
What is tyrosinemia
Tyrosinemia also called hereditary tyrosinemia, refers to a group of genetic disorders characterized by disruptions in the multistep process that breaks down the amino acid tyrosine, a building block of most proteins. If untreated, tyrosine and its byproducts build up in tissues and organs, which can lead to serious health problems.
There are four types of tyrosinemia, which are each distinguished by their symptoms and genetic cause. Tyrosinemia type 1, the most severe form of tyrosinemia, is characterized by signs and symptoms that begin in the first few months of life. Affected infants fail to gain weight and grow at the expected rate (failure to thrive) due to poor food tolerance because high-protein foods lead to diarrhea and vomiting. Affected infants may also have yellowing of the skin and whites of the eyes (jaundice), a cabbage-like odor, and an increased tendency to bleed (particularly nosebleeds). Tyrosinemia type 1 can lead to liver and kidney failure, softening and weakening of the bones (rickets), and an increased risk of liver cancer (hepatocellular carcinoma). Some affected children have repeated neurologic crises that consist of changes in mental state, reduced sensation in the arms and legs (peripheral neuropathy), abdominal pain, and respiratory failure. These crises can last from 1 to 7 days. Untreated, children with tyrosinemia type 1 often do not survive past the age of 10. Worldwide, tyrosinemia type 1 affects about 1 in 100,000 individuals. Tyrosinemia type 1 is more common in Norway where 1 in 60,000 to 74,000 individuals are affected. Tyrosinemia type 1 is even more common in Quebec, Canada where it occurs in about 1 in 16,000 individuals. In the Saguenay-Lac St. Jean region of Quebec, tyrosinemia type 1 affects 1 in 1,846 people.
Tyrosinemia type 2 occurs in fewer than 1 in 250,000 individuals worldwide. Tyrosinemia type 2 can affect the eyes, skin, and mental development. Signs and symptoms often begin in early childhood and include eye pain and redness, excessive tearing, abnormal sensitivity to light (photophobia), and thick, painful skin on the palms of their hands and soles of their feet (palmoplantar hyperkeratosis). About 50 percent of individuals with tyrosinemia type 2 have some degree of intellectual disability.
Tyrosinemia type 3 is the rarest of the three types; only a few cases have been reported. The characteristic features of tyrosinemia type 3 include intellectual disability, seizures, and periodic loss of balance and coordination (intermittent ataxia).
About 10 percent of newborns have temporarily elevated levels of tyrosine (transient tyrosinemia). In these cases, the cause is not genetic. The most likely causes are vitamin C deficiency or immature liver enzymes due to premature birth.
My son is 18 days old and has been diagnosed with tyrosinemia type 1. Is there any hope of curing my son’s illness?
There is currently no cure for tyrosinemia type 1. Individuals with this condition need to be on a special diet restricted in two amino acids, tyrosine and phenylalanine, throughout life. Affected individuals may also be treated with a medication called nitisinone. Treatment should start as soon as the condition is diagnosed. Some individuals require a liver transplant if their liver disease is already advanced before treatment begins 1.
Types of Tyrosinemia
Tyrosinemia refers to a group of genetic disorders that prevent the breakdown of the amino acid tyrosine.
There are four types of tyrosinemia:
- Transient (temporary) Tyrosinemia
- Tyrosinemia Type 1
- Tyrosinemia Type 2
- Tyrosinemia Type 3
Transient tyrosinemia
Transient tyrosinemia of the newborn is a benign disorder of tyrosine metabolism detected upon newborn screening and often observed in premature infants 2. Transient tyrosinemia shows no clinical symptoms. Transient tyrosinemia is characterized by tyrosinemia, moderate hyperphenylalaninemia, and tyrosiluria that usually resolve after 2 months of age.
Tyrosinemia type 1
Tyrosinemia type 1 also called hepatorenal tyrosinemia or fumarylacetoacetate hydrolase (FAH) deficiency, is the most severe form of tyrosinemia, resulting in the accumulation of tyrosine and its metabolites in the liver causing severe liver disease. Kidney function and peripheral nerves also are affected. Patients with Type I may have acute liver crisis, episodes of peripheral neuropathy and chronic liver disease. Effects on the kidneys can range from mild tubular dysfunction to renal failure. Early symptoms can include fever, diarrhea, vomiting, enlarged liver, jaundice, rickets, lethargy and irritability.
Tyrosinemia type 1 is caused by a shortage of the enzyme fumarylacetoacetate hydrolase (FAH), one of the enzymes required for the multi-step process that breaks down tyrosine. This enzyme shortage is caused by mutations in the FAH gene. Tyrosinemia type 1 is inherited in an autosomal recessive manner 3. Fumarylacetoacetase (FAH) is the terminal enzyme in the tyrosine catabolic pathway. In FAH deficiency, fumarylacetoacetate (FAA), the immediate precursor:
- Appears to accumulate in hepatocytes, causing cellular damage and apoptosis as identified in an animal model by Endo and Sun 4;
- Is diverted into succinylacetoacetate and succinylacetone. Succinylacetone interferes with the activity of the following hepatic enzymes:
- Parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), resulting in elevation of plasma tyrosine concentration;
- PBG synthase, resulting in (1) reduced activity of the enzyme δ-ALA dehydratase in liver and circulating red blood cells; (2) reduced heme synthesis; (3) increased δ-aminolevulinic acid (δ-ALA), which may induce acute neurologic episodes; and (4) increased urinary excretion of δ-ALA.
Tyrosinemia type 1 symptoms usually appear in the first few months of life and include failure to thrive, diarrhea, vomiting, jaundice, cabbage-like odor, and increased tendency to bleed (particularly nosebleeds). Untreated tyrosinemia type 1 usually presents either in young infants with severe liver involvement or later in the first year with liver dysfunction and renal tubular dysfunction associated with growth failure and rickets. Tyrosinemia type 1 can lead to liver and kidney failure, softening and weakening of the bones, problems affecting the nervous system, and an increased risk of liver cancer. Untreated children may have repeated, often unrecognized, neurologic crises lasting one to seven days that can include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation. Death in the untreated child usually occurs before age ten years, typically from liver failure, neurologic crisis, or hepatocellular carcinoma.
Treatment should be started as soon as the condition is diagnosed and includes a diet restricted in tyrosine and phenylalanine along with nitisinone, a medication that blocks the second step in the tyrosine degradation pathway 5. Combined treatment with nitisinone and a low-tyrosine diet has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets.
Tyrosinemia type 1 prevalence
In geographic areas without newborn screening, tyrosinemia type 1 affects approximately one in 100,000 to 120,000 births 6. Because of the inconsistent and confusing nature of its clinical presentation, it is estimated that fewer than 50% of affected individuals are diagnosed while alive.
In the general US population, the carrier frequency is estimated at 1:150 to 1:100.
Two regions of the world have a higher than expected frequency of tyrosinemia type 1 due to the increased frequency of certain pathogenic variants resulting from a founder effect:
- In the Scandanavian countries (c.1062+5G>A (IVS 12+5 G>A), p.Gly337Ser, and/or p.249HisfsTer5.5) and in Finland (p.Trp262Ter), the birth prevalence is estimated at 1:74,000 and 1:60,000 live births, respectively 7.
- A founder effect from colonization by French settlers is present in the province of Quebec, Canada. The c.1062+5G>A (IVS12+5 G>A) pathogenic variant accounts for 87% of allelic variants in this population. The birth prevalence in the province of Quebec is 1:16,000. In the Saguenay-Lac Saint-Jean region of Quebec, it is 1:1,846 live births. The overall carrier frequency in Quebec is 1:66 based on newborn screening data. The carrier frequency in the Saguenay-Lac St-Jean region is 1:16-1:20.
Tyrosinemia type 1 diagnosis
Tyrosinemia type 1 should be suspected in individuals with the following newborn screening results, clinical features, and supportive laboratory findings.
Newborn screening
- Presence of succinylacetone, measured directly from the newborn blood spot by tandem mass spectroscopy, is pathognomonic for tyrosinemia type 1.
- Elevated tyrosine or methionine concentration in the blood suggests liver disease, which can be from a variety of causes; the diagnosis of tyrosinemia type 1 should be further evaluated by quantification of plasma or urinary succinylacetone.
- Infants with tyrosinemia type I may have only modestly elevated or normal blood concentrations of tyrosine and methionine when the first newborn screening sample is collected.
- Elevated tyrosine concentration on newborn screening can be the result of transient tyrosinemia of the newborn, tyrosinemia type II or III, or other liver disease.
- Elevated methionine concentration can indicate liver dysfunction, defects in methionine metabolism, or homocystinuria.
- Low delta-ALA-dehydratase (PBG synthase) enzyme activity. This is measured in the newborn screening program 8. Succinylacetone is then measured in the urine of infants with apparent δ-ALA dehydratase deficiency 9.
Supportive laboratory findings
- Increased succinylacetone concentration in the blood and excretion in the urine
- Note: (1) Increased excretion of succinylacetone in the urine of a child with liver failure or severe renal disease is a pathognomonic sign of tyrosinemia type 1. (2) Many laboratories require that measurement of succinylacetone be specifically requested when ordering urine organic acids.
- Elevated plasma concentration of tyrosine, methionine, and phenylalanine
- Note: (1) Plasma tyrosine concentration in affected infants can be normal in cord blood and during the newborn period. (2) Elevated plasma tyrosine concentration can also be a nonspecific indicator of liver damage or immaturity; for example, in infants taking a high-protein formula 10, including undiluted goat’s milk 11.
- Elevated urinary concentration of tyrosine metabolites p-hydroxyphenylpyruvate, p-hydroxyphenyllactate, and p-hydroxyphenylacetate detected on urine organic acid testing
- Increased urinary excretion of the compound δ-ALA secondary to inhibition of the enzyme δ-ALA dehydratase by succinylacetone in the liver and circulating red blood cells 12
- Changes in liver function (in untreated tyrosinemia type 1)
- Markedly elevated serum concentration of alpha-fetoprotein (average 160,000 ng/mL) (normal: <1000 ng/mL for infants age 1-3 months; <12 ng/mL for children age 3 months to 18 years)
- Prolonged prothrombin and partial thromboplastin times
Note: (1) Changes in serum concentration of alpha-fetoprotein (AFP) and prothrombin time/partial thromboplastin time (PT/PTT) are more severe in tyrosinemia type 1 than in nonspecific liver disease and are often the presenting findings in tyrosinemia type 1. (2) Transaminases and bilirubin are only modestly elevated, if at all. (3) An individual with liver disease and normal serum concentration of alpha-fetoprotein (AFP) and normal prothrombin time/partial thromboplastin time (PT/PTT) has a low probability of having tyrosinemia type 1.
Establishing the diagnosis
The diagnosis of tyrosinemia type I is established in a proband with characteristic biochemical findings (increased succinylacetone concentration in the blood and urine; elevated plasma concentrations of tyrosine, methionine, and phenylalanine; and elevated urinary concentration of tyrosine metabolites and the compound δ-ALA) and/or by the identification of biallelic pathogenic variants in fumarylacetoacetase (FAH) on molecular genetic testing 1.
Evaluations following initial diagnosis
To establish the extent of disease and needs of a child diagnosed with tyrosinemia type I on the basis of newborn screening, the following evaluations are recommended:
- Complete blood count (CBC) with platelet count
- Serum concentration of electrolytes
- Assessment of liver function (PT, PTT, AST, ALT, GGT, serum bilirubin concentration, alkaline phosphatase, and serum AFP).
For children ascertained on the basis of clinical symptoms, testing should include the evaluations listed above in addition to:
- Baseline abdominal imagining by CT or MRI (with contrast) to evaluate for liver adenomas or nodules (see Dubois et al [1996]) and renal size;
- X-ray of wrist to document presence or absence of rickets.
Clinical genetics consultation is indicated for all affected individuals.
Tyrosinemia type 1 treatment
Management guidelines have been published. US recommendations include Chinsky et al 13; European recommendations include de Laet et al 14.
Acute management of liver failure: Children may require respiratory support, appropriate fluid management, and blood products for correction of bleeding diathesis.
Nitisinone (Orfadin®): 2-(2-nitro-4-trifluoro-methylbenzyol) 1,3 cyclohexanedione (NTBC) blocks parahydroxyphenylpyruvic acid dioxygenase (p-HPPD), the second step in the tyrosine degradation pathway, and prevents the accumulation of fumarylacetoacetic acid and its conversion to succinylacetone.
- Nitisinone should be prescribed as soon as the diagnosis of tyrosinemia type 1 is confirmed.
- Nitisinone is generally prescribed at 1.0 mg/kg/day; individual requirements may vary. Dosage should be adjusted to maintain blood nitisinone levels between 40 and 60 µmol/L, which theoretically blocks more than 99% of p-HPPD activity. Rarely, an individual may require higher blood levels of nitisinone (70 µmol/L) to suppress succinylacetone excretion. As long as blood concentration of nitisinone is within the therapeutic range, urine succinylacetone does not need to be measured.
- Nitisinone is typically given in two divided doses; however, because of the long half-life (50-60 hours), affected individuals who are older than one year and stable may maintain adequate therapy with 1x/day dosing 15.
- Rare side effects of nitisinone have included: transient low platelet count and transient low neutrophil count that resolved without intervention; and photophobia that resolved with stricter dietary control and subsequent lowering of blood tyrosine concentrations.
Low-tyrosine diet
Nitisinone increases blood concentration of tyrosine, necessitating a low-tyrosine diet to prevent tyrosine crystals from forming in the cornea.
- Dietary management should be started immediately upon diagnosis and should provide a nutritionally complete diet with controlled intakes of phenylalanine and tyrosine using a vegetarian diet with low-protein foods and a medical formula such as Tyrex® (Ross) or Tyros-1® (Mead Johnson).
- Phenylalanine and tyrosine requirements are interdependent and vary from individual to individual and within the same individual depending on growth rate, adequacy of energy and protein intakes, and state of health. With appropriate dietary management, plasma tyrosine concentration should be 300-600 µmol/L, regardless of age; plasma phenylalanine concentration should be 20-80 µmol/L (0.3-1.3 mg/dL). If the blood concentration of phenylalanine is too low (<20 µmol/L), additional protein should be added to the diet from milk or foods.
Liver transplantation
Prior to the availability of nitisinone for the treatment of tyrosinemia type 1, the only definitive therapy was liver transplantation.
- Liver transplantation should be reserved for those children who (1) have severe liver failure at clinical presentation and fail to respond to nitisinone therapy or (2) have documented evidence of malignant changes in hepatic tissue 16.
- Transplant recipients require long-term immunosuppression. Mortality associated with liver transplantation in young children is approximately 10%.
- Transplant recipients may also benefit from low-dose (0.1mg/kg/day) nitisinone therapy to prevent continued renal tubular and glomerular dysfunction resulting from persistence of succinylacetone in the plasma and urine 17.
Prevention of primary manifestations
- Treatment with nitisinone (Orfadin®) should begin as soon as the diagnosis is confirmed.
Prevention of secondary complications
Because carnitine deficiency secondary to the renal tubular Fanconi syndrome can cause skeletal muscle weakness, serum concentration of carnitine should be measured so that carnitine deficiency, if identified, can be treated 18.
Osteoporosis and rickets resulting from renal tubular damage are treated by correction of acidosis, restoring of calcium and phosphate balance, and administration of 25-hydroxy-vitamin D.
Surveillance
Frequent evaluation of the following parameters is typical in the management of individuals with tyrosinemia type 1 (Table 1)
Table 1. Suggested guidelines for monitoring in individuals with Tyrosinemia Type 1 diagnosed by newborn screening
| Evaluation | Initiation of Therapy (Baseline) | First 6 Months: | After 6 Mos of Rx: Every 6-12 months | After 2 Yrs of Rx: Every 6-12 months | As Clinically Indicated | ||
|---|---|---|---|---|---|---|---|
| 1x/month | Every 3 months | ||||||
| Tyrosinemia type I markers | Plasma concentration of methionine, phenylalanine, tyrosine | X | x | X | X | or X | |
| Blood / urine succinylacetone | X | X (urine) | X | or X | |||
| Blood nitisinone concentration | X | X | X | or X | |||
| CBC | Hemoglobin, hematocrit, WBC, platelet count | X | X | X | X | or X | |
| Liver evaluation | Serum AFP concentration | X | X | X | X | X | |
| PT/PTT | X | X (until normal) | |||||
| Bilirubin | X | ||||||
| ALT/AST/GGT | X | X (until normal) | X | ||||
| Alkaline phosphatase | X | X (until normal) | X | X | |||
| CT or MRI 1 | X | ||||||
| Renal studies | BUN / creatinine | X | |||||
| Urine: PO4, Ca, Prot/Cr ratio | X | ||||||
| Skeletal evaluation | X-ray of wrist (for rickets) | X | |||||
Footnote:
1 MRI with contrast to evaluate for liver adenomas or nodules and for kidney size
Abbreviations: AFP = alpha-fetoprotein; ALT/AST = alanine transaminase/aspartate transaminase; BUN = blood urea nitrogen; CBC = complete blood count; GGT = gamma-glutamyl transferase; PT/PTT = prothrombin time/partial thromboplastin time
[Source 1 ]Pregnancy management
Little data exist on the use of nitisinone during human pregnancy. Speculation would assume that the pregnant woman remains safe from untoward events; however, the developing fetus may be at risk because of alterations in tyrosine metabolism.
At least two women have given birth to healthy infants while receiving therapeutic doses of nitisinone 19.
- In one instance the affected mother gave birth to an unaffected infant who is reported to be healthy and developing normally at age 2.5 years 20.
- In another instance, an affected woman gave birth to an affected child. The child is reported to have normal growth and development at age seven months 19. The authors speculate that the affected child was protected from in utero liver damage by maternal treatment with nitisinone during pregnancy.
Tyrosinemia type 1 life expectancy
Survival in untreated children. Untreated infants diagnosed before age two months had a two-year survival rate of 29% 21.
- Those diagnosed between ages two and six months had a 74% two-year survival rate; those diagnosed after age six months had a 96% two-year survival rate.
- After more than five years the survival rate of the group diagnosed between ages two and six months dropped to approximately 30% and that of the group diagnosed after age six months dropped to approximately 60%
The natural history in children who are treated with nitisinone is different from that in untreated children. Affected children younger than age two years who are treated with a combination of nitisinone and low-tyrosine diet are markedly improved compared to those children treated with low-tyrosine diet alone. The combined nitisinone and low-tyrosine diet treatment has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets 22.
- Neurologic crises observed in treated children have always been associated with a prolonged interruption in nitisinone treatment.
- Children with acute liver failure require support prior to and during the initiation of treatment with nitisinone. Improvement generally occurs within one week of starting nitisinone treatment.
- Corneal crystals. Nitisinone blocks the tyrosine catabolic pathway such that succinylacetone is not produced but tissue tyrosine levels are raised. Blood tyrosine concentration greater than 600 mol/L confers risk of precipitation of tyrosine as bilateral, linear, branching subepithelial corneal opacities 23, causing photophobia and itchy, sensitive eyes. The crystals resolve once tyrosine levels are reduced.
- Hepatocellular carcinoma. Although Holme and Lindstedt 24 and van Spronsen et al 25 reported hepatocellular carcinoma in individuals after years of nitisinone therapy, it is estimated that fewer than 5% of children placed on nitisinone therapy before age two years develop hepatocellular carcinoma by age ten years 22.
- Where tyrosinemia type 1 is included in the newborn screening program, no affected individuals have been hospitalized for manifestations of tyrosinemia type 1 and hepatocellular carcinoma has not been reported in individuals who were placed on nitisinone therapy prior to age 30 days. The longest period of treatment reported in this group is 12 years 22.
Tyrosinemia type 2
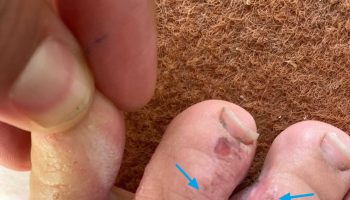
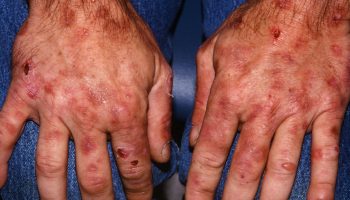
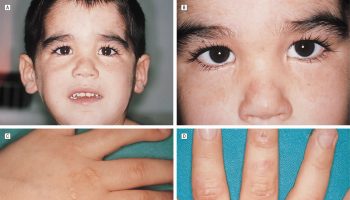
Tyrosinemia type 2 is caused by a defect in tyrosine aminotransferase (TAT), one of the enzymes required for the multi-step process that breaks down tyrosine. This enzyme shortage is caused by mutations in the TAT gene (16q22.1). Tyrosinemia type 2 is inherited in an autosomal recessive manner 3. Affected individuals have painful, non-pruritic, and hyperkeratotic plaques on the soles and palms. The plantar surface of the digits may show marked yellowish thickening associated with the hyperkeratosis. Ophthalmologic involvement is recalcitrant pseudodendritic keratitis 26. Symptoms of tyrosinemia type 2 often begin in early childhood and include excessive tearing, abnormal sensitivity to light (photophobia), eye pain and redness, and painful skin lesions on the palms and soles (palmoplantar hyperkeratosis). Although developmental delay appears to be common, it is unclear if ascertainment bias accounts for this and the reports of neurologic symptoms. Findings improve on a diet restricted in tyrosine and phenylalanine 27.
Prevalence is unknown but less than 150 cases have been reported in the literature so far. Tyrosinemia type 2 appears to be more common in Arab and Mediterranean populations 28.
Tyrosinemia type 2 causes
Tyrosinemia type 2 is caused by mutations in the TAT gene (16q22.1) encoding tyrosine aminotransferase (TAT). The elevated levels of tyrosine caused by TAT deficiency appear to result in deposition of tyrosine crystals leading to an inflammatory response and the oculocutaneous findings. It has also been suggested that there is a correlation between the extent of the central nervous system (CNS) involvement and the levels of tyrosine in the plasma.
Tyrosinemia type 2 signs and symptoms
Tyrosinemia type 2 tends to affect the skin and eyes. Signs of tyrosinemia type 2 usually begin in the first year of life. These signs include:
- Increased tear production
- Sensitivity to light (called photophobia)
- Eye redness
- Skin lesions on the hands and feet
- Behavior changes
- Poor coordination
Many of these signs may occur when your baby eats foods that their body cannot break down. They can be triggered by long periods of time without eating, illnesses, and infections.
If your baby shows any of these signs, be sure to contact your baby’s doctor immediately.
Tyrosinemia type 2 diagnosis
Establishing the diagnosis of tyrosinemia type 2 relies on the following:
- Plasma tyrosine concentration typically greater than 500 µmol/L that may exceed 1000 µmol/L (The concentration of other amino acids is normal.)
- Increased excretion of 4-hydroxyphenylpyruvate, 4-hydroxyphenyllactate, 4-hydroxyphenylacetate and presence of small quantities of N-acetyltyrosine and 4-tyramine on urine organic acid analysis
- Some patients with tyrosinemia type 2 may be identified through neonatal screening program studies.
Antenatal diagnosis:
- Molecular genetic prenatal diagnosis has been reported in families in which the TAT mutation had already been identified.
There is no cure for tyrosinemia type 2; however, some of the symptoms may be managed with a diet that limits certain amino acids, such as phenylalanine and tyrosine 28. A medication called NTBC may also be used to help control the amount of tyrosine in the body 29.
Tyrosinemia type 2 treatment
Management revolves around dietary restriction of phenylalanine and tyrosine. Oral retinoids may also be administered for treatment of the skin lesions.
Dietary treatments
Your baby may need to be on a restricted diet in order to avoid certain proteins that their body cannot break down. Babies with tyrosinemia type II (tyrosinemia type 2) may need to limit certain amino acids (phenylalanine, tyrosine, and methionine) in their diet. Amino acids are the building blocks of protein. These are all found in many proteins, and phenylalanine is also found in artificial sweeteners.
Special foods and formulas for children with tyrosinemia type 2 are available. These formulas will likely need to be continued through adulthood.
Medications
Your baby’s doctor might prescribe a drug called NTBC, which helps lower amino acid levels in the blood. Everyone has some amino acids in their blood, but the high levels associated with tyrosinemia type 2 can be toxic.
Tyrosinemia type 2 prognosis
The controlled diet results in lowering of plasma tyrosine levels and rapid resolution of the oculocutaneous manifestations. Children who receive early and ongoing treatment for tyrosinemia type 2 can have healthy growth and development. This is why newborn screening for tyrosinemia type 2 is so important. However, the extent to which this controlled diet prevents the CNS involvement is unclear.
About half of the individuals who have been diagnosed with tyrosinemia type 2 have intellectual disabilities. Early treatment can reduce the risk of developing intellectual disabilities.
Tyrosinemia type 3
Tyrosinemia type 3, the rarest of the tyrosine disorders with fewer than twenty reported cases, is caused by a deficiency of 4-hydroxyphenylpyruvate dioxygenase, one of the enzymes required for the multi-step process that breaks down tyrosine. 4-hydroxyphenylpyruvate dioxygenase enzyme shortage is caused by mutations in the HPD gene. Plasma concentration of tyrosine ranges from 350 to 650 µmol/L. Excretion of 4-hydroxyphenylpyruvic acid, 4-hydroxyphenyllactate, and 4-hydroxyphenylacetate is increased. The precise quantities vary with protein intake.
Few individuals with tyrosinemia type 3 have been identified, and the clinical phenotype remains ill defined. The first affected individuals came to medical attention because of intellectual disability, seizures, and periodic loss of balance and coordination (intermittent ataxia); another was detected on routine screening 6. These individuals, like those with tyrosinemia type 2, have no liver involvement but have skin or ocular changes. It remains unclear if tyrosinemia type 3 is truly associated with cognitive delays or if the association has resulted from ascertainment bias 27.
A diet low in phenylalanine and tyrosine can lower plasma tyrosine concentration.
Tyrosinemia type 3 causes
Tyrosinemia type 3 is caused by a defect in the HGD gene, which encodes the 4-hydroxyphenylpyruvate dioxygenase (HGD) enzyme. Babies with tyrosinemia type 3 do not make enough 4-hydrocyphenylpyruvate dioxygenase. When 4-hydrocyphenylpyruvate dioxygenase is not working correctly, the body cannot break down tyrosine. This causes tyrosine and other amino acids (methionine and phenylalanine) to build up in the body, which can be toxic. When 4-hydrocyphenylpyruvate dioxygenase cannot break down tyrosine correctly, tyrosine may be converted into a harmful acid called homogentisic acid. If homogentisic acid builds up in the body, it can be dangerous. Everyone has some acid and amino acids in their blood, but the high levels associated with tyrosinemia type 3 can be toxic.
Tyrosinemia type 3 is an autosomal recessive genetic condition. This means that a child must inherit two copies of the non-working gene for Tyrosinemia type 3, one from each parent, in order to have the condition. The parents of a child with an autosomal recessive condition each carry one copy of the non-working gene, but they typically do not show signs and symptoms of the condition. While having a child with Tyrosinemia type 3 is rare, when both parents are carriers, they can have more than one child with the condition.
Tyrosinemia type 3 signs and symptoms
Signs of tyrosinemia type 3 are highly variable and not yet well known. Signs of tyrosinemia type 3 may include:
- Poor coordination and balance
- Seizures (Epilepsy)
These signs may occur when your baby eats foods that their body cannot break down. They can be triggered by long periods of time without eating, illnesses, and infections.
If your baby shows any of these signs, be sure to contact your baby’s doctor immediately.
Tyrosinemia type 3 treatment
Although the signs and symptoms of tyrosinemia, type III (tyrosinemia type 3) are not well known, treatment is still recommended to try to avoid any possible health complications for your child.
Dietary treatments
Your baby may need to be on a restricted diet in order to avoid certain proteins that their body cannot break down. Babies with tyrosinemia type 3 may need to limit certain amino acids (phenylalanine, tyrosine, and methionine) in their diet. Amino acids are the building blocks of protein. These are all found in many proteins, and phenylalanine is also found in artificial sweeteners.
Special foods and formulas for children with tyrosinemia type 3 are available. These formulas will likely need to continue through adulthood.
Medications
Your baby’s doctor might prescribe a medication called NTBC. NTBC helps lower amino acid levels in the blood. Everyone has some amino acids in their blood, but the high levels associated with tyrosinemia type 3 can be toxic.
Tyrosinemia type 3 prognosis
Babies who are treated early for tyrosinemia type III (tyrosinemia type 3) may never develop any signs. This is why newborn screening for tyrosinemia type 3 is so important.
Children with tyrosinemia type 3 do have a risk of having an intellectual disability, but this risk is lower when the child receives treatment.
Tyrosinemia causes
Mutations in the FAH, TAT, and HPD genes can cause tyrosinemia types 1, 2 and 3, respectively. In the liver, enzymes break down tyrosine in a five step process, resulting in molecules that are either excreted by the kidneys or used to produce energy or make other substances in the body. The FAH gene provides instructions for the fumarylacetoacetate hydrolase enzyme, which is responsible for the final step of tyrosine breakdown. The enzyme produced from the TAT gene, called tyrosine aminotransferase enzyme, is involved at the first step in the process. The HPD gene provides instructions for making the 4-hydroxyphenylpyruvate dioxygenase enzyme, which is responsible for the second step.
Mutations in the FAH, TAT, or HPD gene cause a decrease in the activity of one of the enzymes in the breakdown of tyrosine. As a result, tyrosine and its byproducts accumulate to toxic levels, which can cause damage and death to cells in the liver, kidneys, nervous system, and other organs.
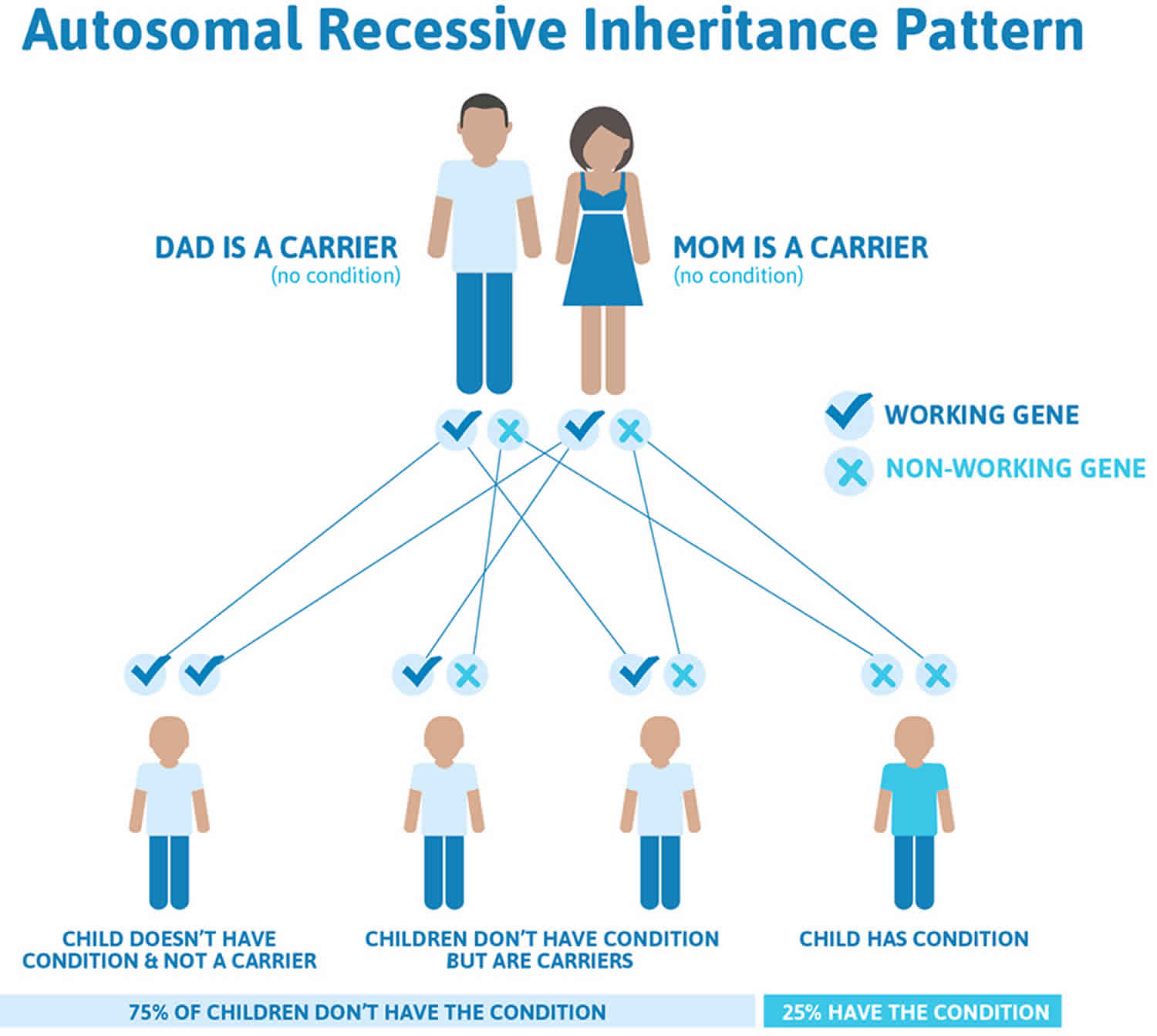
Tyrosinemia inheritance pattern
Tyrosinemia is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 1 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 1. Tyrosinemia autosomal recessive inheritance pattern
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://abgc.learningbuilder.com/Search/Public/MemberRole/Verification) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (https://clinics.acmg.net/) has a searchable database of medical genetics clinic services in the United States.
Tyrosinemia symptoms
Tyrosinemia type 1 clinical features (in untreated individuals):
- Severe liver disease in young infants
- Signs of renal disease, rickets, and/or neurologic crises in children older than age six months
- Untreated children may have repeated neurologic crises lasting one to seven days that can include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation.
For children who were not detected by newborn screening, tyrosinemia type 1 usually presents either in young infants with severe liver involvement or later in the first year with liver dysfunction and significant renal involvement, growth failure, and rickets. Growth failure results from chronic illness with poor nutritional intake, liver involvement, and/or chronic renal disease. Death in the undetected or untreated child usually occurs before age ten years, typically from liver failure, neurologic crisis, or hepatocellular carcinoma.
- Liver involvement. Undetected or untreated children presenting before age six months typically have acute liver failure with initial loss of synthetic function for clotting factors. PT and PTT are markedly prolonged and not corrected by vitamin K supplementation; factor II, VII, IX, XI, and XII levels are decreased; factor V and factor VIII levels are preserved. Paradoxically, serum transaminase levels may be only modestly elevated; serum bilirubin concentration may be normal or only slightly elevated, in contrast to most forms of severe liver disease in which marked elevation of transaminases and serum bilirubin concentration occur concomitantly with prolongation of PT and PTT. Resistance of affected liver cells to cell death may explain the observed discrepancy in liver function 30. This early phase can progress to liver failure with ascites, jaundice, and gastrointestinal bleeding. Children may have a characteristic odor of “boiled cabbage” or “rotten mushrooms.” Infants occasionally have persistent hypoglycemia; some have hyperinsulinism 31. Others have chronic low-grade acidosis. Untreated affected infants may die from liver failure within weeks or months of first symptoms 32.
- Renal tubular involvement. In the more chronic form of the untreated disorder, symptoms develop after age six months; renal tubular involvement is the major manifestation. The renal tubular dysfunction involves a Fanconi-like renal syndrome with generalized aminoaciduria, phosphate loss, and, for many, renal tubular acidosis. The continued renal loss of phosphate is believed to account for rickets; serum calcium concentrations are usually normal.
- Neurologic crises. Untreated children may have repeated neurologic crises similar to those seen in older individuals with acute intermittent porphyria. These crises include change in mental status, abdominal pain, peripheral neuropathy, and/or respiratory failure requiring mechanical ventilation. Crises can last one to seven days. Repeated neurologic crises often go unrecognized:
- Hepatocellular carcinoma. Those children who are not treated with nitisinone and a low-tyrosine diet and who survive the acute onset of liver failure are at high risk of developing and succumbing to hepatocellular carcinoma.
Tyrosinemia type 2 symptoms
Skin lesions occur in 80% of cases, ocular involvement in 75% of cases and neurologic findings and some degree of intellectual deficit in up to 60% of cases. Onset is variable but the ocular symptoms (redness, photophobia, excessive tearing and pain) usually develop in the first year of life. Ocular signs include corneal clouding with bilateral dendritiform corneal lesions (pseudodendritic keratitis), neovascularization, corneal ulceration and scarring, which may lead to decreased visual acuity. Cutaneous manifestations usually begin after the first year of life but may develop at the same time as the ocular symptoms. The skin lesions consist of nonpruritic, hyperkeratotic papules and plaques principally located on the palms and soles (palmoplantar hyperkeratosis). These lesions are painful and progressive and are often associated with hyperhidrosis. Central nervous system (CNS) involvement is highly variable with intellectual deficit (ranging from mild to severe) being the most common manifestation. Other signs of CNS involvement include behavioral problems, nystagmus, tremor, ataxia, and convulsions.
Tyrosinemia type 3 signs and symptoms
Signs of tyrosinemia type 3 are highly variable and not yet well known. Signs of tyrosinemia type 3 may include:
- Poor coordination and balance
- Seizures (Epilepsy)
These signs may occur when your baby eats foods that their body cannot break down. They can be triggered by long periods of time without eating, illnesses, and infections.
Tyrosinemia diagnosis
Newborn screening (heel-stick blood draw) is essential to detect all forms of tyrosinemia, including the transient tyrosinemia. The transient tyrosinemia is detected most frequently in premature infants, and usually resolves after 2 months of age. Transient tyrosinemia is characterized by elevated tyrosine (and sometimes elevated phenylalanine) in the blood. If your baby’s newborn screening result for tyrosinemia was above the normal range, your baby’s doctor will contact you to arrange for your baby to have additional testing. It is important to remember that an out-of-range screening result does not necessarily mean that your child has a life long condition. An out-of-range result may occur because the initial blood sample was too small, the test was performed too early, or may be due to transient (temporary) tyrosinemia. It is very important that you go to your follow-up appointment for a confirmatory test to determine the cause.
Tyrosinemia type 1 is detected on newborn screening if the most accurate test (succinylacetone measurement) is used to screen the heel stick blood. Measurement of tyrosine alone is often not sensitive enough to detect tyrosinemia type 1. Measurement of succinylacetone is the most accurate way to identify tyrosinemia type 1 in a newborn. Early detection of tyrosinemia type 1 is critical, since it is considered the most severe form of tyrosinemia. If not detected early and treated with nitisinone, diet, and medical food, tyrosinemia type 1 can result in neurologic crisis, liver failure, liver cancer, renal damage, and other severe complications. If treated early and managed well, children with tyrosinemia type 1 can lead much healthier lives.
Tyrosinemia type 2 and type 3 are both detected on newborn screening by elevations in blood tyrosine. Both are treated by managing blood tyrosine levels with a low protein diet and medical food. A dietician works with the families at every stage of growth and development to optimize health and well-being for the child and family.
Tyrosinemia treatment
There is currently no cure for tyrosinemia type 1. Individuals with this condition need to be on a special diet restricted in two amino acids, tyrosine and phenylalanine, throughout life. Affected individuals may also be treated with a medication called nitisinone. Treatment should start as soon as the condition is diagnosed. Some individuals require a liver transplant if their liver disease is already advanced before treatment begins.
The management of tyrosinemia type 2 revolves around dietary restriction of phenylalanine and tyrosine. This controlled diet typically lowers the blood levels of tyrosine, resulting in rapid resolution of the skin and eye symptoms. However, the effects of this controlled diet on central nervous system involvement (mental development) remains unclear. In some cases, skin lesions may be treated with oral retinoids 28.
Tyrosinemia type 3 can also be managed with low protein diet and medical food.
Tyrosinemia prognosis
The prognosis for tyrosinemia, specifically tyrosinemia type 1 has significantly improved with the introduction of nitisinone and a low-tyrosine diet. Combined treatment with nitisinone and a low-tyrosine diet has resulted in a greater than 90% survival rate, normal growth, improved liver function, prevention of cirrhosis, correction of renal tubular acidosis, and improvement in secondary rickets 34, 35, 36, 37. One study in which 5 tyrosinemia type 1 patients were started on nitisinone and dietary restriction within 28 days of life resulted in no death or liver failure during 15 years of follow-up 38. Early diagnosis and treatment are crucial for a better outcome. Untreated, tyrosinemia type 1 can be fatal, often before the age of 10. However, with proper management, individuals can live into adulthood and experience a fairly normal life.
- Sniderman King L, Trahms C, Scott CR. Tyrosinemia Type I. 2006 Jul 24 [Updated 2017 May 25]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1515[↩][↩][↩]
- Transient tyrosinemia of the newborn. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=en&Expert=3402[↩]
- Tyrosinemia. https://ghr.nlm.nih.gov/condition/tyrosinemia[↩][↩]
- Endo F, Sun MS. Tyrosinaemia type I and apoptosis of hepatocytes and renal tubular cells. J Inherit Metab Dis. 2002;25:227–34.[↩]
- Tyrosinemia. http://www.idph.state.il.us/HealthWellness/fs/tyrosinemia.htm[↩]
- Mitchell GA, Grompe M, Lambert M, Tanguay RM. Hypertyrosinemia. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw Hill; 2001:1777-806.[↩][↩]
- Bliksrud YT, Brodtkorb E, Backe PH, Woldseth B, Rootwelt H. Hereditary tyrosinaemia type I in Norway: incidence and three novel small deletions in the fumarylacetoacetase gene. Scand J Clin Lab Invest. 2012;2012;72:369–73.[↩]
- Giguère Y, Ruel J, Belanger N, Grenier A, Laberge C, Quebec Neonatal Blood Screening Programme, CHUL du CHUQ, Quebec, Canada. Neonatal mass screening for hereditary tyrosinemia type 1 in Quebec: a historical perspective (1970-2005). Portland, OR: Newborn Screening and Genetic Testing Symposium. 2005[↩]
- Schulze A, Frommhold D, Hoffmann GF, Mayatepek E. Spectrophotometric microassay for delta-aminolevulinate dehydratase in dried-blood spots as confirmation for hereditary tyrosinemia type I. Clin Chem. 2001;47:1424–9.[↩]
- Techakittiroj C, Cunningham A, Hooper PF, Andersson HC, Thoene J. High protein diet mimics hypertyrosinemia in newborn infants. J Pediatr. 2005;146:281–2.[↩]
- Hendriksz CJ, Walter JH. Feeding infants with undiluted goat’s milk can mimic tyrosinaemia type 1. Acta Paediatr. 2004;93:552–3[↩]
- Sassa S, Kappas A. Hereditary tyrosinemia and the heme biosynthetic pathway. Profound inhibition of delta-aminolevulinic acid dehydratase activity by succinylacetone. J Clin Invest. 1983;71:625–34.[↩]
- Chinsky JM, Singh R, Ficicioglu C, et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19(12):. doi:10.1038/gim.2017.101 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5729346[↩]
- de Laet C, Dionisi-Vici C, Leonard JV, et al. Recommendations for the management of tyrosinaemia type 1. Orphanet J Rare Dis. 2013;8:8. Published 2013 Jan 11. doi:10.1186/1750-1172-8-8 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3558375[↩]
- Schlune A, Thimm E, Herebian D, Spiekerkoetter U. Single dose NTBC-treatment of hereditary tyrosinemia type I. J Inherit Metab Dis. 2012;2012;35:831–6.[↩]
- Bartlett DC, Lloyd C, McKiernan PJ, Newsome PN. Early nitisinone treatment reduces the need for liver transplantation in children with tyrosinaemia type 1 and improves post-transplant renal function. J Inherit Metab Dis. 2014;37:745–52.[↩]
- Bartlett DC, Preece MA, Holme E, Lloyd C, Newsome PN, McKiernan PJ. Plasma succinylacetone is persistently raised after liver transplantation in tyrosinaemia type 1. J Inherit Metab Dis. 2013;36:15–20.[↩]
- Nissenkorn A, Korman SH, Vardi O, Levine A, Katzir Z, Ballin A, Lerman-Sagie T. Carnitine-deficient myopathy as a presentation of tyrosinemia type I. J Child Neurol. 2001;16:642–4.[↩]
- Garcia Segarra N, Roche S, Imbard A, Benoist JF, Grenèche MO, Davit-Spraul A, Ogier de Baulny H. Maternal and fetal tyrosinemia type I. J Inherit Metab Dis. 2010;33 Suppl 3:S507–10.[↩][↩]
- Vanclooster A, Devlieger R, Meersseman W, Spraul A, Kerckhove KV, Vermeersch P, Meulemans A, Allegaert K, Cassiman D. Pregnancy during nitisinone treatment for tyrosinaemia type I: first human experience. JIMD Rep. 2012;5:27–33.[↩]
- van Spronsen FJ, Thomasse Y, Smit GP, Leonard JV, Clayton PT, Fidler V, Berger R, Heymans HS. Hereditary tyrosinemia type I: a new clinical classification with difference in prognosis on dietary treatment. Hepatology. 1994;20:1187–91[↩][↩]
- Larochelle J, Alvarez F, Bussières JF, Chevalier I, Dallaire L, Dubois J, Faucher F, Fenyves D, Goodyer P, Grenier A, Holme E, Laframboise R, Lambert M, Lindstedt S, Maranda B, Melançon S, Merouani A, Mitchell J, Parizeault G, Pelletier L, Phan V, Rinaldo P, Scott CR, Scriver C, Mitchell GA. Effect of nitisinone (NTBC) treatment on the clinical course of hepatorenal tyrosinemia in Québec. Mol Genet Metab. 2012;107:49–54.[↩][↩][↩]
- Ahmad S, Teckman JH, Lueder GT. Corneal opacities associated with NTBC treatment. Am J Ophthalmol. 2002;134:266–8.[↩]
- Holme E, Lindstedt S. Nontransplant treatment of tyrosinemia. Clin Liver Dis. 2000;4:805–14.[↩]
- van Spronsen FJ, Bijleveld CM, van Maldegem BT, Wijburg FA. Hepatocellular carcinoma in hereditary tyrosinemia type I despite 2-(2 nitro-4-3 trifl). J Pediatr Gastroenterol Nutr. 2005;40:90–3.[↩]
- Macsai MS, Schwartz TL, Hinkle D, Hummel MB, Mulhern MG, Rootman D. Tyrosinemia type II: nine cases of ocular signs and symptoms. Am J Ophthalmol. 2001;132:522–7.[↩]
- Ellaway CJ, Holme E, Standing S, Preece MA, Green A, Ploechl E, Ugarte M, Trefz FK, Leonard JV. Outcome of tyrosinaemia type III. J Inherit Metab Dis. 2001;24:824–32.[↩][↩]
- Tyrosinemia type 2. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=28378[↩][↩][↩]
- Tyrosinemia Type II. https://www.babysfirsttest.org/newborn-screening/conditions/tyrosinemia-type-ii[↩]
- Vogel A, van Den Berg IE, Al-Dhalimy M, Groopman J, Ou CN, Ryabinina O, Iordanov MS, Finegold M, Grompe M. Chronic liver disease in murine hereditary tyrosinemia type 1 induces resistance to cell death. Hepatology. 2004;39:433–43.[↩]
- Baumann U, Preece MA, Green A, Kelly DA, McKiernan PJ. Hyperinsulinism in tyrosinaemia type I. J Inherit Metab Dis. 2005;28:131–5.[↩]
- Hegarty R, Hadzic N, Gissen P, Dhawan A. Inherited metabolic disorders presenting as acute liver failure in newborns and young children: King’s College Hospital experience. Eur J Pediatr. 2015;174:1387–92.[↩]
- Mitchell G, Larochelle J, Lambert M, Michaud J, Grenier A, Ogier H, Gauthier M, Lacroix J, Vanasse M, Larbrisseau A, et al. Neurologic crises in hereditary tyrosinemia. N Engl J Med. 1990;322:432–7.[↩]
- Tyrosinemia. https://medlineplus.gov/genetics/condition/tyrosinemia/[↩]
- Sniderman King L, Trahms C, Scott CR. Tyrosinemia Type I. 2006 Jul 24 [Updated 2017 May 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1515[↩]
- Das AM, Ballhausen D, Haas D, et al. Diagnosis, treatment, management and monitoring of patients with tyrosinaemia type 1: Consensus group recommendations from the German-speaking countries. J Inherit Metab Dis. 2025 Jan;48(1):e12824. doi: 10.1002/jimd.12824[↩]
- Äärelä L, Hiltunen P, Soini T, Vuorela N, Huhtala H, Nevalainen PI, Heikinheimo M, Kivelä L, Kurppa K. Type 1 tyrosinemia in Finland: a nationwide study. Orphanet J Rare Dis. 2020 Oct 12;15(1):281. doi: 10.1186/s13023-020-01547-w[↩]
- Introne WJ. Nitisinone: two decades treating hereditary tyrosinaemia type 1. Lancet Diabetes Endocrinol. 2021 Jul;9(7):409-411. doi: 10.1016/S2213-8587(21)00121-2[↩]


{kind=link}