Transmissible spongiform encephalopathies
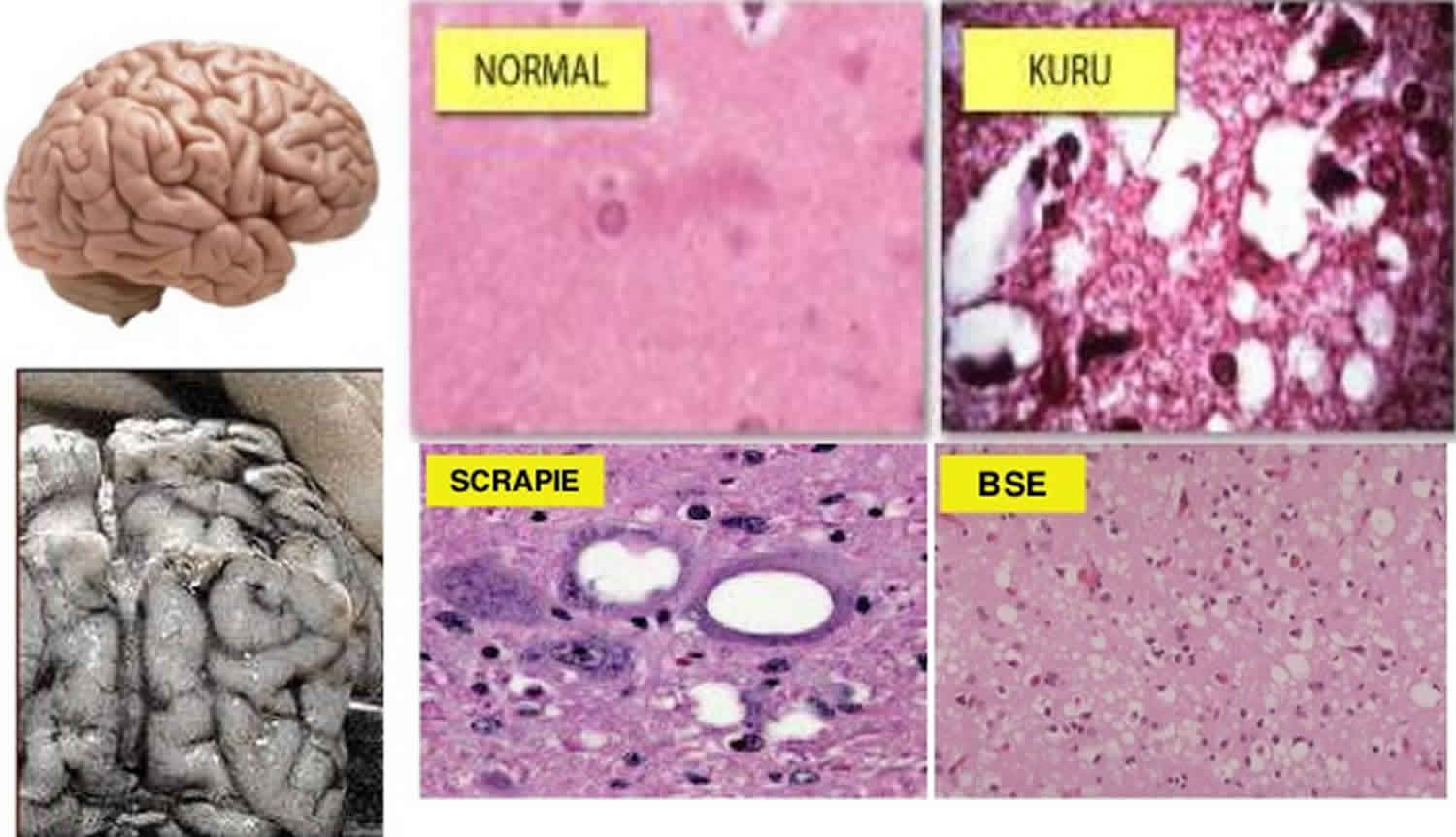
Transmissible spongiform encephalopathies also known as prion diseases, are a group of rare degenerative brain disorders that affect both humans and animals, that are characterized by tiny holes that give the brain a “spongy” appearance 1. These holes can be seen when brain tissue is viewed under a microscope. Research suggests that transmissible spongiform encephalopathies are caused by an abnormal version of a protein called a prion (prion is short for proteinaceous infectious particle). Prion proteins occur in both a normal form, which is a harmless protein found in the body’s cells, and in an infectious form, which causes disease. The harmless and infectious forms of the prion protein are nearly identical, but the infectious form takes on a different folded shape from the normal protein. Although these infections usually remain asymptomatic for years, the disease is always progressive and fatal once the clinical signs develop.
Human transmissible spongiform encephalopathies can occur three ways: sporadically; as hereditary diseases; or through transmission from infected individuals. Sporadic transmissible spongiform encephalopathies may develop because some of a person’s normal prions spontaneously change into the infectious form of the protein and then alter the prions in other cells in a chain reaction. Inherited cases arise from a change, or mutation, in the prion protein gene that causes the prions to be shaped in an abnormal way. This genetic change may be transmitted to an individual’s offspring. Transmission of transmissible spongiform encephalopathies from infected individuals is relatively rare. Transmissible spongiform encephalopathies cannot be transmitted through the air or through touching or most other forms of casual contact. However, they may be transmitted through contact with infected tissue, body fluids, or contaminated medical instruments. Normal sterilization procedures such as boiling or irradiating materials do not prevent transmission of transmissible spongiform encephalopathies.
Symptoms of transmissible spongiform encephalopathies vary, but they commonly include personality changes, psychiatric problems such as depression, lack of coordination, and/or an unsteady gait. Patients also may experience involuntary jerking movements called myoclonus, unusual sensations, insomnia, confusion, or memory problems. In the later stages of the disease, patients have severe mental impairment and lose the ability to move or speak.
Creutzfeldt-Jakob disease (CJD) is the most well-known of the human transmissible spongiform encephalopathies. Creutzfeldt-Jakob disease (CJD) is a rare type of dementia that affects about one in every one million people each year. CJD has several subcategories: Sporadic (sCJD, accounting for 80–90% of CJD cases); Familial (hereditary CJD); Iatrogenic (associated with treatment and transplant, such as use of infected dura mater patches or administration of human pituitary-derived hormones) 2. A new type of Creutzfeldt-Jakob disease, called variant Creutzfeldt-Jakob disease (vCJD), was first described in 1996 and has been found in Great Britain and several other European countries. The initial symptoms of variant Creutzfeldt-Jakob disease (vCJD) are different from those of classic Creutzfeldt-Jakob disease and the disorder typically occurs in younger patients. Research suggests that variant Creutzfeldt-Jakob disease (vCJD) may have resulted from human consumption of beef from cattle with a transmissible spongiform encephalopathy disease called bovine spongiform encephalopathy (BSE), also known as “mad cow disease.”
Other human transmissible spongiform encephalopathies include kuru, fatal familial insomnia (FFI) and Gerstmann-Straussler-Scheinker disease (GSS) 2. Kuru was identified in people of an isolated tribe in Papua New Guinea and has now almost disappeared. Fatal familial insomnia and Gerstmann-Straussler-Scheinker disease are extremely rare hereditary diseases, found in just a few families around the world.
Although some prion diseases usually occur in one or a few closely related species, other prions can cross species barriers. Bovine spongiform encephalopathy (mad cow disease) has a particularly wide host range. Cattle are the primary hosts for this disease, but some other ruminants, cats, lemurs and humans can also be affected; in cats, the disease is known as feline spongiform encephalopathy (FSE) and in humans, it is called variant Creutzfeldt-Jakob disease (vCJD). Some evidence also suggests that transmissible mink encephalopathy (mink scrapie) might be a caused by an atypical variant of the bovine spongiform encephalopathy (BSE) agent called L-BSE. The discovery that bovine spongiform encephalopathy (mad cow disease) can cross species barriers and is zoonotic has raised concerns about all transmissible spongiform encephalopathies. Prion diseases in livestock can also result in trade sanctions. For these reasons, many countries are conducting control or eradication programs.
Other transmissible spongiform encephalopathies found in animals include feline spongiform encephalopathy (FSE); scrapie (tremblante de mouton, rida), which affects sheep and goats; chronic wasting disease (CWD), which affects elk and deer; and transmissible mink encephalopathy (mink scrapie). In a few rare cases, transmissible spongiform encephalopathies have occurred in other mammals such as zoo animals. These cases are probably caused by contaminated feed. Creutzfeldt-Jakob disease (CJD) and other transmissible spongiform encephalopathies also can be transmitted experimentally to mice and other animals in the laboratory.
Transmissible spongiform encephalopathies tend to progress rapidly and usually culminate in death over the course of a few months to a few years.
Transmissible spongiform encephalopathies causes
Transmissible spongiform encephalopathies are caused by prions (proteinaceous infectious particle), infectious proteins that appear to replicate by converting a normal cellular protein into copies of the prion 3. The cellular protein, which is called PrPc, is found on the surface of neurons. Pathogenic isoforms of PrPc are designated PrPres (‘res’ refers to the proteinase K-resistant nature of prions, compared to normal PrPc). Alternative names for this protein are PrPTSE, PrPSc (Sc stands for scrapie, the first animal prion disease to be recognized) and PrPd (for disease-associated). Different prions or more accurately different strains of PrPres cause scrapie, bovine spongiform encephalopathy (BSE) and chronic wasting disease (CWD). Bovine spongiform encephalopathy (BSE) in felids is called feline spongiform encephalopathy (FSE), and BSE in zoo ruminants was formerly called ‘spongiform encephalopathy of exotic ruminants’. There are both classical and atypical BSE and scrapie prions. Atypical prions were first recognized during the last 20 years, as the result of enhanced surveillance for transmissible spongiform encephalopathies. The term ‘classical’ is used to distinguish the usual BSE and scrapie strains from these newly recognized forms. There are at least two atypical BSE prions. One has higher molecular mass fragments than classical BSE in an immunoblot and is called H-type BSE or H-BSE; the other has a lower molecular mass and is called L-type BSE or L-BSE. Currently, the most likely hypothesis is that L-BSE and H-BSE arise spontaneously in cattle, as genetic diseases. One of them may have given rise to the classical BSE epidemic after amplification through the food chain. Atypical/ Nor98 scrapie might, similarly, be a spontaneously occurring prion disease of small ruminants; however, this is not yet certain. Which prion causes transmissible mink encephalopathy (TME) is still uncertain; however, it is expected to be an agent that occurs in meat fed to ranched mink. Current evidence suggests that it may be atypical L-BSE, although other possibilities, such as certain strains of the scrapie prion, have not been entirely ruled out.
Prions can persist for long periods in the environment. Infectious scrapie and chronic wasting disease (CWD) prions have been found in soil for 1.5 to 3 years or more in laboratory experiments. Evidence from Iceland, where scrapie-affected premises are not restocked for at least 2 years after depopulation and decontamination, suggests that this prion can infect animals up to 16 years later, under some conditions. Soil-bound prions remain infectious for animals. Repeated cycles of wetting and drying are reported to decrease, though not necessarily eliminate, soil infectivity. Prions can also remain infectious after passage through the digestive tracts of mammals and birds that eat infected animals.
Transmissible spongiform encephalopathies transmission
Transmissible spongiform encephalopathies are thought to be acquired primarily by ingestion, although some prions may also enter the body by other routes. Prions occur mainly in the central nervous system (CNS) of TSE-infected animals, but they can also be found in lymphoid tissues, peripheral nerves and various organs, to a greater or lesser extent depending on the prion and host species. Animals carry prions for life, and can transmit them even if they remain asymptomatic. Prions have also been transmitted iatrogenically in some species (e.g., via contaminated surgical instruments or in blood transfusions).
Bovine spongiform encephalopathy (BSE), feline spongiform encephalopathy (FSE) and transmissible mink encephalopathy (TME) do not seem to be transmitted horizontally between animals. Animals including cats with feline spongiform encephalopathy (FSE) become infected with classical bovine spongiform encephalopathy (BSE) prions when they eat contaminated tissues from infected animals. Bovine spongiform encephalopathy (BSE) prions caused epidemics in cattle when they were amplified by recycling tissues from infected cattle into ruminant feed supplements. Young animals appear to be infected more readily than cattle over the age of 6 months. Vertical transmission has been reported in experimentally infected sheep, but it has not been documented in cattle. However, the offspring of bovine spongiform encephalopathy (BSE)-infected cows seem to have an elevated risk of developing bovine spongiform encephalopathy (BSE), for reasons that are still unclear. There is one report of possible maternal transmission of feline spongiform encephalopathy (FSE) in a cheetah. Transmissible mink encephalopathy (TME) is, likewise, thought to spread primarily by ingestion, probably when mink are fed tissues from infected animals; however, some evidence suggests that this prion might also enter the body through breaks in the mucous membranes or skin. Vertical transmission does not seem to occur in mink.
Scrapie and chronic wasting disease (CWD) can be transmitted directly between animals, or indirectly via the environment. Chronic wasting disease (CWD) prions have been found, though sometimes at very low levels, in the saliva, blood, urine, feces and antler velvet of cervids. Most animals are thought to acquire this disease from the environment, probably during grazing. Vertical transmission has been documented in some cervid species. Most small ruminants seem to acquire scrapie from their dam during the neonatal period, although they can also become infected from unrelated animals or the environment. Older animals remain susceptible, though to a lesser extent. The placenta can contain scrapie prions, with particularly large amounts in sheep and newborns may be infected by licking fetal membranes and fluids. The milk and colostrum of small ruminants are also infectious. In utero infection appears possible in sheep, though rare. Highly sensitive techniques have found low levels of scrapie prions in the urine, saliva and feces of sheep, but how much these sources contribute to transmission is still uncertain. The epidemiology of atypical or Nor98 scrapie suggests that it is not transmitted efficiently (or perhaps at all) between animals in nature, although laboratory experiments have demonstrated that newborn lambs are susceptible to oral inoculation.
Transmissible spongiform encephalopathies have incubation periods of months or years. Transmissible mink encephalopathy (TME) has the shortest estimated incubation period, at 6-12 months, while scrapie, bovine spongiform encephalopathy (BSE), feline spongiform encephalopathy (FSE) and chronic wasting disease (CWD) usually become apparent after 2 years or more in their natural hosts. Clinical cases can occur before this time, but are uncommon.
Zoonotic transmission
In humans, variant Creutzfeldt-Jakob disease (vCJD) usually results from the ingestion of BSE prions, but a few people have been infected in blood transfusions from asymptomatic individuals. Other potential routes of infection include organ/tissue transplantation and surgical transmission via contaminated equipment. Prions do not spread between people during casual contact.
Transmissible spongiform encephalopathies prevention
Bovine spongiform encephalopathy (BSE), feline spongiform encephalopathy (FSE) and transmissible mink encephalopathy (TME) can be prevented by not feeding tissues that may contain prions to susceptible species. Complete avoidance is necessary, as cooking or rendering cannot completely inactivate these agents. Banningruminant (or mammalian) tissues from ruminant (or animal) feed has significantly reduced the number of new cases of BSE and controlled the epidemics in cattle.The specific bans, and protein sources prohibited, vary with the country. Surveillance and tracing of infected animals can reveal cohorts that may have received the same feed. Countries may place trade bans on the importation of live animals and certain animal proteins from TSE-affected nations.
Diseases that can spread horizontally, such as scrapie and chronic wasting disease (CWD), require additional methods of control. The risk of introducing these diseases can be reduced by maintaining a closed herd/ flock or minimizing outside purchases of stock. If replacement animals must be added, they should be from herds that test negative for the disease and are managed in a way that makes them unlikely to become infected. Milk and colostrum from potentially infected sheep or goats should not be fed to scrapie-free flocks. Reducing exposure to high concentrations of prions (e.g., in the placenta of sheep) may reduce transmission within a herd or flock that has become infected. The risk that a sheep will develop scrapie is strongly influenced by its genotype, and selecting genetically resistant animals can aid control efforts. Whether genetic control methods are feasible in scrapie-infected goat herds or chronic wasting disease (CWD)-infected cervid herds is still uncertain. Complete depopulation, followed by cleaning and disinfection, is sometimes used on prion-infected farms; however, decontamination may bedifficult and the disease may recur. Some countries have developed voluntary and/or mandatory programs to control chronic wasting disease (CWD) and scrapie, with eradication from domestic herds/ flocks as the ultimate goal.
Controlling chronic wasting disease (CWD) in wild cervids is very difficult. Infected captive cervids should be kept from contact with wild cervids, to avoid transmitting this disease to uninfected areas. Many states and provinces also have restrictions on transporting tissues from hunter-killed cervids in chronic wasting disease (CWD)-endemic areas. Attempts to reduce transmission in wild populations include banning practices that encourage cervids to congregate in certain areas (e.g., feeding or baiting),and culling to reduce animal densities. Other transmissible spongiform encephalopathies are not known to be an issue in wild animals.
There are no established control methods for L-BSE, H-BSE or atypical/ Nor98 scrapie, which could be spontaneous genetic diseases. These prions do not currently affect a nation’s BSE or scrapie status for international trade.
Transmissible spongiform encephalopathies disinfection
Complete decontamination of prion-contaminated tissues, surfaces and environments can be difficult. These agents are very resistant to most disinfectants, including formalin and alcohol. They are also resistant to heat, or ultraviolet, microwave and ionizing radiation, particularly when they are protected in organic material or preserved with aldehyde fixatives, or when the prion titer is high. Prions can bind tightly to some surfaces, including stainless steel and plastic, without losing infectivity. Prions bound to metal seem to be highly resistant to decontamination.
Relatively few prion decontamination techniques have been published and confirmed to be effective for routine use. Some laboratories pre-treat tissues with formic acid to decrease infectivity before sectioning tissue blocks. A 1-2 N sodium hydroxide solution, or a sodium hypochlorite solution containing at least 2% (20,000 ppm) available chlorine, has traditionally been recommended for equipment and surfaces. Surfaces should be treated for more than 1 hour at 20°C (68°F). Overnight disinfection is recommended for equipment. Cleaning before disinfection removes organic material that may protect prions. Experimentally, some milder treatments have also been effective against certain prions, under some conditions. They include a specific phenolic disinfectant, various alkaline and enzymatic detergents (although the efficacy of specific agents within these classes varies), hydrogen peroxide gas plasma, radiofrequency gas plasma, and sodium dodecyl sulfate plus acetic acid. These agents might be useful for items that cannot withstand harsher decontamination procedures.
Physical inactivation of prions can be carried out by porous load autoclaving at 134°C (273°F) for 18 minutes at 30 lb/in². Resistance to heat may vary with the specific prion, the degree of contamination and type of sample. Tissue films containing prions are more difficult to decontaminate by steam after they have dried, and human guidelines for surgical instruments recommend that, after use, they be kept moist or wet until decontamination is performed. The cleaning agent used before autoclaving should also be chosen with care, as certain agents (e.g., some enzymatic treatments) can increase the resistance of prions to steam sterilization.Dry heat is less effective than moist heat; some prions can survive dry heat at temperatures as high as 360°C (680°F) for an hour, and one group even reported that infectivity survived incineration at 600°C (1112°F). A combination of chemical and physical decontamination can be more effective than either procedure alone, and effective combinations of chemical agents (e.g., NaOH) and autoclaving have been published. It should be noted that even the harshest combination of chemical and physical disinfection is not guaranteed to destroy all prions in all types of samples.
Decontaminating contaminated facilities, especially sites such as animal pens, may be very difficult. In one study, genetically susceptible sheep became infected with scrapie after being placed in pens that had been pressure washed and decontaminated with high concentrations of sodium hypochlorite (20,000 ppm free chorine solution) for one hour, followed by painting and full re-galvanization or replacement of metalwork. Decontaminating soil contaminated with prions is currently impractical, although some agents, including an aqueous subtilisin-based enzymatic treatment (effective at ambient temperatures), appear promising in the laboratory. Incineration is commonly used for carcasses, but two studies found that composting may reduce or eliminate scrapie and other prions in tissues, while another suggested that soil microorganisms might degrade prions in buried carcasses.
Transmissible spongiform encephalopathies symptoms
Bovine spongiform encephalopathy (mad cow disease)
Bovine spongiform encephalopathy (BSE or mad cow disease) is the only animal transmissible spongiform encephalopathy (TSE) known to affect humans; people who have eaten BSE prions may develop variant Creutzfeldt-Jakob disease (vCJD). The majority of cases have occurred in the United Kingdom and France 4. As of May 2016, 228 cases of variant Creutzfeldt-Jakob disease (vCJD) have been reported worldwide. With the exception of 27 cases in France, most of these people had resided in the U.K. for more than 6 months during the peak of the BSE epidemic, and are likely to have been infected there. In the U.K., the incidence of variant Creutzfeldt-Jakob disease (vCJD) peaked in 2000, when 28 cases were diagnosed,and gradually fell to 2-5 cases per year between 2006 and 2011. Only two additional cases were recognized between 2012 and 2016. Since variant Creutzfeldt-Jakob disease (vCJD) was first reported in 1996, a total of only 231 patients with this disease, including 3 secondary, blood transfusion-related cases, have been reported worldwide 5. The risk to human health from BSE in the United States is extremely low. The number of people who have been infected asymptomatically, and the percentage of those likely to develop vCJD, is still unclear. Based on the decreasing number of clinical cases, some sources suggest that few additional cases may be seen. However, some studies that have examined lymphoid tissues, such as the tonsils or appendix, suggest that from 1 in 2000 to 1 in 10,000 people in the U.K. may be infected subclinically with BSE prions. Whether these people will ever develop vCJD is not known.
The symptoms of variant Creutzfeldt-Jakob disease (vCJD) are broadly similar to the sporadic (genetic) form of Creutzfeldt-Jakob disease, but usually appear in younger patients. The median age of onset is 26 years, although cases have been reported in people from 12 to 74 years, with a median duration of illness between 13 to 14 months and the median age at death is 28 years 4. The first signs are usually psychiatric symptoms such as anxiety, depression and social withdrawal, and/or persistent painful sensory symptoms. In most patients, frank neurological signs (e.g., ataxia, tremors) appear a few months later; however, neurological signs coincide with or precede psychiatric symptoms in a minority of patients. Cognitive function gradually deteriorates, and involuntary movements and visual disturbances may develop late in the course of the disease. There is no known treatment, and most patients die within 2 years. To date, all individuals with confirmed clinical cases have been homozygous for methionine at codon 129 in the PrPC protein.
Prevention of variant Creutzfeldt-Jakob disease (vCJD) is based on the avoidance of high-risk tissues from cattle. Many countries exclude animals with BSE-like clinical signs, as well as high risk tissues from asymptomatic cattle (specified risk materials or SRM), from human food. Specified risk materials generally include the brain, spinal cord, associated bones and some nerve ganglia; tonsils; and various portions of the intestinal tract (e.g., currently, the distal ileum in the U.S., and the last 4 meters of the small intestine, the cecum and mesentery in the E.U.). Some nations conduct active surveillance of cattle at slaughter,using rapid tests, to detect cases of BSE. Positive carcasses are destroyed. Slaughter and processing techniques that have a high risk of contaminating muscle tissues with central nervous system (brain and spinal cord) have also been prohibited in many nations. Many countries restrict blood donations from people with a significant risk of having been infected during the BSE epidemics. Some may also take other measures to prevent transmission in blood transfusions or during high-risk surgical procedures. Although laboratory or abattoir-related cases have not been reported, veterinarians and laboratory workers should always take precautions when conducting necropsies on BSE-suspects or handling tissues; BSL-3 is the recommended level of protection for handling these prions.
Chronic wasting disease
Although no human infections with chronic wasting disease (CWD) have ever been reported, the possibility that this disease could be zoonotic has not been ruled out. Hunters should consider having carcasses tested for chronic wasting disease (CWD); information on this program is available from most state wildlife agenciesin the U.S. Meat from cervids that appear to be ill, as well as meat from apparently healthy animals that test positive for chronic wasting disease (CWD), should not be eaten or fed to any animals. Gloves should be worn when field-dressing a cervid carcass. Boning-out the meat and minimizing the handling of the brain, spinal cord and lymphoid tissues associated with the gastrointestinal tract (e.g. tonsils) from cervids may reduce the risk of exposure, but will not necessarily remove all prions.
Transmissible spongiform encephalopathies diagnosis
Transmissible spongiform encephalopathies diagnosis is mainly through history and physical examination. The characteristic progression of symptoms in the vulnerable populations leads to high suspicion of the diagnosis. However, Creutzfeldt-Jakob disease is often a diagnostic challenge for physicians as it presents similarly to rapidly progressing dementias 6.
Recommended initial screening tests for evaluation of a rapidly progressive dementia are complete blood count (CBC), basic metabolic panel including magnesium level, liver function tests, rapid plasma reagin, erythrocyte sedimentation rate, antinuclear antibody, C-reactive protein, thyroid function tests, vitamin B-12, HIV, Lyme disease titer, autoimmune antibodies, urinalysis, cerebrospinal (CSF) studies including glucose, oligoclonal bands, cell count and differential, VDRL, MRI brain (including FLAIR and DWI) with and without contrast, and electroencephalogram (EEG). With summation of clinical presentation and supportive diagnostic studies, Creutzfeldt-Jakob disease can be diagnosed.
In 1998, the WHO published diagnostic criteria for Creutzfeldt-Jakob disease (CJD) with the diagnosis relying on clinical examination, electroencephalogram (EEG) and cerebrospinal fluid (CSF) findings. However, due to advances in medicine with newer testing like MRI, genetic testing, and other modern laboratory tests, the diagnostic criteria perhaps need to be updated.
Several tests can help diagnose Creutzfeldt-Jakob disease (CJD) including MRI brain, CSF based tests, and EEG.
For variant Creutzfeldt-Jakob disease (vCJD) MRI brain is a more sensitive and specific test than CSF 14–3–3 protein and found to be accurate in about 90% of cases. Brain MRI with T2-weighted, diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) sequences often reveal abnormalities in the cortical gray matter (cortical ribboning) and deep nuclei in sporadic Creutzfeldt-Jakob disease (sCJD). MRI with DWI/FLAIR imaging has a sensitivity of 98% and specificity of 93%. DWI typically demonstrates hyperintensities within the basal ganglia, thalamus, and cortex. The “hockey stick” or “pulvinar” sign is indicative of variant (infectious acquired) CJD (vCJD), but also seen in other forms of CJD. For Creutzfeldt-Jakob disease (CJD), CSF 14-3-3 is more sensitive followed by typical EEG findings.
CSF protein biomarkers including 14–3–3 protein, total tau (t-tau) and neuron-specific enolase (NSE) are markers of rapid neurodegeneration, so they assist in CJD diagnosis, but these are not CJD specific. Routine CSF studies including glucose, total protein, white blood cell count, total cell count, and oligoclonal IgG are generally unremarkable in CJD patients. In 2012, the American Academy of Neurology recommended ordering CSF 14-3-3 only when CJD is strongly suspected. A recent comparison of these three non-prion-specific CSF biomarkers and MRI found that DWI MRI had a higher diagnostic accuracy of 97%, more than any or all of these three CSF biomarkers like t-tau (79.6%) or 14-3-3 protein (70.4%) or neuron-specific enolase (NSE) (71.4%). Detection of these traditional surrogate marker proteins is accurate in approximately three-fourths of cases.
The National Prion Disease Pathology Surveillance Center in April 2015 developed a new diagnostic test called second-generation Real Time-Quaking-Induced Conversion (RT-QuIC) which is very sensitive and specific for CJD. RT-QuIC can detect pathogenic prion protein in the cerebrospinal fluid of CJD patients with high accuracy. RT-QuIC directly detects the pathogenic prion protein, whereas the existing indirect markers of rapid neurodegeneration like protein 14-3-3 and tau proteins cannot do this.
A few studies have demonstrated modest sensitivity (>80%) but high specificity (approximately 98%) of CSF RT-QuIC for sporadic Creutzfeldt-Jakob disease (sCJD). Although it does not have as high sensitivity as MRI, it is often positive in many forms of genetic prion disease, some of which usually do not show the classic MRI findings identified in most sporadic CJD cases. RT-QuIC in CSF is more specific than protein 14-3-3, probably NSE and t-tau also. RT-QuIC appears to be a highly specific test for human prion disease and might be more sensitive using olfactory epithelium (from nasal brushings) than CSF. Recent studies have shown that the RT-QuIC technique is the most sensitive and specific diagnostic test that can replace brain biopsy for accurate diagnosis of CJD. Because Real Time-Quaking-Induced Conversion (RT-QuIC) is less invasive compared to brain biopsy, this should be the first test to do in the workup of a patient with suspected CJD. However, the existing CJD guidelines do not include newer less invasive diagnostic modalities, and probably need to be updated.
EEG is the least sensitive test compared to MRI brain or CSF studies, and typical periodic sharp wave complexes can be seen.
Brain tissue biopsy or postmortem exam of the brain confirms the diagnosis of CJD. However not all areas in the brain are affected by the disease, so neurosurgeons target the areas that are abnormal on imaging studies which are most often in deep-seated subcortical structures. The surgery can be risky, and it may not always obtain the affected brain tissue. As the confirmatory diagnosis of CJD does not change the clinical outcome of the patient, a brain biopsy is only indicated when a reversible condition is suspected in the differential.
Few societies and organizations including CDC have proposed updated diagnostic criteria for CJD.
Transmissible spongiform encephalopathies treatment
There is currently no treatment that can halt progression of any of the transmissible spongiform encephalopathies. The mainstay of treatment is symptomatic and supportive care and making the patient as comfortable as possible. A few drug trials were done on Creutzfeldt-Jakob disease (CJD), but none of them have shown any clear benefit so far. More research is needed to find the treatment for this fatal condition.
Transmissible spongiform encephalopathies prognosis
Creutzfeldt-Jakob disease (CJD) is a degenerative neurological disorder that is incurable and invariably fatal. It is the most common among the types of transmissible spongiform encephalopathy found in humans. Despite all the advances that have helped in understanding this disease, the prognosis is extremely poor. Death often occurs within 1 year of symptom onset 4.
- Transmissible Spongiform Encephalopathies Information Page. https://www.ninds.nih.gov/Disorders/All-Disorders/Transmissible-Spongiform-Encephalopathies-Information-Page[↩]
- Dudhatra G. B., Avinash Kumar, Modi C. M., Awale M. M., Patel H. B., Mody S. K., “Transmissible Spongiform Encephalopathies Affecting Humans”, International Scholarly Research Notices, vol. 2013, Article ID 387925, 11 pages, 2013. https://doi.org/10.5402/2013/387925[↩][↩]
- Transmissible Spongiform Encephalopathiesof Animals. http://www.cfsph.iastate.edu/Factsheets/pdfs/transmissible_spongiform_encephalopathy.pdf[↩]
- Sitammagari KK, Masood W. Creutzfeldt Jakob Disease. [Updated 2020 Apr 5]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2020 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK507860[↩][↩][↩]
- BSE in North America. https://www.cdc.gov/prions/bse/bse-north-america.html[↩]
- Rudge P, Jaunmuktane Z, Hyare H, Ellis M, Koltzenburg M, Collinge J, Brandner S, Mead S. Early neurophysiological biomarkers and spinal cord pathology in inherited prion disease. Brain. 2019 Mar 01;142(3):760-770.[↩]





{kind=link}