occurs in systemic mastocytosis associated with eosinophilia and predicts response to imatinib mesylate therapy. Blood. 2003 Nov 1. 102(9):3093-6.
- Gotlib J, Kluin-Nelemans HC, George TI, Akin C, Sotlar K, Hermine O, et al. Efficacy and Safety of Midostaurin in Advanced Systemic Mastocytosis. N Engl J Med. 2016 Jun 30. 374 (26):2530-41.
- Systemic Mastocytosis. https://emedicine.medscape.com/article/203948-overview
- Pieri L, Bonadonna P, Elena C, Papayannidis C, Grifoni FI, et al. Clinical presentation and management practice of systemic mastocytosis. A survey on 460 Italian patients. Am J Hematol. 2016 Apr 7.




{kind=link}