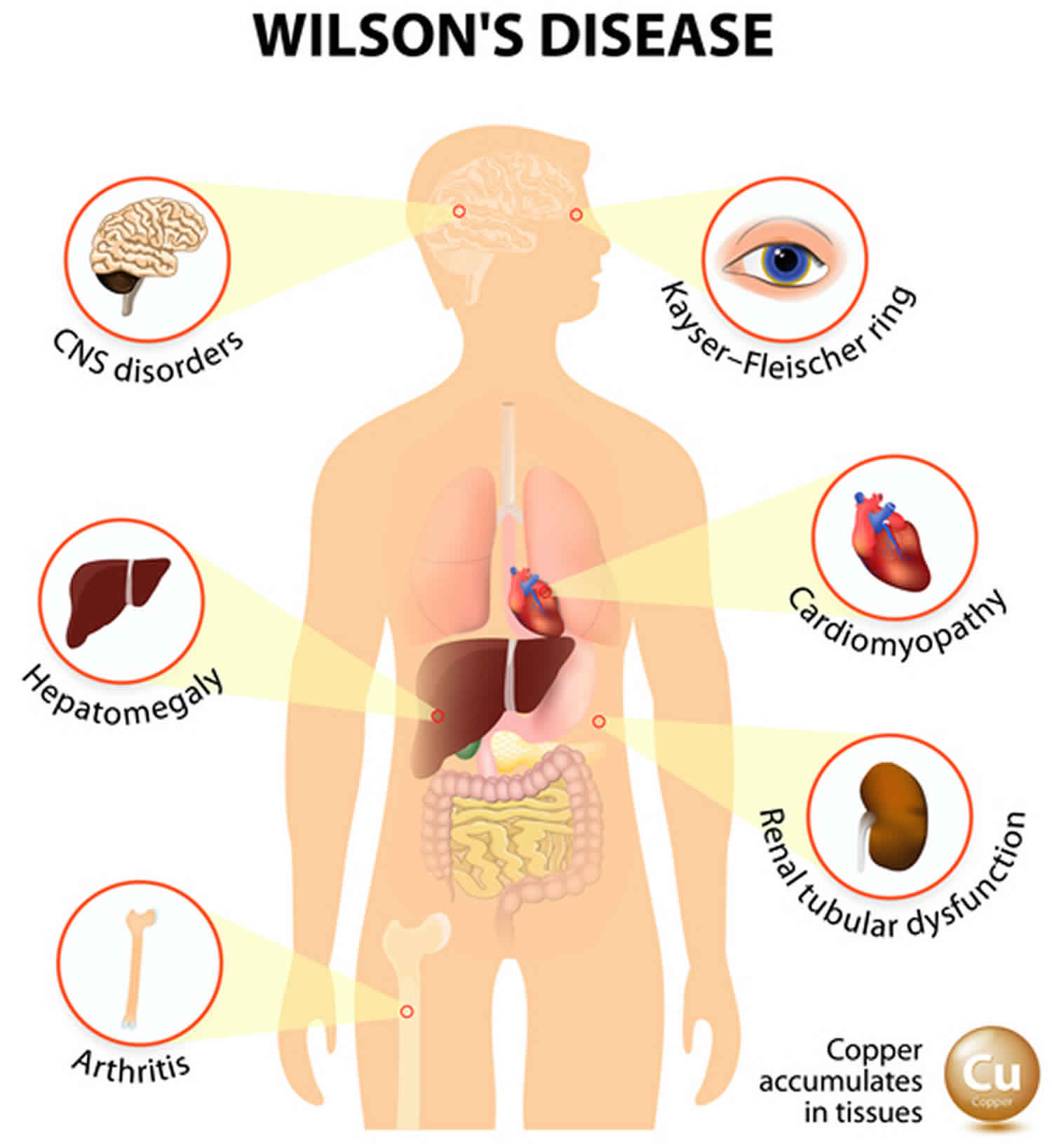
Wilson disease
Wilson disease also called hepatolenticular degeneration, is a rare inherited disorder (autosomal recessive disease) due to impaired biliary copper excretion that causes copper to accumulate in your liver, basal ganglia of the brain, kidneys, eyes and other vital organs. Because high levels of copper are toxic to tissues and organs, the buildup of copper can lead to damage of the liver, brain and eyes. Signs and symptoms of Wilson disease include chronic liver disease, central nervous system abnormalities or psychiatric (mental health-related) disturbances, or a combination of these, in individuals ranging from age three years to older than 50 years; symptoms vary among and within families 1, 2.
Wilson disease is a rare genetic disorder affecting 1 per 30,000 people with fewer than 50,000 people in the U.S. have this disease. Wilson disease affects female as well as male individuals and manifests mostly in late childhood or adolescence. Wilson disease is caused by a mutation of the ATP7B gene located on the long arm of chromosome 13 at position 14.3 (13q14) and is inherited in an autosomal recessive manner 3, 4, 5.
Copper plays a key role in the development of healthy nerves, bones, collagen and the skin pigment melanin. You need a small amount of copper from food to stay healthy. Too much copper is poisonous. Normally, copper is absorbed from your food, and excess is excreted through a substance produced in your liver called bile (a digestive fluid). But in people with Wilson’s disease, copper isn’t eliminated properly and instead it accumulates, possibly to a life-threatening level. When diagnosed early, Wilson’s disease is treatable to reduce or control the amount of copper that accumulates in your body, and many people with Wilson’s disease live normal lives. With early detection and proper treatment, you can enjoy good health.
Wilson disease is present at birth, but symptoms usually start between ages 5 and 35. It first attacks the liver, the central nervous system (brain and spinal cord) or both. The most characteristic sign is a rusty brown ring around the cornea of the eyes called the Kayser-Fleischer ring (Figure 1). A physical exam and laboratory tests can diagnose it.
- Liver disease includes recurrent jaundice, simple acute self-limited hepatitis-like illness, autoimmune-type hepatitis, fulminant hepatic failure, or chronic liver disease.
- Neurologic presentations include movement disorders (tremors, poor coordination, loss of fine-motor control, chorea, choreoathetosis) or rigid dystonia (mask-like facies, rigidity, gait disturbance, pseudobulbar involvement).
- Psychiatric disturbance includes depression, neurotic behaviors, disorganization of personality, and, occasionally, intellectual deterioration.
- Kayser-Fleischer rings, frequently present, result from copper deposition in Descemet’s membrane of the cornea and reflect a high degree of copper storage in the body.
There is no cure for Wilson disease, patients are typically placed on a low-copper diet (no liver, shellfish, nuts, chocolate or mushrooms) for the rest of your life and started on lifelong chelating agents (drugs to remove the extra copper from your body). Of the chelating agents, trientine (Syprine) is the drug of choice. Trientine is preferred because of fewer side effects. Oral zinc also may be given as it prevents the intestines from absorbing copper from the food you eat. At the beginning of treatment, you’ll also need to avoid chocolate, mushrooms, and nuts. Have your drinking water checked for copper content and don’t take multivitamins that contain copper. Routine monitoring of serum copper and ceruloplasmin is indicated along with liver enzymes, blood clotting factor, complete blood count with differential and urinalysis while on chelators. Also a yearly physical with 24-hour urinary copper should be performed while on medication 6. Patients will have a normal life expectancy if diagnosed early, however, they will develop fulminant hepatic failure if left untreated. Fulminant hepatic failure is treated with liver transplantation.
Figure 1. Kayser-Fleischer ring (the right eye showing a heavily pigmented red brown Kayser-Fleischer ring)
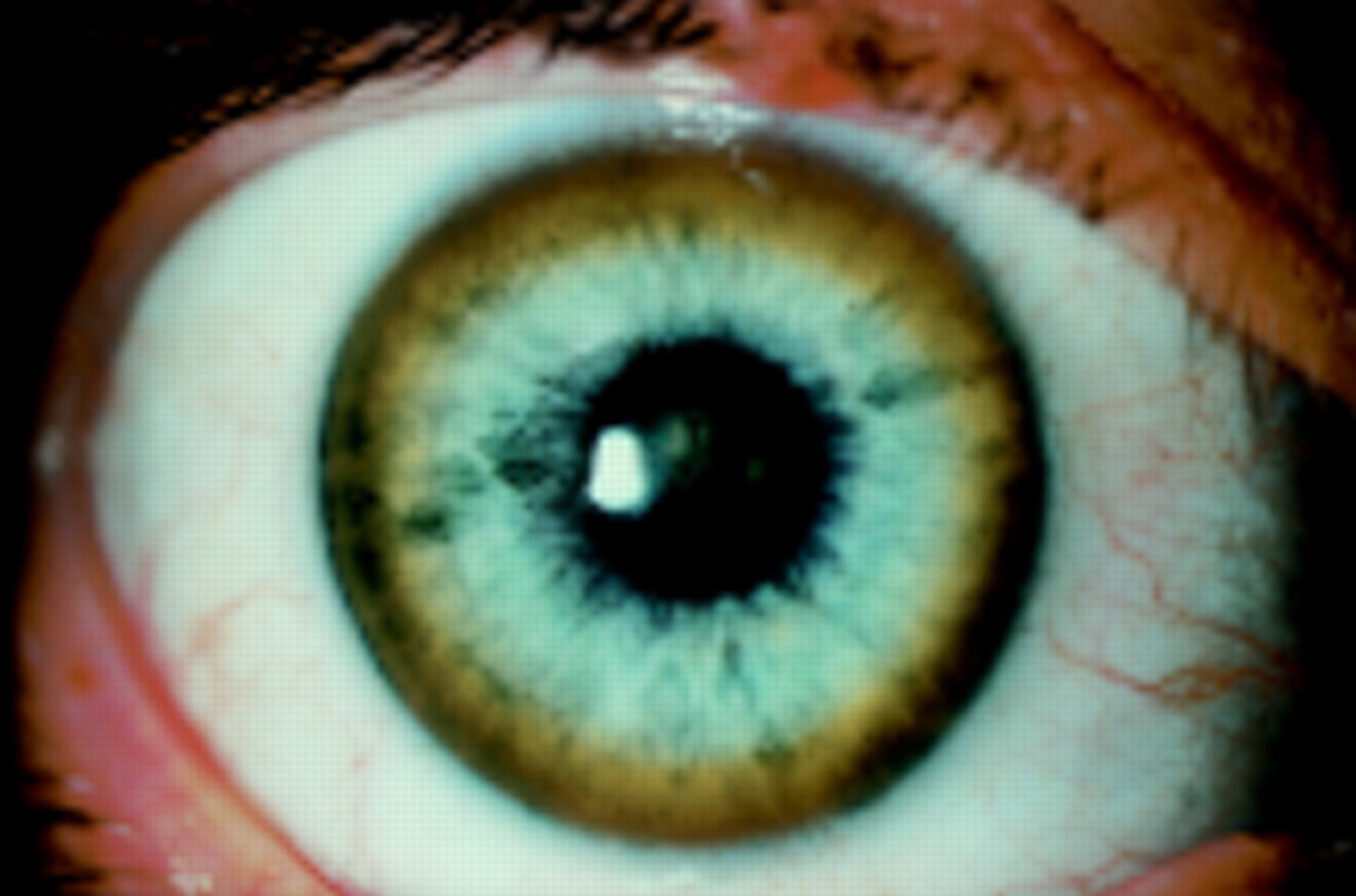
Figure 2. Wilson’s disease symptoms
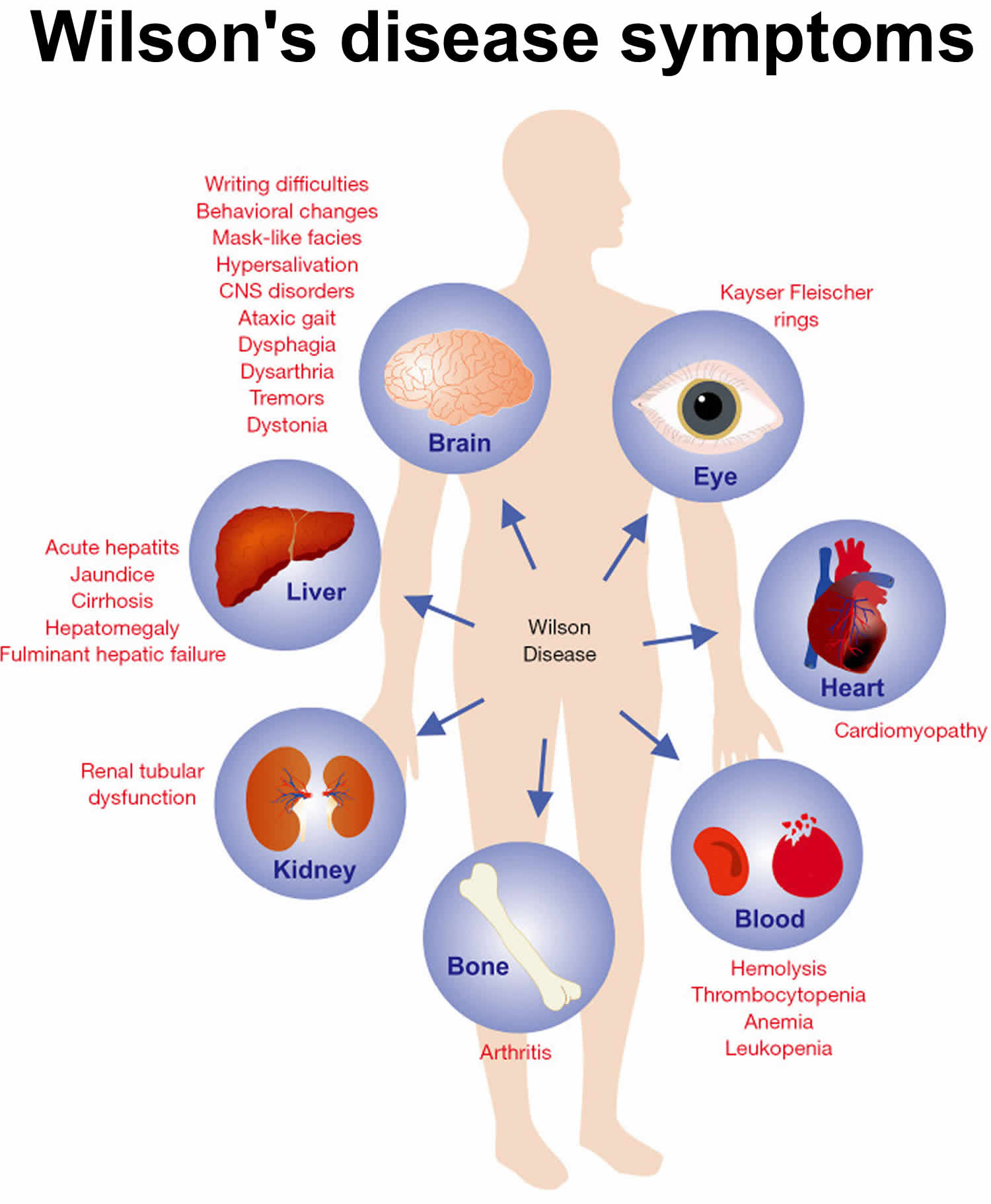
Footnote: In Wilson disease the symptoms are highly variable and can affect liver, brain, kidney, eyes, heart, bone, blood system, and many other organs or tissues.
[Source 8 ]Is Wilson disease curable?
No. A abnormal gene cannot be corrected – it is present for life. Wilson’s disease a genetic disease that is inherited as an autosomal recessive trait. Wilson disease is caused by mutations in the ATP7B gene on chromosome 13, which codes for a membrane-bound copper transporting ATPase found mostly in the liver 9. ATP7B gene provides instructions for making a protein called copper-transporting ATPase 2, which plays a role in the transport of copper from the liver to other parts of the body. Copper is necessary for many cellular functions, but it is toxic when present in excessive amounts. The copper-transporting ATPase 2 protein is particularly important for the elimination of excess copper from the body. Mutations in the ATP7B gene prevent the transport protein from functioning properly. With a shortage of functional protein, excess copper is not removed from the body. As a result, copper accumulates to toxic levels that can damage tissues and organs, particularly the liver and brain.
The ATP7B mutations that cause Wilson disease are inherited, meaning they are passed from parent to child. These mutations are autosomal recessive , meaning that a person must inherit two ATP7B genes with mutations, one from each parent, to have Wilson disease. People who have one ATP7B gene without a mutation and one ATP7B gene with a mutation do not have Wilson disease, but they are carriers of the disease.
People can inherent Wilson disease if both parents are carriers who don’t have the disease. Figure 2 below shows the chance of inheriting Wilson disease from parents who are carriers.
People with specific questions about genetic risks or genetic testing for themselves or family members should speak with a genetics professional.
Resources for locating a genetics professional in your community are available online:
- The National Society of Genetic Counselors (https://www.findageneticcounselor.com/) offers a searchable directory of genetic counselors in the United States and Canada. You can search by location, name, area of practice/specialization, and/or ZIP Code.
- The American Board of Genetic Counseling (https://www.abgc.net/about-genetic-counseling/find-a-certified-counselor/) provides a searchable directory of certified genetic counselors worldwide. You can search by practice area, name, organization, or location.
- The Canadian Association of Genetic Counselors (https://www.cagc-accg.ca/index.php?page=225) has a searchable directory of genetic counselors in Canada. You can search by name, distance from an address, province, or services.
- The American College of Medical Genetics and Genomics (http://www.acmg.net/ACMG/Genetic_Services_Directory_Search.aspx) has a searchable database of medical genetics clinic services in the United States.
How common is Wilson disease?
Experts are still studying how common Wilson disease is. Older studies suggested that about 1 in 30,000 people have Wilson disease 10. These studies were conducted before researchers discovered the gene mutations that cause Wilson disease.
Newer studies of people’s genes suggest that Wilson disease may be more common. A study in the United Kingdom found that about 1 in 7,000 people have gene mutations that cause Wilson disease 11.
Experts aren’t sure why gene studies suggest that Wilson disease is more common than previously thought. One reason might be that some people with Wilson disease are not diagnosed. Another reason might be that some people have gene mutations for Wilson disease but don’t develop the disease 12.
Who is more likely to have Wilson disease?
People have a higher chance of having Wilson disease if they have a family history of Wilson disease, especially if a first-degree relative, a parent, sibling, or child, has the disease.
People who have Wilson disease typically develop symptoms when they are between ages 5 and 40 13. However, some people develop symptoms at younger or older ages. Doctors have found the first symptoms of Wilson disease in infants as young as 9 months and in adults older than 70 years 10, 14.
How or where is the brain affected by copper accumulation?
Generally, the brain is affected symmetrically with excess copper deposition, although symptoms can be worse on one side of the body than another. This may have to do with factors of asymmetric neurologic development, such as being right or left-handed. The copper is often seen most prominently in the basal ganglia, the area deep within the brain that coordinates movements. The face of the giant panda sign refers to a characteristic appearance of the basal ganglia in advanced Wilson’s disease. This is a description of the appearance of the basal ganglia wherein one can get an impressionists image of the face of a giant panda.
Can Wilson’s disease result in permanent brain damage leading to memory loss?
Even though Wilson’s disease is primarily classified and considered as a “movement disorder,” cognitive decline is very common. The analogy is with Parkinson disease, where some patients develop “subcortical dementia.” The subject of dementia/cognitive decline in Wilson’s disease has not been studied that well but some papers about this have been published. Basically, what is going on is the disruption of projections of the basal ganglia to the prefrontal cortex. This may be reversible if Wilson disease is treated and the patient responds to the treatment.
My doctor says I have high copper in my blood (serum copper). Could I have Wilson disease?
High serum copper is not an indication of Wilson disease (see diagnosis below). Since most Wilson patients have a low ceruloplasmin they actually have a lower than normal serum copper. Ceruloplasmin is the protein that binds with copper to remove it from the body. It is the unbound (to ceruloplasmin) copper that is free to roam around the body and accumulate in organs causing Wilson disease damage.
An elevated serum copper is more often due to an elevation of the level of serum ceruloplasmin since it contains ~90% of the circulating copper bound to it. Elevations of ceruloplasmin can occur with inflammation, in response to estrogen therapy and in pregnancy. Note: The exception to this is when there is severe liver injury (acute liver failure) caused by Wilson disease. This causes very large amounts of copper to be released into circulation and causes markedly elevated serum copper. When this occurs, patients are very ill and usually have jaundice (yellow eyes and skin color) and very abnormal lab results with respect to liver function and blood coagulation.
Are copper IUDs safe for Wilson’s disease patients?
The IUD is plastic with copper wound around it. Experts would not advise it in women with Wilson’s disease. There are progesterone containing IUDs, one of which was just released, that are good for five years instead of one to two years.
It is not uncommon for symptomatic women with Wilson’s disease to suffer irregular periods and multiple miscarriages. These are due to malfunction of the liver causing hormonal changes which are reversible with successful treatment of the underlying disorder.
With what medication should the Wilson’s disease patient begin treatment?
There are now new options for the medical treatment of patients with Wilson’s disease. Penicillamine is no longer the “treatment of choice,” as there is a growing experience with safer and effective alternatives. Trientine may be the best first choice amongst currently approved drugs as initial therapy for symptomatic patients requiring chelation therapy, and may be even more effective when used in combination with zinc treatment. Zinc is an effective medication for maintenance therapy. Further studies are needed to determine the best therapy for pregnant patients with Wilson’s disease, and whether combination therapy using trientine and zinc will be the next “treatment of choice” for all symptomatic patients with liver or neurologic disease.
What happens if too much copper is removed through chelation?
When too much copper is removed, then there can be reductions in blood counts and inhibition of wound healing, amongst other possible effects. This is the reason that blood counts should be periodically monitored. The amount of copper can be gauged by reviewing the patterns of the 24- hour urine copper excretion as well as looking for the “non-ceruloplasmin” copper in the blood– this being done by a simultaneous measure of ceruloplasmin and serum copper. When these values are too low, excluding the possibility of non-compliance which can alter the interpretation of the urine copper excretion, then the dosages must be adjusted.
Can Zinc and Trientine be used in combination?
The combination of zinc and trientine can be used for the first four months of therapy in patients with significant liver or neurologic disease. How often you check lab values depends on the extent of Wilson’s disease, the range can be from weekly checks of urine copper and zinc to every three months after initiating treatment. Subsequently, the urine copper and zinc could be checked every six months for two years, then annually with more frequent checks if compliance is in doubt. Urine copper levels should be in the range expected for treatment with trientine alone, in other words 1-2 mg /24 hours initially and then reducing to 0.5-1.0 mg after several months of treatment.
Doctors look at the trend in urine values over time as much as the absolute numbers at any one time to gauge compliance and treatment effectiveness at reducing copper. You also can monitor the non-ceruloplasmin copper. This frequently falls to less than 10 micrograms/dl on the combination therapy. Monitoring for compliance: The urine zinc should be over 2.0 mg with adequate compliance with zinc treatment. Compliance with trientine is gauged by an initial increase in urine copper when the medicine is first started followed by a drop in urine copper values over time.
Monitoring for over treatment: Be concerned if the urine copper is less than 35, consider in the presence of anemia or decreased white blood count. On combination therapy, the urine copper may be higher than 35 in the presence of copper deficiency, so watch for unexplained anemia or decreased white blood count.
Monitoring for toxicity: Watch for gastric side effects from zinc such as nausea. Watch for proteinuria, bone marrow suppression and autoimmune disease with trientine.
Summary
Monitoring patients taking the combination of trientine and zinc is most like monitoring those taking trientine alone. The main difference is an increase in urine zinc should be seen to demonstrate compliance with zinc treatment.
How long should Zinc and Trientine be used in combination?
In our experience, the combination of zinc and trientine is not necessary beyond the initial four to six month period of removing free copper stores. Zinc maintenance therapy works well for most people thereafter. Obviously, treatment programs and decisions need to be individualized. Nevertheless, experts are not aware of compelling data to suggest that there is any need for taking the combination of zinc and trientine long term for the majority of people.
Will Galzin dissolve and remove the already accumulated copper in the brain or is it only effective controlling the further accumulation of copper?
With zinc acetate (Galzin) treatment there is a negative copper balance. More copper goes out than in, so in time all extra stores of copper, even in the brain, are removed. Proof of this is the loss of Kayser-Fleischer rings in treated patients with time on zinc. However, if there has been damage done to the brain cells by copper, this may [or may not] be reversible.
Are there any problems with zinc collecting in the body over time?
The zinc levels in the body increase slightly initially but zinc does not accumulate over time. Experts know this from repeat measurements including repeat liver biopsies which were used to measure liver copper and zinc to answer this very question. In short, the main concern with long-term treatment with zinc (as well as a concern with all other forms of treatment of Wilson’s Disease) would be the potential for over treatment which may lead to copper deficiency. Fortunately, this is relatively unusual but doctors monitor 24-hour urine copper and zinc levels to avert this from happening on at least an annual basis. If copper levels get too low, zinc doses can be reduced before the symptoms of copper deficiency, primarily manifested as anemia, ensue. The best approach is keep taking your zinc and check your urine copper and zinc at least once a year.
How long does Galzin last?
Galzin capsules should last indefinitely, so long as they do not physically fall apart. Zinc itself is an element, so it does not deteriorate over time.
How can I reduce the possible nausea and indigestion when taking zinc?
Experts suggest that you take a small piece of lunch meat (turkey, bologna, ham, etc.) with your first morning dose of zinc. This should help settle your stomach. Do not take any carbohydrate (bread, etc.) with the dose–this is very important! The nausea usually abates as treatment persists. Occasionally, someone finds that nausea to persist, but lunchmeat generally helps.
Does zinc cause ulcers?
Zinc does not cause ulcers, but ulcers are common problems. Patients experiencing abdominal pain and nausea should consult with their gastroenterologist.
What does high urine zinc mean?
High urine zinc proves the patient is taking his or her zinc conscientiously.
Is Wilson’s disease treatment safe for the unborn baby?
Review of the literature and experts experience indicate that women successfully treated with either penicillamine, trientine or zinc uninterruptedly, have excellent chances for carrying through uncomplicated pregnancies and for delivering normal babies. However, precautions are indicated in women with dilated veins in the stomach or esophagus, which may rupture and bleed because of the increased abdominal pressure caused by the enlarging uterus. There is no report of an untoward reaction to a baby nursed while the mother continued on an anti-copper regimen.
Will Tizanidine help control or reduce dystonia?
Medications doctors commonly use to control the symptoms of dystonia are Klonipin and Artane. Tizanidine may be reasonable to try as well. Obviously, an examining physician must decide what is best to try for each patient’s individual condition. Doctors also use zinc acetate or at times trientine in preference to penicillamine due to the risk of making neurologic disease worse on penicillamine. Physical therapy is also quite important for your treatment plan.
Should Wilson’s disease patients use weight-gaining supplements to help maintain their weight?
Metabolic enhancers like pro-kinetic are dangerous for people with liver disease, they can cause liver failure even in healthy people. Many dietary supplements like Ensure, etc., also contain copper supplements. If you are trying to gain weight and have been on appropriate therapy, drinking one can of Vanilla Boost Plus per day should not give you too much copper. You also can make your own high calorie drinks by mixing protein supplements with a milk shake.
What copper levels in drinking water are potentially hazardous for Wilson’s disease patients?
If the water copper level is over 0.1 ppm (parts per million) (which is 0.1 mg/L), experts recommend you drink water from an alternative source. While 0.1 ppm isn’t particularly hazardous, it indicates that significant copper is coming from somewhere, and at certain times or under certain circumstances the level might be quite a bit higher.
Should Wilson’s patients receive Hepatitis A or Hepatitis B vaccine?
Yes. Since Wilson’s disease often affects the liver, many Wilson’s disease patients cannot afford additional injury to the liver. Hepatitis A or Hepatitis B vaccine is as safe for Wilson’s disease patients as it is for others.
What percentage of Wilson’s disease patients need liver transplants?
Only about 5% of patients with Wilson’s disease need transplants. Two thirds are those presenting with liver failure. The remainder are those discovered with severe liver disease that doesn’t respond well to medical therapy, those who stop therapy and deteriorate, or those that experience severe complications of cirrhosis such as frequent gastrointestinal bleeding from varices due to portal hypertension or low oxygenation due to hepatopulmonary syndrome.
Do patients taking penicillamine have a difficult time in general healing from surgery?
For all surgical procedures, it is recommended that the dosage of penicillamine be temporarily reduced to 250-500 mg per day until wound healing is achieved. Similarly, the dosage should be reduced during the last trimester pregnancy and until wound healing after childbirth is achieved. No such dosage reduction is needed for zinc treatment.
What level of liver recovery can I expect with treatment?
The course of liver disease in Wilson’s disease stands in contrast to other forms of cirrhosis for many people. The chronic liver injury in Wilson’s disease is caused by excess free copper, and the liver disease often stabilizes or even improves once the excess copper is treated with zinc acetate maintenance therapy. While some people do progress to need liver transplantation, others may actually see long-term improvement in their liver function over time. It is important to be attentive to issues such as immunizations for viral hepatitis, avoiding excess alcohol consumption, and treating complications of portal hypertension in order to give the liver its best chance to mend.
What genetic testing is currently available for Wilson disease?
The sequence analysis of ATP7B gene (Wilson disease gene) to identify the mutations is clinically available as a test. Although this is the most updated and thorough test, please be aware that some alterations such as large deletion or duplication may not be detected with this method. It is important that the biochemical testing must be performed prior to genetic tests.
Wilson disease causes
Wilson disease is caused by mutations in the ATP7B gene on the long arm of chromosome 13 at position 14.3 (13q14), which codes for a membrane-bound copper transporting ATPase found mostly in the liver 15, 16, 2. In Wilson disease almost 800 different mutations of the ATP7B gene are known 3, 17, 18. ATP7B gene provides instructions for making a protein called copper-transporting ATPase 2, which plays a role in the transport of copper from the liver to other parts of the body 19. Copper is necessary for many cellular functions, but it is toxic when present in excessive amounts. The copper-transporting ATPase 2 protein is particularly important for getting rid of excess copper from the body 4. Mutations in the ATP7B gene prevent the transport protein from functioning properly. With a shortage of functional protein, excess copper is not removed from the body. As a result, copper accumulates to toxic levels that can damage tissues and organs, particularly the liver and brain.
Normally, the liver releases extra copper into bile. Bile carries the copper, along with other toxins and waste products, out of the body through the digestive tract. In Wilson disease, the liver releases less copper into bile, and extra copper stays in the body.
Research indicates that a normal variation in the PRNP gene may modify the course of Wilson disease. The PRNP gene provides instructions for making prion protein, which is active in the brain and other tissues and appears to be involved in transporting copper. Studies have focused on the effects of a PRNP gene variation that affects position 129 of the prion protein. At this position, people can have either the protein building block (amino acid) methionine or the amino acid valine. Among people who have mutations in the ATP7B gene, it appears that having methionine instead of valine at position 129 of the prion protein is associated with delayed onset of symptoms and an increased occurrence of neurological symptoms, particularly tremors. Larger studies are needed, however, before the effects of this PRNP gene variation on Wilson disease can be established.
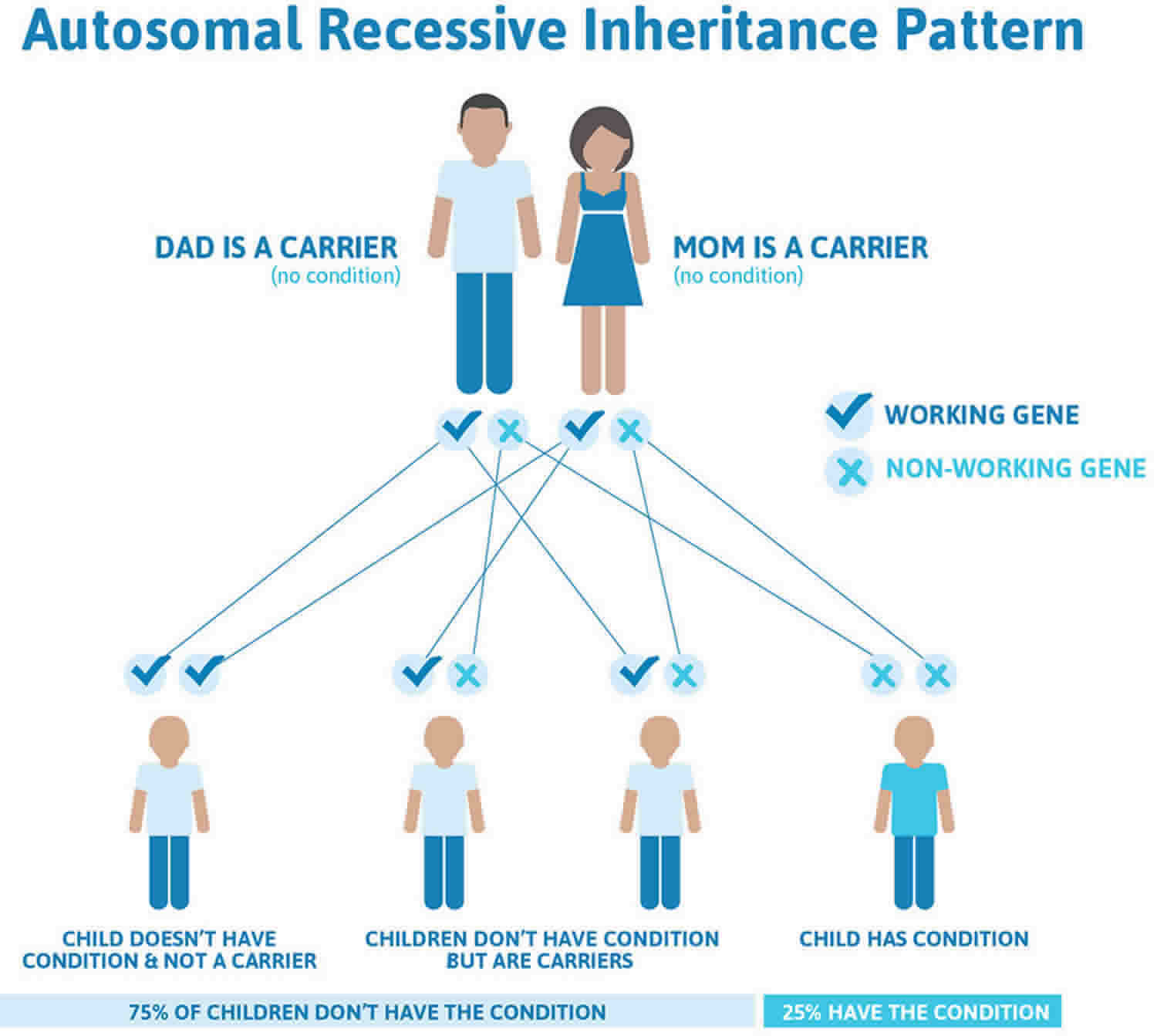
Wilson disease inheritance pattern
Wilson disease is inherited in an autosomal recessive pattern, meaning that a person must inherit two ATP7B genes with mutations, one from each parent, to have Wilson disease. The parents of an individual with Wilson disease (an autosomal recessive condition) each carry one copy of the mutated ATP7B gene, but they typically do not show signs and symptoms of the condition and do not have Wilson disease, but they are carriers of the disease.
People can inherent Wilson disease if both parents are carriers who don’t have the disease. The mutations that cause Wilson disease are autosomal recessive, meaning that a person must inherit two ATP7B genes with mutations to have Wilson disease. The chart below shows the chance of inheriting Wilson disease from parents who are carriers.
It is rare to see any history of autosomal recessive conditions within a family because if someone is a carrier for one of these conditions, they would have to have a child with someone who is also a carrier for the same condition. Autosomal recessive conditions are individually pretty rare, so the chance that you and your partner are carriers for the same recessive genetic condition are likely low. Even if both partners are a carrier for the same condition, there is only a 25% chance that they will both pass down the non-working copy of the gene to the baby, thus causing a genetic condition. This chance is the same with each pregnancy, no matter how many children they have with or without the condition.
- If both partners are carriers of the same abnormal gene, they may pass on either their normal gene or their abnormal gene to their child. This occurs randomly.
- Each child of parents who both carry the same abnormal gene therefore has a 25% (1 in 4) chance of inheriting a abnormal gene from both parents and being affected by the condition.
- This also means that there is a 75% ( 3 in 4) chance that a child will not be affected by the condition. This chance remains the same in every pregnancy and is the same for boys or girls.
- There is also a 50% (2 in 4) chance that the child will inherit just one copy of the abnormal gene from a parent. If this happens, then they will be healthy carriers like their parents.
- Lastly, there is a 25% (1 in 4) chance that the child will inherit both normal copies of the gene. In this case the child will not have the condition, and will not be a carrier.
These possible outcomes occur randomly. The chance remains the same in every pregnancy and is the same for boys and girls.
Figure 3 illustrates autosomal recessive inheritance. The example below shows what happens when both dad and mum is a carrier of the abnormal gene, there is only a 25% chance that they will both pass down the abnormal gene to the baby, thus causing a genetic condition.
Figure 3. Wilson disease autosomal recessive inheritance pattern
Can I prevent Wilson disease?
No, you can’t prevent Wilson disease. If you have a first-degree relative—a parent, sibling, or child—with Wilson disease, talk with your doctor about testing you and other family members for Wilson disease. A doctor may be able to diagnose and begin treating Wilson disease before symptoms appear. Early diagnosis and treatment can reduce or prevent organ damage.
Wilson disease signs and symptoms
The symptoms of Wilson disease vary. Wilson disease is present at birth, but the symptoms don’t appear until the copper builds up in the liver, the brain, or other organs.
Some people do not have symptoms of Wilson disease before they are diagnosed with the disease and treated. If you do have symptoms, the symptoms may be related to your liver, nervous system and mental health, eyes, or other organs.
Liver symptoms
People with Wilson disease may develop symptoms of hepatitis, or inflammation of the liver. In some cases, people develop these symptoms when they have acute liver failure. These symptoms may include:
- feeling tired
- nausea and vomiting
- poor appetite
- pain over the liver, in the upper part of the abdomen
- darkening of the color of urine
- lightening of the color of stool
- yellowish tint to the whites of the eyes and skin, called jaundice
Some people with Wilson disease have symptoms only if they develop chronic liver disease and complications from cirrhosis. These symptoms may include:
- feeling tired or weak
- losing weight without trying
- bloating from a buildup of fluid in the abdomen, called ascites
- swelling of the lower legs, ankles, or feet, called edema
- itchy skin
- jaundice
Nervous system and mental health symptoms
People with Wilson disease may develop nervous system and mental health symptoms after copper builds up in their body. These symptoms are more common in adults but sometimes occur in children. Nervous system symptoms may include:
- problems with speech, swallowing, or physical coordination
- stiff muscles
- tremors or uncontrolled movements
Mental health symptoms may include:
- anxiety
- changes in mood, personality, or behavior
- depression
- psychosis
Eye symptoms
Many people with Wilson disease have Kayser-Fleischer rings, which are greenish, gold, or brownish rings around the edge of the corneas . A buildup of copper in the eyes causes Kayser-Fleischer rings. A doctor can see these rings during a special eye exam called a slit-lamp exam .
Among people who have nervous system symptoms of Wilson disease, more than 9 out of 10 have Kayser-Fleischer rings. However, among people who have only liver symptoms, 5 or 6 out of 10 have Kayser-Fleischer rings 20.
Other symptoms and health problems
Wilson disease can affect other parts of your body and cause symptoms or health problems, including:
- a type of anemia called hemolytic anemia
- bone and joint problems, such as arthritis or osteoporosis
- heart problems, such as cardiomyopathy
- kidney problems, such as renal tubular acidosis and kidney stones
Wilson disease liver
Wilson disease may lead to liver complications, but early diagnosis and treatment can lower your chances of developing them.
Acute liver failure
Wilson disease can cause acute liver failure, a condition in which your liver fails rapidly without warning. About 5 percent of people with Wilson disease have acute liver failure when they are first diagnosed 21. Acute liver failure most often requires a liver transplant.
Acute kidney failure and a type of anemia called hemolytic anemia often occur in people who have acute liver failure due to Wilson disease.
Cirrhosis
In cirrhosis, scar tissue replaces healthy liver tissue and prevents your liver from working normally. Scar tissue also partly blocks the flow of blood through the liver. As cirrhosis gets worse, the liver begins to fail.
Among people who are diagnosed with Wilson disease, 35 to 45 percent already have cirrhosis at the time of diagnosis 20
Cirrhosis increases your chance of getting liver cancer. However, doctors have found that liver cancer is less common in people who have cirrhosis due to Wilson disease than in people who have cirrhosis due to other causes.
Liver failure
Cirrhosis may eventually lead to liver failure. With liver failure, your liver is badly damaged and stops working. Liver failure is also called end-stage liver disease. This condition may require a liver transplant.
Wilson disease complications
People who have Wilson disease that is not treated or diagnosed early can have serious complications, such as:
- Cirrhosis—scarring of the liver. As liver cells try to make repairs to damage done by excess copper, scar tissue forms in the liver, making it more difficult for the liver to function.
- Kidney damage—as liver function decreases, the kidneys may be damaged. Wilson’s disease can damage the kidneys, leading to problems such as kidney stones and an abnormal number of amino acids excreted in the urine.
- Persistent nervous system problems when nervous system symptoms do not resolve. Tremors, involuntary muscle movements, clumsy gait and speech difficulties usually improve with treatment for Wilson’s disease. However, some people have persistent neurological difficulty despite treatment.
- Psychological problems. These might include personality changes, depression, irritability, bipolar disorder or psychosis.
- Liver cancer—hepatocellular carcinoma is a type of liver cancer that can occur in people with cirrhosis
- Liver failure—a condition in which the liver stops working properly. This can occur suddenly (acute liver failure), or it can develop slowly over years. A liver transplant might be a treatment option.
- Blood problems. These might include destruction of red blood cells (hemolysis) leading to anemia and jaundice.
- Death, if left untreated
Other complications may include:
- Anemia (hemolytic anemia is rare)
- Fatty liver
- Hepatitis
- Increased number of bone fractures
- Increased number of infections
- Injury caused by falls
- Jaundice
- Joint contractures or other deformity
- Loss of ability to care for self
- Loss of ability to function at work and home
- Loss of ability to interact with other people
- Loss of muscle mass (muscle atrophy)
- Psychological complications
- Side effects of penicillamine and other medicines used to treat the disorder
- Spleen problems
Liver failure and damage to the central nervous system (brain, spinal cord) are the most common and dangerous effects of the disorder. If Wilson disease is not caught and treated early, it can be fatal.
Wilson disease diagnosis
Doctors diagnose Wilson disease based on your medical and family history, a physical exam, an eye exam, and tests. However, diagnosing Wilson’s disease can be challenging because its signs and symptoms are often hard to tell from those of other liver diseases, such as hepatitis. Also, symptoms can evolve over time. Behavioral changes that come on gradually can be especially hard to link to Wilson disease. Data from different Wilson disease population groups show that diagnosing this rare disease remains difficult as the mean time between the first symptoms and the diagnosis usually exceeds 2 years 22, 23, 24.
Doctors rely on a combination of symptoms and test results to make the diagnosis. Tests and procedures used to diagnose Wilson’s disease include:
- Blood and urine tests. Blood tests can monitor your liver function and check the level of a protein that binds copper in the blood (ceruloplasmin) and the level of copper in your blood. Your doctor also might want to measure the amount of copper excreted in your urine during a 24-hour period.
- Eye exam. Using a microscope with a high-intensity light source (slit lamp), an ophthalmologist checks your eyes for Kayser-Fleischer rings, which is caused by excess copper in the eyes. Wilson’s disease also is associated with a type of cataract, called a sunflower cataract, that can be seen on an eye exam.
- Removing a sample of liver tissue for testing (biopsy). Your doctor inserts a thin needle through your skin, into your liver and draws a small sample of tissue. A laboratory tests the tissue for excess copper.
- Genetic testing. A blood test can identify the genetic mutations that cause Wilson’s disease. Knowing the mutations in your family allows doctors to screen siblings and begin treatment before symptoms arise.
There is no completely reliable test for Wilson disease, but levels of ceruloplasmin (a protein that carries copper in the bloodstream) and copper in the blood, as well as copper excreted in urine during a 24-hour period, are used to form an impression of the amount of copper in the body 2. The gold standard is a liver biopsy 2.
According to the most relevant diagnostic features a Wilson disease-specific score has been developed (Table 2) 25.
Table 1. Diagnosis of Wilson disease
| Parameter | Criteria to diagnose Wilson disease |
|---|---|
| Ceruloplasmin | <20 mg/dL |
| Serum copper | <70 mcg/dL (12 µmol/L) |
| Urinary copper | >60 mcg/day (>0.94 µmol/day) |
| Free copper | >15 mcg/dL (0.23 µmol/dL) |
| D-penicillamine 500 | Urinary copper increase to >1,600 mcg/day |
| Liver copper | >250 mcg/g dry weight |
| Kayser-Fleischer-rings | Sensitivity 50%; Specificity 95% |
| Genetic analysis | Sensitivity 90%; Specificity 100% |
Table 2. Scoring system developed at the 8th International Meeting on Wilson’s disease, Leipzig 2001
| Typical clinical symptoms and signs | Score |
|---|---|
| Kayser-Fleischer rings | • Present: 2 |
| • Absent: 0 | |
| Neurologic symptoms or typical imaging at brain magnetic resonance imaging | • Severe: 2 |
| • Mild: 1 | |
| • Absent: 0 | |
| Serum ceruloplasmin | • Normal (>0.2 g/L): 0 |
| • Slightly reduced (0.1–0.2 g/L): 1 | |
| • Drastically reduced (<0.1 g/L): 2 | |
| Coombs negative hemolytic anemia | • Present: 1 |
| • Absent: 0 | |
| Liver copper (in the absence of cholestasis) | • >5× upper limit of normal (>4 µmol/g): 2 |
| • 0.8-4 µmol/g: 1 | |
| • Normal (<0.8 µmol/g): −1 | |
| • Rhodamine-positive granules: +1 (if no quantitative liver copper is available) | |
| Urinary copper (in the absence of acute hepatitis) | • Normal: 0 |
| • 1–2× upper limit of normal: 1 | |
| • >2× upper limit of normal: 2 | |
| • Normal, but >5× upper limit of normal after one day challenge with 2×0.5 g D-penicillamine: 2 |
Medical and family history
Your doctor will ask about your family and personal medical history of Wilson disease and other conditions that could be causing your symptoms.
Physical exam
During a physical exam, your doctor will check for signs of liver damage such as:
- changes in the skin
- enlargement of the liver or spleen
- tenderness or swelling in the abdomen
- swelling in the lower legs, feet, or ankles, called edema
- yellowish color of the whites of the eyes
Eye exam
During a slit-lamp exam , a doctor will use a special light to look for Kayser-Fleischer rings in your eyes. The presence of Kayser Fleischer rings in the presence of neuropsychiatric symptoms is suggestive of Wilson disease.
Doctors typically use blood tests and a 24-hour urine collection test to diagnose Wilson disease. Doctors may also use a liver biopsy and imaging tests.
Blood tests
For a blood test, a health care professional will take a blood sample from you and send the sample to a lab.
Your doctor may order one or more blood tests, including tests that check amounts of:
- Ceruloplasmin, a protein that carries copper in the bloodstream. People with Wilson disease often have low ceruloplasmin levels, but not always. Ceruloplasmin is decreased in 85-90% of individuals. Ceruloplasmin level will be less than 20 mg/dL (normal 20 mg/dL to 40 mg/dL) 2.
- Copper. People with Wilson disease may have lower than normal blood copper levels. Acute liver failure due to Wilson disease may cause high blood copper levels.
- Liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST). People with Wilson disease may have abnormal ALT and AST levels 27, 28.
- Red blood cells to look for signs of anemia. Copper inhibits glycolytic enzymes causing a Coombs’ negative hemolytic anemia
- Genetic testing. A blood test can identify the genetic mutations that cause Wilson’s disease. Knowing the mutations in your family allows doctors to screen siblings and begin treatment before symptoms arise.
Doctors may order a blood test to check for the gene mutations that cause Wilson disease if other medical tests don’t confirm or rule out a diagnosis of the disease.
How to calculate serum free copper
The amount of serum free copper is the amount of copper circulating in the blood which is unbound by ceruloplasmin. This is the copper which is “free” to accumulate in the liver and other organs. Most reference labs do not automatically calculate the amount of serum free copper in a Wilson’s disease patient’s lab report. To calculate serum free copper, use the following formula:
(Total Serum Copper in μg/dl) – (Ceruloplasmin in mg/dl x 3) = Free Copper (Normal range is 5 – 15 μg/dl)
Example:
- Serum Copper of 22.2 (μg/dl) 22.2 (μg/dl)
- Ceruloplasmin of 4.7 (mg/dl) x 3 -14.1 (μg/dl)
- Free Copper =8.1(μg/dl)
OR - (Total Serum Copper in μmole/L x 63.55) – (Ceruloplasmin in mg/L x 3) = (Free Copper in μg/L ÷ 10) = Free Copper in μg/dl (Normal range is 5 – 15 μg/dl)
Example:
- Serum Copper of 6.3(μmole/L) x 63.55 400.3 (μg/L)
- Ceruloplasmin of 60 (mg/L) x 3 -180 (μg/L)
- Free Copper =220.3 (μg/L)
- Free Copper in (μg/L) ÷ 10 =20.3(μg/dl)
NOTE: There are often lab variations in the determination of ceruloplasmin and sometimes negative values are found. Some labs will not report the low serum coppers. If a patient has a very low ceruloplasmin, this formula may not be useable to determine free copper.
24-hour urine collection test
For 24 hours, you will collect your urine at home in a special container that is copper-free, provided by a health care professional. A health care professional will send the urine to a lab, which will check the amount of copper in your urine. Copper levels in the urine are often higher than normal (> 100 mcg/24 hour if symptomatic) in people who have Wilson disease 2.
Liver biopsy
If the results of blood and urine tests don’t confirm or rule out a diagnosis of Wilson disease, your doctor may order a liver biopsy. During a liver biopsy, a doctor will take small pieces of tissue from your liver. A pathologist will examine the tissue under a microscope to look for features of specific liver diseases, such as Wilson disease, and check for liver damage and cirrhosis. A piece of liver tissue will be sent to a lab, which will check the amount of copper in the tissue. A positive result is a copper level greater than 250 mcg/g of dry liver tissue 2.
Genetic testing
Genetic testing is specialized testing that is available from a limited number of reference or research laboratories. Genetic testing is used to diagnose Wilson disease and identify mutation(s) and identify carriers. Some prediction of disease severity can be established based upon the mutations present, but testing cannot determine the severity, complications, or organ involvement that will be experienced by a specific individual. Severity can vary significantly, even between family members with the same mutations.
- ATP7B gene: panels of the most prevalent mutations in a region or ethnic population may be performed.
- If the mutations have been identified in a person with Wilson disease, then the family members of that person can be tested for those specific mutations.
- Gene sequencing can be performed to examine the entire gene for mutations. This is the most thorough test.
- Linkage analysis: this requires blood from parents, siblings, and an affected family member. It compares genetic information present close to the ATP7B gene.
Imaging tests
In people who have nervous system symptoms, doctors may use imaging tests to check for signs of Wilson disease or other conditions in the brain. Doctors may use
- Magnetic resonance imaging (MRI) , which uses radio waves and magnets to produce detailed images of organs and soft tissues without using x-rays
- Computed tomography (CT) scan , which uses a combination of x-rays and computer technology to create images
Wilson disease treatment
Your doctor might recommend medications called chelating agents, which bind copper and then prompt your organs to release the copper into your bloodstream. The copper is then filtered by your kidneys and released into your urine. And zinc, which prevents the intestines from absorbing copper.
Treatment then focuses on preventing copper from building up again. For severe liver damage, a liver transplant might be necessary.
In many cases, treatment can improve or prevent symptoms and organ damage. Doctors may also recommend changing your diet to avoid foods that are high in copper.
People who have Wilson disease need lifelong treatment. Stopping treatment may cause acute liver failure. Doctors regularly perform blood and urine tests to check how the treatment is working.
Chelating agents
Penicillamine (Cupramine, Depen) and trientine (Syprine) are two chelating agents used to treat Wilson disease. These medicines remove copper from the body.
Penicillamine is more likely to cause side effects than trientine. Side effects of penicillamine may include fever, rash, kidney problems, or bone marrow problems. Penicillamine may also reduce the activity of vitamin B6, and doctors may recommend taking a vitamin B6 supplement along with penicillamine. In some cases, when people with nervous system symptoms begin taking chelating agents, their symptoms get worse.
When treatment begins, doctors gradually increase the dose of chelating agents. People take higher doses of chelating agents until the extra copper in the body has been removed. When Wilson disease symptoms have improved and tests show that copper is at safe levels, doctors may prescribe lower doses of chelating agents as maintenance treatment. Lifelong maintenance treatment prevents copper from building up again.
Chelating agents may interfere with wound healing, and doctors may prescribe a lower dose of chelating agents for people who are planning to have surgery.
Zinc
Zinc acetate (Galzin) prevents the intestines from absorbing copper from the food you eat. Doctors may prescribe zinc as a maintenance treatment, after chelating agents (penicillamine or trientine) have removed extra copper from the body. Doctors may also prescribe zinc for people who have Wilson disease but do not yet have symptoms. Zinc acetate might be used as primary therapy if you can’t take penicillamine or trientine. It is important that patients taking zinc acetate use the prescription version of zinc (Galzin) because nutritional supplements containing zinc may not be bioequivalent and may be ineffective. The most common side effect of zinc is stomach upset.
Treatment monitoring
Long term monitoring is vital with measurements of urinary copper, complete blood counts, free copper, renal and liver function at 4-8 week intervals 2. The eyes should also be examined. Most patients need lifelong chelation therapy with follow up 2. Patients need to avoid alcohol and all hepatotoxic medications 2.
Table 3. Wilson disease treatment monitoring
| Treatment | Treatment Goal | Initial Treatment | Undertreatment or Nonadherence | Overtreatment |
|---|---|---|---|---|
| Penicillamine | 250 – 500 mcg/day | >1000 | >500 | <150 |
| Trientine | 150 – 250 mcg/day | >1000 | >500 | <100 |
| Zinc | 30 – 120 mcg/day | 30 – 120 | >120 (or higher than baseline) | <30 |
Footnote: Based on 24 hour urine copper excretion in micrograms/day
[Source 29 ]Physical therapy
Physical therapy restores function for individuals who have neuromuscular or skeletal problems like dystonia, arthritis, osteoporosis, joint and muscle pain, or balance or coordination issues. Therapeutic treatment may include:
- Training in mobility, gait stability, posture and positioning
- Exercise programs to increase muscle function, coordination, balance and endurance
- Joint and soft tissue mobilization to increase range-of-motion
- Pain management
Occupational therapy
Occupational therapy assists individuals with adapting to their social and physical environment.
Therapists help improve function through:
- Education and training in areas such as dressing, bathing, eating and grooming
- Activities to help maintain memory, orientation and cognitive integration
- Adaptive techniques or equipment to overcome physical disabilities
- Upper body strengthening and fine motor coordination exercises
- Exercises to reduce the effects of arthritis or other conditions to maintain normal upper body joint movement
Speech and language therapy
Speech therapy addresses communication as well as difficulty with swallowing and eating.
Treatment plans are designed for individual needs, such as:
- Recovery of speech, language and memory skills
- Verbal and non-verbal communication, including programs for the hearing impaired
- Oral muscle functioning and strength required for speaking and swallowing
- Appropriate diet recommendations to minimize choking and aspiration of food into the lungs
Liver transplant
A liver transplant may be necessary in people when:
- cirrhosis leads to liver failure
- acute liver failure happens suddenly
- treatment is not effective
If your liver damage is severe, you might need a liver transplant. During a liver transplant, a surgeon removes your diseased liver and replaces it with a healthy liver from another person, called a donor. A successful transplant is a life-saving treatment for people with liver failure.
Most transplanted livers come from donors who have died. But in some cases a liver can come from a living donor, such as a family member. In that case, the surgeon removes your diseased liver and replaces it with a portion of the donor’s liver.
The success rate of liver transplantation in Wilson disease is high compared to other causes of acute hepatic failure 30. About 85 percent of transplanted livers are functioning after 1 year 31.
Most liver transplants are successful. Liver transplant surgery provides a cure for Wilson disease in most cases. Still a mortality rate of 10–20% within the first year after transplantation is too high to recommend this procedure for the general Wilson disease population, particularly because good drug therapy options are available which are associated with an unimpaired prognosis compared to the normal age adapted population 32. Challenging is the observation that with transplantation the liver disease is cured and does not require further copper controlling therapy 26. The question arises whether neurologic symptoms also disappear due to normalization of ceruloplasmin in blood, while the genetic defect in brain persists. The observations of neurologic improvement after liver transplantation are ambiguous 33.
Wilson disease in women who are pregnant
Pregnant women should continue treatment for Wilson disease throughout pregnancy. Doctors may prescribe a lower dose of chelating agents for women who are pregnant. Since the fetus needs a small amount of copper, lowering the dose may keep copper at safe levels without removing too much copper.
In most cases, doctors recommend that women continue to take the full dose of zinc during pregnancy. Experts recommend that women with Wilson disease do not breastfeed if they are taking chelating agents. Penicillamine is present in breast milk and can be harmful to a baby. Experts have little information about the safety of trientine and zinc in breast milk.
How do doctors treat the complications of Wilson disease?
If Wilson disease leads to cirrhosis, doctors can treat health problems and complications related to cirrhosis with medicines, surgery, and other medical procedures.
If Wilson disease causes acute liver failure or liver failure due to cirrhosis, you may need a liver transplant. A liver transplant cures Wilson disease in most cases.
Wilson disease diet
If you have Wilson’s disease, your doctor will likely recommend that you limit the amount of copper you consume in your diet. You might also want to have your tap water’s copper levels tested if you have copper pipes in your home. And be sure to avoid multivitamins that contain copper.
When you start treatment for Wilson disease, your doctor may recommend avoiding foods that are high in copper, such as:
- chocolate
- liver
- mushrooms
- nuts
- shellfish
After treatments have lowered your copper levels and you begin maintenance treatment, talk with your doctor about whether you can safely eat moderate amounts of these foods.
If your tap water comes from a well or runs through copper pipes, have the copper levels in your water checked. Water sitting in copper pipes may pick up copper. Run the water to flush the pipes before you drink the water or use it for cooking. You may need to use a water filter to remove copper from your tap water.
For safety reasons, talk with your doctor before using dietary supplements , such as vitamins, or any complementary or alternative medicines or medical practices. Some dietary supplements may contain copper.
Table 4. Copper content of selected foods
| Food | Micrograms (mcg) per serving |
|---|---|
| Beef, liver, pan fried (3 ounces) | 12400 |
| Oysters, eastern, wild, cooked, 3 ounces | 4850 |
| Baking chocolate, unsweetened, 1 ounce | 938 |
| Potatoes, cooked, flesh and skin, 1 medium potato | 675 |
| Mushrooms, shiitake, cooked, cut pieces, ½ cup | 650 |
| Cashew nuts, dry roasted, 1 ounce | 629 |
| Crab, Dungeness, cooked, 3 ounces | 624 |
| Sunflower seed kernels, toasted, ¼ cup | 615 |
| Turkey, giblets, simmered, 3 ounces | 588 |
| Chocolate, dark, 70%-85% cacao solids, 1 ounce | 501 |
| Tofu, raw, firm, ½ cup | 476 |
| Chickpeas, mature sees, ½ cup | 289 |
| Millet, cooked, 1 cup | 280 |
| Salmon, Atlantic, wild, cooked, 3 ounces | 273 |
| Pasta, whole wheat, cooked, 1 cup (not packed) | 263 |
| Avocado, raw, ½ cup | 219 |
| Figs, dried, ½ cup | 214 |
| Spinach, boiled, drained, ½ cup | 157 |
| Asparagus, cooked, drained, ½ cup | 149 |
| Seseame seeds, ¼ cup | 147 |
| Turkey, ground, cooked, 3 ounces | 128 |
| Cereals, Cream of Wheat, cooked with water, stove-top, 1 cup | 104 |
| Tomatoes, raw, chopped, ½ cup | 53 |
| Yogurt, Greek, plain, lowfat, 7-ounce container | 42 |
| Milk, nonfat, 1 cup | 27 |
| Apples, raw, with skin, ½ cup slices | 17 |
Wilson disease prognosis
The prognosis of untreated Wilson disease patients is poor with a median life expectancy of 40 years 8. Today with all of the available therapeutic regimens, a normal life expectancy of patients with Wilson disease is achievable 32. In addition clinical, neurological, and hematological symptoms improve during treatment to a significant extent 32. Exceptions are those indicating impaired liver function due to cirrhosis and complications of portal hypertension 35. Most challenging are neurologic disorders, which are only partial reversed and in rare cases may even deteriorate which may be due to inefficient access of present therapies to brain copper and iron or due to the mutated neuronal ATP7B 8.
Various scoring systems have been put forward for Wilson disease prognostication. Some of these incorporate levels of AST, bilirubin, and prothrombin time, etc. on presentation 36. Patients with a prognostic score of 7 or more should be referred for a liver transplant. Patients with this score are generally dead within 8 weeks without treatment. After liver transplant, the prognosis is good. A survival rate of 87% at 15 years has been reported 37.
Wilson’s disease life expectancy
The oldest newly diagnosed Wilson’s disease patient is over 70 years old.
The chronic liver injury in Wilson’s disease is caused by excess free copper, and the liver disease often stabilizes or even improves once the excess copper is treated with zinc acetate maintenance therapy. While some people do progress to need liver transplantation, others may actually see long-term improvement in their liver function over time. It is important to be attentive to issues such as immunizations for viral hepatitis, avoiding excess alcohol consumption, and treating complications of portal hypertension in order to give the liver its best chance to mend.
- Weiss KH. Wilson Disease. 1999 Oct 22 [Updated 2016 Jul 29]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1512[↩]
- Chaudhry HS, Anilkumar AC. Wilson Disease. [Updated 2023 Jan 21]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441990[↩][↩][↩][↩][↩][↩][↩][↩][↩][↩]
- Park S, Park JY, Kim GH, et al. Identification of novel ATP7B gene mutations and their functional roles in Korean patients with Wilson disease. Hum Mutat 2007;28:1108-13. 10.1002/humu.20574[↩][↩]
- Wu F, Wang J, Pu C, Qiao L, Jiang C. Wilson’s disease: a comprehensive review of the molecular mechanisms. Int J Mol Sci. 2015 Mar 20;16(3):6419-31. doi: 10.3390/ijms16036419[↩][↩]
- Xiong X, Wei H, Zhu Y, Zhou X, Zhao Z, Chen Q. Wilson disease in pregnancy: A case series. Medicine (Baltimore). 2023 Feb 17;102(7):e32968. doi: 10.1097/MD.0000000000032968[↩]
- Roberts EA, Schilsky ML; Division of Gastroenterology and Nutrition, Hospital for Sick Children, Toronto, Ontario, Canada. A practice guideline on Wilson disease. Hepatology. 2003 Jun;37(6):1475-92. doi: 10.1053/jhep.2003.50252. Erratum in: Hepatology. 2003 Aug;38(2):536.[↩]
- Sullivan CA, Chopdar A, Shun-Shin GA. Dense Kayser-Fleischer ring in asymptomatic Wilson’s disease (hepatolenticular degeneration). British Journal of Ophthalmology 2002;86:114. https://bjo.bmj.com/content/86/1/114.1[↩]
- Stremmel W, Weiskirchen R. Therapeutic strategies in Wilson disease: pathophysiology and mode of action. Ann Transl Med. 2021 Apr;9(8):732. doi: 10.21037/atm-20-3090[↩][↩][↩][↩][↩]
- Tanzi RE, Petrukhin K, Chernov I, et al. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet 1993;5:344-50.[↩]
- Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015 Jan;14(1):103-13. doi: 10.1016/S1474-4422(14)70190-5[↩][↩]
- Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, Klaffke S, Joyce CJ, Dhawan A, Hadzic N, Mieli-Vergani G, Kirk R, Elizabeth Allen K, Nicholl D, Wong S, Griffiths W, Smithson S, Giffin N, Taha A, Connolly S, Gillett GT, Tanner S, Bonham J, Sharrack B, Palotie A, Rattray M, Dalton A, Bandmann O. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013 May;136(Pt 5):1476-87. doi: 10.1093/brain/awt035[↩]
- Definition & Facts for Wilson Disease. https://www.niddk.nih.gov/health-information/liver-disease/wilson-disease/definition-facts[↩]
- Kanwar P, Kowdley KV. Metal storage disorders: Wilson disease and hemochromatosis. Med Clin North Am. 2014 Jan;98(1):87-102. doi: 10.1016/j.mcna.2013.09.008[↩]
- Kim JW, Kim JH, Seo JK, Ko JS, Chang JY, Yang HR, Kang KH. Genetically confirmed Wilson disease in a 9-month old boy with elevations of aminotransferases. World J Hepatol. 2013 Mar 27;5(3):156-9. doi: 10.4254/wjh.v5.i3.156[↩]
- Tanzi R.E., Petrukhin K., Chernov I., Pellequer J.L., Wasco W., Ross B., Romano D.M., Pavone L., Brzustowicz L.M. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. J. Nat. Genet. 1993;5:344–350. doi: 10.1038/ng1293-344[↩]
- Gouider-Khouja N. Wilson’s disease. Parkinsonism Relat. Disord. 2009;15:126–129. doi: 10.1016/S1353-8020(09)70798-9[↩]
- Terada K, Schilsky ML, Miura N, et al. ATP7B (WND) protein. Int J Biochem Cell Biol 1998;30:1063-7. 10.1016/S1357-2725(98)00073-9[↩]
- Parisi S, Polishchuk EV, Allocca S, et al. Characterization of the most frequent ATP7B mutation causing Wilson disease in hepatocytes from patient induced pluripotent stem cells. Sci Rep 2018;8:6247. 10.1038/s41598-018-24717-0[↩]
- Dong Q.Y., Wu Z.Y. Advance in the pathogenesis and treatment of Wilson disease. Transl. Neurodegener. 2012 doi: 10.1186/2047-9158-1-23[↩]
- Schilsky ML. Wilson disease: diagnostic tests. UpToDate. https://www.uptodate.com/contents/wilson-disease-diagnostic-tests.[↩][↩]
- Schilsky ML. Liver transplantation for Wilson’s disease. Annals of the New York Academy of Sciences. 2014:1315;45–49.[↩]
- Merle U, Schaefer M, Ferenci P, et al. Clinical presentation, diagnosis and long-term outcome of Wilson disease: a cohort study. Gut 2007;56:115-20. 10.1136/gut.2005.087262[↩]
- Bruha R, Marecek Z, Pospisilova L, et al. Long-term follow-up of Wilson disease: natural history, treatment, mutations analysis and phenotypic correlation. Liver Int 2011;31:83-91. 10.1111/j.1478-3231.2010.02354.x[↩]
- Litwin T, Gromadzka G, Członkowska A. Gender differences in Wilson disease. J Neurol Sci 2012;312:31-5. 10.1016/j.jns.2011.08.028[↩]
- Ferenci P, Stremmel W, Członkowska A, et al. Age and sex but not ATP7B genotype effectively influence the clinical phenotype of Wilson disease. Hepatology 2019;69:1464-76. 10.1002/hep.30280[↩]
- Schilsky ML, Scheinberg IH, Sternlieb I. Liver transplantation for Wilson’s disease: indications and outcome. Hepatology 1994;19:583-7. 10.1002/hep.1840190307[↩][↩]
- Nagral A, Sarma MS, Matthai J, et al. Wilson’s Disease: Clinical Practice Guidelines of the Indian National Association for Study of the Liver, the Indian Society of Pediatric Gastroenterology, Hepatology and Nutrition, and the Movement Disorders Society of India. J Clin Exp Hepatol. 2019 Jan-Feb;9(1):74-98. doi: 10.1016/j.jceh.2018.08.009. Epub 2018 Sep 3. Erratum in: J Clin Exp Hepatol. 2020 Jan-Feb;10(1):99.[↩]
- Capone K, Azzam RK. Wilson’s Disease: A Review for the General Pediatrician. Pediatr Ann. 2018 Nov 1;47(11):e440-e444. doi: 10.3928/19382359-20181026-01[↩]
- Monitoring Treatment. https://wilsondisease.org/medical-professionals/monitoring-treatment[↩]
- Weiss KH, Schäfer M, Gotthardt DN, et al. Outcome and development of symptoms after orthotopic liver transplantation for Wilson disease. Clin Transplant 2013;27:914-22. 10.1111/ctr.12259[↩]
- Organ Procurement and Transplantation Network (OPTN) and Scientific Registry of Transplant Recipients (SRTR). OPTN/SRTR 2011 annual data report. Department of Health and Human Services. Health Resources and Services Administration. Healthcare Systems Bureau, Division of Transplantation https://srtr.transplant.hrsa.gov/annual_reports/2011/[↩]
- Stremmel W, Meyerrose KW, Niederau C, et al. Wilson disease: clinical presentation, treatment and survival. Ann Intern Med 1991;115:720-6. 10.7326/0003-4819-115-9-720[↩][↩][↩]
- Lankarani KB, Malek-Hosseini SA, Nikeghbalian S, et al. Fourteen Years of Experience of Liver Transplantation for Wilson’s Disease; a Report on 107 Cases from Shiraz, Iran. PLoS One 2016;11:e0167890. 10.1371/journal.pone.0167890[↩]
- U.S. Department of Agriculture, Agricultural Research Service. FoodData Central. https://fdc.nal.usda.gov[↩]
- Beinhardt S, Leiss W, Stättermayer AF, et al. Long-term outcomes of patients with Wilson disease in a large Austrian cohort. Clin Gastroenterol Hepatol 2014;12:683-9. 10.1016/j.cgh.2013.09.025[↩]
- Nazer H, Ede RJ, Mowat AP, Williams R. Wilson’s disease: clinical presentation and use of prognostic index. Gut. 1986 Nov;27(11):1377-81. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1434058/pdf/gut00371-0105.pdf[↩]
- Guillaud O, Dumortier J, Sobesky R, Debray D, Wolf P, Vanlemmens C, Durand F, Calmus Y, Duvoux C, Dharancy S, Kamar N, Boudjema K, Bernard PH, Pageaux GP, Salamé E, Gugenheim J, Lachaux A, Habes D, Radenne S, Hardwigsen J, Chazouillères O, Trocello JM, Woimant F, Ichai P, Branchereau S, Soubrane O, Castaing D, Jacquemin E, Samuel D, Duclos-Vallée JC. Long term results of liver transplantation for Wilson’s disease: experience in France. J Hepatol. 2014 Mar;60(3):579-89. doi: 10.1016/j.jhep.2013.10.025[↩]





{kind=link}