Thanatophoric dysplasia
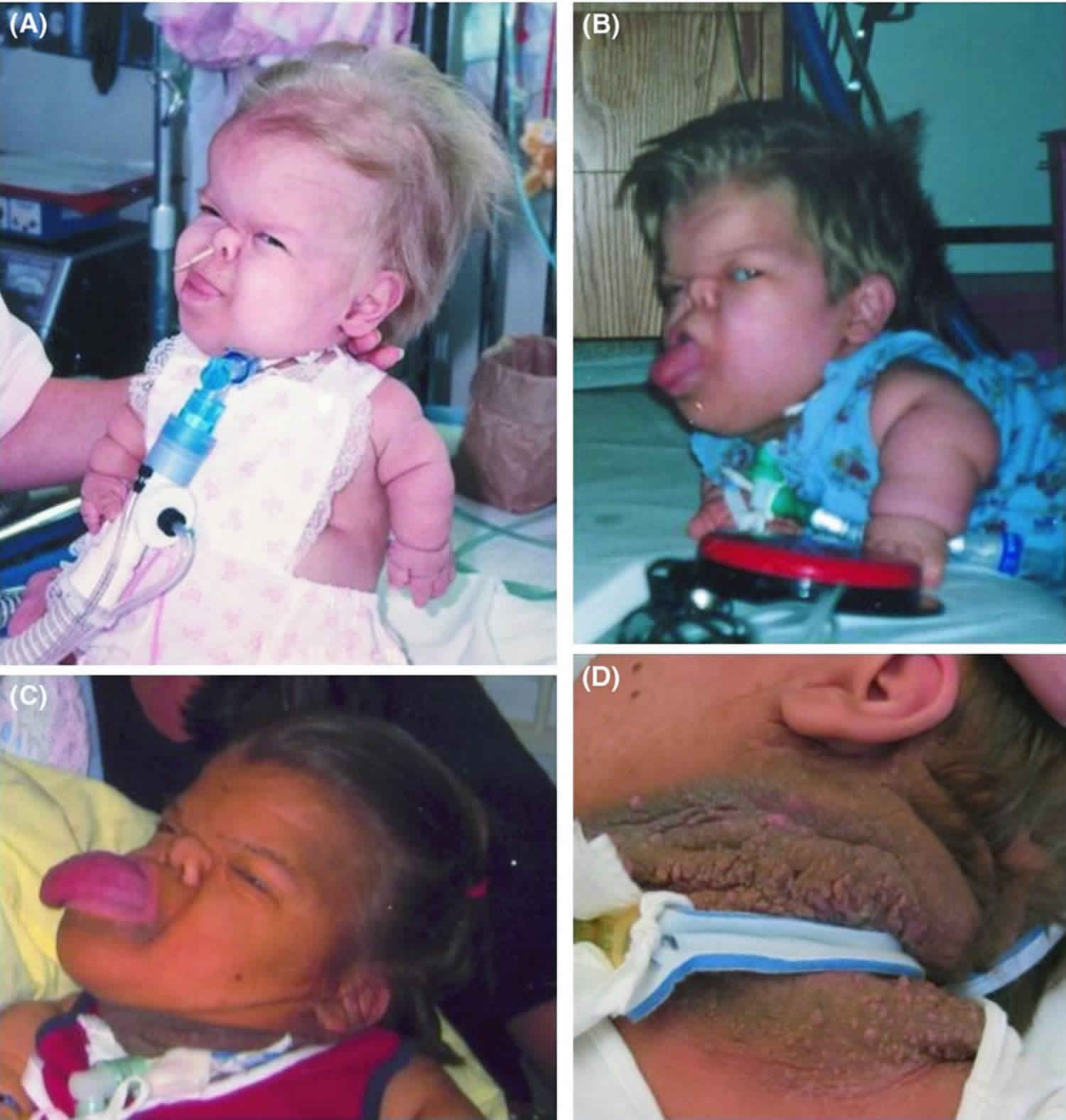
The term thanatophoric derives from the Greek word thanatophorus, which means “death bringing” or “death bearing”. Infants with thanatophoric dysplasia are usually stillborn or die shortly after birth from respiratory failure; however, a few affected individuals have survived into childhood with extensive medical help 1. Thanatophoric dysplasia also called thanatophoric dwarfism or thanatophoric short stature, is a severe skeletal disorder characterized by severe shortening of the limbs and folds of extra (redundant) skin on the arms and legs. Other features of thanatophoric dysplasia dwarfism include a narrow bell-shaped chest, short ribs, underdeveloped lungs, normal trunk length and an enlarged head with a large forehead (macrocephaly) and prominent, wide-spaced eyes.
Researchers have described two major forms of thanatophoric dysplasia, type 1 and type 2 1.
- Thanatophoric dysplasia type 1 is distinguished by the presence of curved thigh bones and flattened bones of the spine (platyspondyly).
- Thanatophoric dysplasia type 2 is characterized by straight thigh bones and a moderate to severe skull abnormality called a cloverleaf skull.
Thanatophoric dysplasia occurs in 1 in 20,000 to 50,000 newborns. Type 1 thanatophoric dysplasia is more common than type 2.
Thanatophoric dysplasia is caused by mutations in the FGFR3 gene. While thanatophoric dysplasia is considered to be autosomal dominant, virtually all cases have occurred in people with no history of the disorder in their family.
Most individuals with thanatophoric dysplasia die in the perinatal period because of the multisystem complications of the disorder. Management goals should be established with the family, and may focus on provision of comfort care. Newborns require long-term respiratory support (typically with tracheostomy and ventilation) to survive. Anesthetic management guidelines for skeletal dysplasias are applicable to individuals with thanatophoric dysplasia. Other treatment measures may include shunt placement for hydrocephalus, suboccipital decompression for relief of craniocervical junction constriction, antiepileptic drugs to control seizures, and hearing aids.
Thanatophoric dysplasia type 1
Thanatophoric dysplasia type 1 is a form of thanatophoric dysplasia characterized by short, bowed femurs, micromelia, narrow thorax, and brachydactyly.
The prevalence is unknown but it is more common than thanatophoric dysplasia type 2.
Thanatophoric dysplasia type 1 presents in the prenatal period (in the first to second trimester) with growth deficiency of the limbs of less than 5%, bowed femurs (like a telephone receiver), shortened ribs, and platyspondyly of the vertebrae. Distinctive facial features include macrocephaly, large anterior fontanel, frontal bossing, proptosis and low nasal bridge. Neonates usually die shortly after birth due to respiratory insufficiency and/or spinal cord/brain stem compression. Platyspondylic lethal skeletal dysplasia, San Diego type (PTSD-SD) is now thought to be an earlier fetal phenotype of thanatophoric dysplasia type 1, and is no longer characterized as a distinct dysplasia.
Thanatophoric dysplasia type 2
Thanatophoric dysplasia type 2 is a form of thanatophoric dysplasia characterized by micromelia, straight long-bones, macrocephaly, brachydactyly, shortened ribs and a clover-leaf skull (kleeblattschaedel).
The prevalence is unknown but it is less common than thanatophoric dysplasia type 1
Thanatophoric dysplasia type 2 presents in the prenatal period (in the first to second trimester) with micromelia, long-bones (femurs) that are straight and not as short as those seen in thanatophoric dysplasia type 1, platyspondyly of the vertebrae, narrow thorax, and a cloverleaf (trilobed) skull. Distinctive facial features include macrocephaly, frontal bossing, low nasal bridge, large anterior fontanel and proptosis. Polyhydramnios is common. Neonates usually die shortly after birth due to respiratory insufficiency and/or spinal cord/brain stem compression.
Thanatophoric dysplasia causes
Mutations in the FGFR3 gene located to chromosome 4p16.3 cause thanatophoric dysplasia. Both types of this condition result from mutations in the FGFR3 gene. The FGFR3 gene provides instructions for making a protein that is involved in the development and maintenance of bone and brain tissue. Mutations in this gene cause the FGFR3 protein to be overly active, which leads to the severe disturbances in bone growth that are characteristic of thanatophoric dysplasia. It is not known how FGFR3 mutations cause the brain and skin abnormalities associated with thanatophoric dysplasia.
Thanatophoric dysplasia inheritance pattern
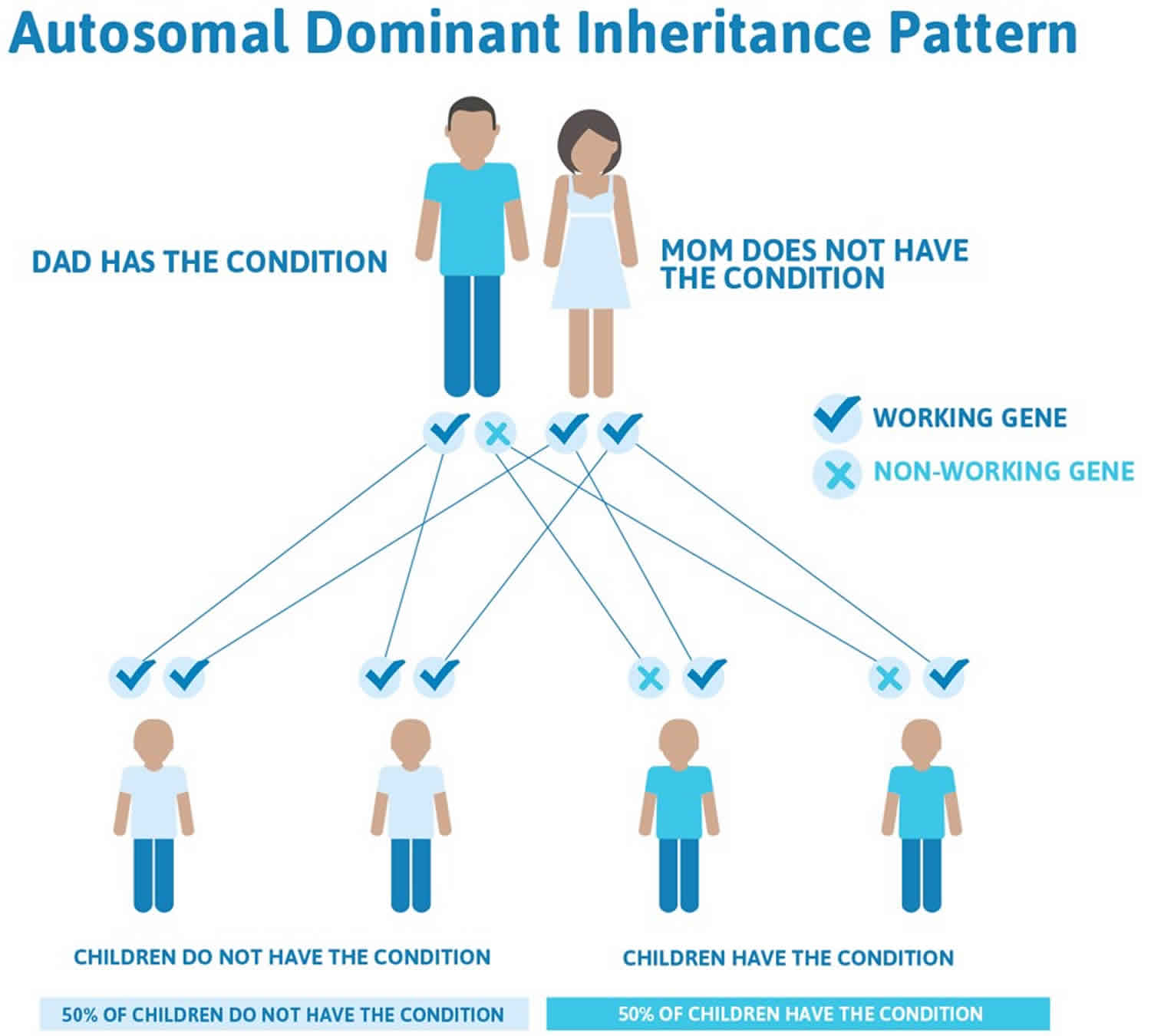
Thanatophoric dysplasia is considered an autosomal dominant disorder because one mutated copy of the FGFR3 gene in each cell is sufficient to cause the condition. Virtually all cases of thanatophoric dysplasia are caused by new mutations (de novo mutation) in the FGFR3 gene and occur in people with no history of the disorder in their family. No affected individuals are known to have had children; therefore, the disorder has not been passed to the next generation.
Figure 1. Thanatophoric dysplasia autosomal dominant inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
Thanatophoric dysplasia symptoms
Newborns with thanatophoric dysplasia are stillborn or die shortly after birth. Death occurs usually within 48 hours and is due to severe respiratory insufficiency from a reduced thoracic capacity and hypoplastic lungs and/or respiratory failure due to brainstem compression. Survival into early childhood has been rarely reported 2.
Salient physical features in an thanatophoric dysplasia newborn are outlined below:
- Severe growth deficiency with an average length of 40 cm (about 16 in) at term
- Generalized hypotonia
- Macrocephalic head with frontal bossing and large anterior fontanel
- Cloverleaf-shaped skull due to premature closure of the cranial sutures
- Flat facies with low nasal bridge and proptotic eyes
- Narrow, bell-shaped thorax with short ribs
- Normal trunk length
- Protuberant abdomen
- Micromelia (marked bilateral shortening of the limbs) with redundant skin folds
- Brachydactyly with a trident hand configuration
To date, more than 200 individuals with thanatophoric dysplasia have been identified with a pathogenic variant in FGFR3 3. The following description of the phenotypic features associated with this condition is based on these reports.
Respiratory insufficiency. Most affected infants die of respiratory insufficiency in the first hours or days of life. Respiratory insufficiency may be secondary to a small chest with lung hypoplasia and/or compression of the brain stem due to a small foramen magnum. Some affected children have survived into childhood, universally requiring tracheostomy and aggressive ventilatory support. Nikkel et al 4 described a long-term survivor with thanatophoric dysplasia at age 28 years. Her course was exceptional, as she did not require invasive ventilatory support until age four months and required full-time ventilatory support from age 15 years.
Neurologic complications include small foramen magnum with brain stem compression, brain malformations (predominantly involving the temporal lobe), hydrocephalus, seizures, and profound developmental impairment in rare long-term survivors.
One infant reported at age 11 months required suboccipital decompression due to clonus and decreased limb movements secondary to a narrow foramen magnum 5. One individual reported at age 28 years underwent surgical decompression of a small foramen magnum and insertion of a ventriculoperitoneal shunt 4. Despite this intervention, the individual developed cervical spinal cord compression and quadriplegia.
Temporal lobe malformations and megalencephaly are likely universal 6. Temporal lobe abnormalities include enlargement, abnormal gyration and sulcation, polymicrogyria, and hippocampal abnormalities. Hydrocephalus is also common 7.
Brain malformations are the most likely etiology of seizures in individuals with thanatophoric dysplasia; however, additional complications such as hypoxia related to respiratory insufficiency may also play a role.
Severe developmental delay is reported, with a stall in developmental progress at a developmental age of 12-20 months. Motor skills may be more significantly impaired due to the skeletal features and micromelia. Later deterioration in abilities following complications such as cord compression is described 4.
Less common neurologic findings include hypoplasia or agenesis of the corpus callosum. Encephalocele has been reported, likely as a secondary consequence of raised intracranial pressure and abnormal skull formation 8.
Craniofacial. Relative macrocephaly is present at birth. Craniosynostosis with cloverleaf skull in individuals with thanatophoric dysplasia type 2 contributes to hydrocephalus and neurologic complications. Dysmorphic facial features including frontal bossing, flat facies, depressed nasal bridge, and ocular proptosis are present.
Musculoskeletal complications in long-term survivors include kyphosis, osteopenia, and both joint hypermobility and joint contractures.
Growth deficiency. Prenatal short-limb short stature is present in all individuals. Growth deficiency persists with micromelia and redundant skin folds. Global growth deficiency is present in long-term survivors.
Integument. Extensive acanthosis nigricans has been reported with development of seborrheic keratoses in adult survivors 4.
Hearing impairment is reported in several long-term survivors, but the cause is not clear. The presence of midface hypoplasia in individuals with thanatophoric dysplasia type I, and recognition that FGFR3 may be implicated in inner ear development 9, suggest that hearing loss in individuals with thanatophoric dysplasia type I may be multifactorial.
Vision. Intermittent exotropia has been reported in a long-term survivor 4.
Other rarely reported findings that do not have a proven association with thanatophoric dysplasia include:
- Cardiac defects. Truncus arteriosus, ventricular septal defect, and patent foramen ovale have been reported 10. Bradycardia has been reported in two individuals 4.
- Renal abnormalities 11. In two long-term survivors, renal calculi were reported 12.
- Protein losing enteropathy with intestinal lymphangestasia, reported in one individual 13. An infant with thanatophoric dysplasia was reported to have chylous ascites 14.
Thanatophoric dysplasia diagnosis
Most cases of thanatophoric dysplasia can be diagnosed by prenatal ultrasonography during the second or third trimester of pregnancy. However, making the conclusive diagnosis of thanatophoric dysplasia using only this imaging tool can be difficult. When thanatophoric dysplasia has been diagnosed prenatally, referral should be made to a maternal-fetal medicine specialist for assessment and management advice.
Imaging studies
Key ultrasonography findings include the following:
- Growth deficiency with limb length of less than 5% (by 20 weeks’ gestation)
- Macrocephaly
- Ventriculomegaly
- Cloverleaf-shaped skull or kleeblattschädel (indicates thanatophoric dysplasia type 2, but also seen in thanatophoric dysplasia type 1) 15
- Well-ossified skull and spine
- Platyspondyly of the vertebrae
- Narrow chest cavity with shortened ribs
- Micromelia
- Bowed femurs with metaphyseal flaring, described as having a “telephone receiver’ appearance (usually indicates thanatophoric dysplasia type 1)
- Polyhydramnios
Postnatal radiography and other imaging studies (CT, MRI) may reveal the following:
- Enlarged skull and a small foramen magnum with potential evidence of brain stem compression
- Central nervous system (CNS) abnormalities such as hydrocephalus, brainstem hypoplasia, temporal lobe malformations, neuronal migration abnormalities
- Flattened vertebral bodies (platyspondyly) with wide intervertebral spacing
- Rhizomelic shortening and irregular metaphyses of the long bones and “telephone receiver–shaped” bowed femurs
Laboratory studies
The following studies are indicated in suspected cases of thanatophoric dysplasia:
- Chromosome analysis (karyotype) to determine the presence or absence of chromosomal abnormalities.
- DNA molecular testing for FGFR3 using targeted and sequence mutation analyses.
A study published in 2015 by Chitty et al 16 suggested that next-generation sequencing is more sensitive than polymerase chain reaction assay and restriction enzyme digest (PCR-RED) in the prenatal diagnostic screening for monogenic disorders, including thanatophoric dysplasia and achondroplasia. In the study, which used cell-free DNA from maternal blood, next-generation sequencing and PCR-RED were performed to aid in the prenatal diagnosis of thanatophoric dysplasia and achondroplasia, with next-generation sequencing being 96.2% accurate, compared with 88.6% for PCR-RED 16.
Table 1. Recommended evaluations following initial diagnosis in individuals with thanatophoric dysplasia
| System/Concern | Evaluation | Comment |
|---|---|---|
| Respiratory |
| If tracheostomy & long-term ventilatory support is considered, consult w/respiratory physician & multidisciplinary team w/expertise in tracheostomy care. |
| Foramen magnum narrowing, hydrocephalus, &/or other central nervous system (CNS) abnormalities | Head & spine CT or MRI |
|
| Craniosynostosis | Head CT in persons w/clinical evidence of craniosynostosis | |
| Seizures | Consider assessment w/neurologist &/or EEG if episodes suspicious for seizures. | |
| Audiology | Audiologic assessment | |
| Ophthalmology | Ophthalmology eval for exotropia | |
| Developmental delay | Developmental assessment | |
| Genetic counseling | By genetics professional 1 | To inform affected persons & families re nature, mode of inheritance & implications of thanatophoric dysplasia to facilitate medical & personal decision making |
| Family support/ resources | Assess:
|
Footnote: 1. Medical geneticist, certified genetic counselor, or certified advanced genetic nurse.
Thanatophoric dysplasia treatment
Inpatient care is necessary for newborns diagnosed with thanatophoric dysplasia. If intubation is performed to treat respiratory distress, admission to a neonatal intensive care unit (NICU) is required. If treatment intervention is deferred, palliative care is essential to keep the infant warm, comfortable, and nourished.
Thanatophoric dysplasia prognosis
Newborns with thanatophoric dysplasia are stillborn or die shortly after birth. Death occurs usually within 48 hours and is due to severe respiratory insufficiency from a reduced thoracic capacity and hypoplastic lungs and/or respiratory failure due to brainstem compression. Survival into early childhood has been rarely reported 2. However, long-term survivors have been reported, including rare reports of survival to adulthood with aggressive ventilatory support and surgical management of neurologic complications 17.
- Thanatophoric dysplasia. https://ghr.nlm.nih.gov/condition/thanatophoric-dysplasia[↩][↩]
- Nikkel SM, Major N, King WJ. Growth and development in thanatophoric dysplasia – an update 25 years later. Clin Case Rep. 2013 Dec. 1 (2):75-8.[↩][↩]
- Xue Y, Sun A, Mekikian PB, Martin J, Rimoin DL, Lachman RS, Wilcox WR. FGFR3 mutation frequency in 324 cases from the International Skeletal Dysplasia Registry. Mol Genet Genomic Med. 2014;2:497–503.[↩]
- Nikkel SM, Major N, King WJ. Growth and development in thanatophoric dysplasia – an update 25 years later. Clin Case Rep. 2013;1:75–8.[↩][↩][↩][↩][↩][↩]
- Thompson DR, Browd SR, Sangaré Y, Rowell JC, Slimp JC, Haberkern CM. Anesthetic management of an infant with thanatophoric dysplasia for suboccipital decompression. Paediatr Anaesth. 2011;21:92–4.[↩]
- Itoh K, Pooh R, Kanemura Y, Yamasaki M, Fushiki S. Brain malformation with loss of normal FGFR3 expression in thanatophoric dysplasia type I. Neuropathology. 2013;33:663–6.[↩]
- Hevner RF. The cerebral cortex malformation in thanatophoric dysplasia: neuropathology and pathogenesis. Acta Neuropathol. 2005;110:208–21.[↩]
- Martínez-Frías ML, Egüés X, Puras A, Hualde J, de Frutos CA, Bermejo E, Nieto MA, Martínez S. Thanatophoric dysplasia type II with encephalocele and semilobar holoprosencephaly: Insights into its pathogenesis. Am J Med Genet A. 2011;155A:197–202.[↩]
- Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–7.[↩]
- McBrien A, Sands A, Paterson A, Tharmaratnam S, Thornton C. Common arterial trunk with thanatophoric dysplasia: a unique case. Fetal Pediatr Pathol. 2008;27:259–63.[↩]
- Prontera P, Sensi A, Pilu G, Baldi M, Baffico M, Bonasoni R, Calzolari E. FGFR3 mutation in thanatophoric dysplasia type 1 with bilateral cystic renal dysplasia: coincidence or a new association? Genet Couns. 2006;17:407–12.[↩]
- Kuno T, Fujita I, Miyazaki S, Katsumata N. Markers for bone metabolism in a long-lived case of thanatophoric dysplasia. Endocr J. 2000;47 Suppl:S141–4.[↩]
- Yang C, Dehner LP. Protein-losing enteropathy with intestinal lymphangiectasia in skeletal dysplasia with Lys650Met mutation. Am J Med Genet A. 2016;170:2993–7.[↩]
- Soo-Kyeong J, Lee N, Bae MH, Han YM, Park KH, Byun SY. Chylous ascites in an infant with thanatophoric dysplasia type I with FGFR3 mutation surviving five months. Fetal Pediatr Pathol. 2018;37:363–71.[↩]
- Cohen MM Jr. Cloverleaf skulls: etiologic heterogeneity and pathogenetic variability. J Craniofac Surg. 2009 Mar. 20 Suppl 1:652-6.[↩]
- Chitty LS, Mason S, Barrett AN, et al. Non-invasive prenatal diagnosis of achondroplasia and thanatophoric dysplasia: next-generation sequencing allows for a safer, more accurate, and comprehensive approach. Prenat Diagn. 2015 Jul. 35 (7):656-62.[↩][↩]
- French T, Savarirayan R. Thanatophoric Dysplasia. 2004 May 21 [Updated 2020 Jun 18]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1366[↩]




{kind=link}